
Abstract
Background
Activation of the epidermal growth factor receptor (EGFR) triggers downstream signaling pathways that regulate many cellular processes involved in tumour survival and growth. Gefitinib ('Iressa') is an orally active tyrosine kinase inhibitor (TKI) targeted to the ATP-binding domain of EGFR (HER1; erbB1).
Methods
In this study we have used a standardised ATP-based tumour chemosensitivity assay (ATP-TCA) to measure the activity of gefitinib alone or in combination with different cytotoxic drugs (cisplatin, gemcitabine, oxaliplatin and treosulfan) against a variety of solid tumours (n = 86), including breast, colorectal, oesophageal and ovarian cancer, carcinoma of unknown primary site, cutaneous and uveal melanoma, non-small cell lung cancer (NSCLC) and sarcoma. The IC50 and IC90 were calculated for each single agent or combination. To allow comparison between samples the IndexSUM was calculated based on the percentage tumour growth inhibition (TGI) at each test drug concentration (TDC). Gefitinib was tested at concentrations ranging from 0.0625–2 microM (TDC = 0.446 microg/ml). This study represents the first use of a TKI in the assay.
Results
There was heterogeneity in the degree of TGI observed when tumours were tested against single agent gefitinib. 7% (6/86) of tumours exhibited considerable inhibition, but most showed a more modest response resulting in a low TGI. The median IC50 value for single agent gefitinib in all tumours tested was 3.98 microM. Interestingly, gefitinib had both positive and negative effects when used in combination with different cytotoxics. In 59% (45/76) of tumours tested, the addition of gefitinib appeared to potentiate the effect of the cytotoxic agent or combination (of these, 11% (5/45) had a >50% decrease in their IndexSUM). In 38% of tumours (29/76), the TGI was decreased when the combination of gefitinib + cytotoxic was used in comparison to the cytotoxic alone. In the remaining 3% (2/76) there was no change observed.
Conclusion
The in vitro model suggests that gefitinib may have differential effects in response to concomitant cytotoxic chemotherapy with the agents tested during this study. The mechanism involved may relate to the effect of TKIs on growth rate versus their effect on the ability of the cell to survive the stimulus to apoptosis produced by chemotherapy.
Similar content being viewed by others


Background
The epidermal growth factor receptor (EGFR) is involved in many cellular processes including cell proliferation, motility, adhesion and angiogenesis via the activation of three pathways: phosphatidylinositol-3 kinase (PI3)/Akt pathway, the Jak/STAT pathway and the ras/raf pathway. EGFR is expressed or highly expressed in a variety of human tumours including non-small-cell lung cancer (NSCLC), breast, bladder, ovarian and head and neck [1] and is therefore a promising target for cancer therapy.
Gefitinib ('Iressa') is an EGFR-tyrosine kinase inhibitor (EGFR-TKI) that competitively inhibits binding of ATP at the ATP site on EGFR. It also displays remarkable selectivity for EGFR (IC50 = 0.033 microM) compared with other receptor tyrosine kinases (RTKs) that share sequence homology in the ATP binding domain [2]. In pre-clinical studies, gefitinib has demonstrated in vitro growth inhibition against a variety of human cell lines including NSCLC, ovarian, breast, colon and head and neck and is active in a range of xenograft models, including breast, colon and prostate [3]. Phase II trials with gefitinib monotherapy have produced encouraging results with clinically significant benefits observed, such as disease control rates at 250 mg/day gefitinib of 54% and 42% in IDEAL 1 and IDEAL 2, respectively [4, 5]. Results from Phase III trials investigating gefitinib in combination with cisplatin and gemcitabine (INTACT 1) [6] and gefitinib in combination with paclitaxel and carboplatin (INTACT 2) [7] in NSCLC concluded there was no added benefit in patients receiving chemotherapy plus gefitinib; however the tolerability of gefitinib was confirmed.
At present, there is conflicting evidence relating the activity of gefitinib directly to the levels of EGFR expression. One group found that the concentration of gefitinib required to inhibit ligand-independent growth by 50% (IC50) in four bladder cancer cell lines ranged from 1.8–9.7 microM and correlated with EGFR protein and transcript level [8]. However, another study using human tumour xenografts found that gefitinib caused growth inhibition of tumours and enhancement of the activity of a number of cytotoxic drugs, but neither was dependent on high levels of EGFR expression [9]. Moreover, no consistent association was demonstrated between EGFR expression and clinical outcome in IDEAL 1 and 2 [10]. Alternative explanations for the activity of gefitinib in systems where EGFR is not over expressed include inhibition of EGFR pathway activation mediated by increased levels of receptor ligands e.g. epidermal growth factor (EGF) and transforming growth factor-alpha (TGF-alpha); heterodimerization with HER2 and cross talk with heterologous receptors; and EGFR mutations yielding a constitutively active receptor that is not down-regulated by endocytosis [11]. There is evidence that the ras/raf pathway mediates proliferation [12], whereas the PI3/Akt pathway is essential for cell survival and may be constitutively activated in many tumours by loss of PTEN [13].
We have previously shown that the ATP-based tumour chemosensitivity assay (ATP-TCA) can be used to measure the effects of cytotoxic agents and antibodies against human tumour-derived cells, and that this matches clinical outcome in a number of tumour types [14, 15]. Use of the assay to direct choice of chemotherapy has been shown to improve response rate and progression-free survival in ovarian cancer [16, 17] and a fully randomized trial of assay-directed versus physician's choice of chemotherapy for platinum-resistant ovarian cancer is in progress [18]. The assay system has been used to assist the development of a number of new agents and combinations [19, 20], but this represents the first use of a TKI in the assay.
EGF and TGF-alpha, ligands of EGFR, act as survival factors for many cells as well as growth factors. As many cytotoxic agents induce apoptosis, gefitinib may be able to potentiate their effects by reducing survival stimuli. The current pilot study was undertaken to assess the effect of gefitinib in combination with existing chemotherapeutic agents (cisplatin, gemcitabine, oxaliplatin, treosulfan) against a wide range of tumour types.
Methods
Tumours
A total of 86 tumours (57 females:29 males) were tested in this study, with a median age of 59 years (range 21–90). The samples tested consisted of the following tumour types; breast adenocarcinoma (n = 8), colorectal carcinoma (n = 18), cutaneous melanoma (n = 7), NSCLC (n = 1), oesophageal adenocarcinoma (n = 4), ovarian carcinoma (n = 26), sarcoma (n = 2), squamous cell carcinoma (n = 2), sweat gland carcinoma (n = 1), uveal melanoma (n = 12) and carcinoma of unknown primary site (n = 5). The 26 ovarian carcinomas were all recurrent stage 3/4 cancers and 25/26 were pre-treated (11 with carboplatin and 14 with carboplatin + paclitaxel). Of the remaining samples, 10/86 had been treated with a variety of chemotherapy regimens and some patients had more than one treatment; epirubicin + cisplatin + 5-Fluorouracil (5-FU) (n = 3), epirubicin + cyclophosphamide (4-HC) (n = 1), 4-HC+methotrexate+5-FU (CMF) (n = 2), cisplatin + vinorelbine (n = 1), mitomycin C + 5-FU (n = 1), mitoxantrone + paclitaxel (n = 1), chlorambucil (n = 1), 4-HC (n = 1) and irinotecan (n = 1). The remaining 51 patients had no previous treatment. In each case only tumour material not required for diagnosis was sent for ATP-TCA, and in all cases consent had been obtained and permission had been granted by the local ethics committee.
ATP-TCA
The ATP-TCA was performed as previously published [14, 21]. Solid tumour or ascites samples were transported to the laboratory in transport medium, consisting of Dulbecco's Eagles Media (DMEM) (Sigma, UK; D6171). Solid samples were dissected under sterile conditions in a BioQ Microfuge Class II Hood and placed into a 0.75 mg/ml collagenase solution (Sigma, UK; C8051) for enzymatic dissociation overnight. Following dissociation, the single celled suspension or ascites sample was washed using DMEM supplemented with 1 M HEPES, (Sigma, UK; H0887), 100 IU/mL penicillin, 10 mg/mL streptomycin (Sigma, UK; P0781) and 10 mg/mL gentamicin (Sigma, UK; G1272). The final cell suspension was then plated in 96-well polypropylene plates (Corning Life Sciences, High Wycombe, UK) at 20,000 (solid sample) or 10,000 (ascites sample) cells/well in a serum-free complete assay medium (CAM, DCS Innovative Diagnostik Systeme, Hamburg, Germany). Drugs were added to triplicate wells at serial dilutions corresponding to 200–6.25% of a test drug concentration (TDC) estimated from pharmacokinetic data, which included the degree of protein binding. Two controls were included in each plate: one with no drug and consisting of media only (MO), and a maximum inhibitor (MI) control which killed all cells present. The plates were incubated for 6 days at 37°C with 5% CO2. At the end of the incubation period, remaining cells were lysed by addition of an ATP extraction reagent (DCS Innovative Diagnostik Systeme). An aliquot of the lysate from each well was added to the corresponding wells of a white 96 well microplate (Thermo Life Sciences, Basingstoke, UK), followed by addition of luciferin-luciferase reagent. The light output corresponding to the level of ATP present was measured in a luminometer (MPLX, Berthold Diagnostic Systems, Hamburg, Germany). These data were transferred automatically to an Excel spreadsheet where the % inhibition achieved at each concentration tested was calculated using the equation; 1-(test-MI)/(MO-MI) × 100. Several parameters of efficacy can be calculated e.g. IC50 and IC90, however previous ATP-TCA studies have found that a natural logarithmic sum index (IndexSUM) calculated by direct addition of the percentage survival at each concentration tested (Index = 600-Σb3;%Inhibition6.25...200) provides a better indication of sensitivity or resistance to different drugs in different tumour types [22]. The total inhibition of growth resulted in an index of 0, and no inhibition of growth at any concentrations produces an index of 600 [23]. Area under the concentration-inhibition curve (IndexAUC) was calculated from the data using the trapezoidal rule.
Data Analysis
The results were entered into an Access 2000 database for further analysis. Statistical tests were performed using non-parametric methods.
Drugs
The cytotoxic drugs used in the assay were obtained as vials for injection and made up according to manufacturers' instructions. Gemcitabine, oxaliplatin and treosulfan were stored in aliquots at -20°C, while cisplatin was stored at room temperature. Table 1 shows the 100% TDC for each of the drugs used. Drug combinations were tested by combining single agents. The EGFR-TKI, gefitinib (kindly provided by AstraZeneca) was tested at concentrations ranging from 0.06–2 microM (100% TDC = 0.99 microM).
Immunohistochemistry
Tissue was available for EGFR immunohistochemical staining in 31/86 (36%) cases comprising of 4 breast carcinomas, 12 colon carcinomas, 2 oesophageal carcinomas, 2 ovarian carcinomas, 1 sarcoma, 4 skin melanomas, 5 uveal melanomas and 1 carcinoma of unknown primary site. Paraffin embedded sections of 4 μm thick were dewaxed and rehydrated in preparation for immunohistochemical staining. Endogenous peroxidase was blocked using 3% hydrogen peroxide in methanol. The sections were pretreated with 0.1% Trypsin (CaCl2/Tris buffer pH8.0) for 10 minutes at 37°C for antigen retrieval. Immunohistochemical studies were performed according to manufacturer's instructions of the Vectastain Universal ABC-AP kit (Vector Laboratories, Burlingame, California, U.S.A), which uses an avidin-biotin complex method and Vector red as the chromogen. Monoclonal antibody for EGFR, Clone E30 (Dakocytomation, Cambridgeshire, U.K) was used at a dilution of 1:20 and incubated with sections for 18 hours at 4°C. Positive (squamous cell carcinoma tissue) and negative controls were included in each staining procedure. Samples were assessed by a pathologist using the H-score. Intensity was graded on a scale ranging between 0, 1+, 2+ or 3+, (where 1+ equals weak staining, 2+ equals moderate and 3+ equals intense) and the proportion of cells stained at the highest intensity. The two values were then multiplied together to give the final value.
The same tissue available for EGFR staining was also available for pAkt staining. Paraffin embedded sections of 4 μm thick were dewaxed and rehydrated in preparation for immunohistochemical staining. Endogenous peroxidase was blocked using 3% hydrogen peroxide in methanol. The sections were pretreated with 0.1 M citrate buffer in a pressure cooker for 2.5 minutes for antigen retrieval. Immunohistochemical studies were performed according to manufacturer's instructions of the Vectastain Universal ABC-AP kit (Vector Laboratories, Burlingame, California, U.S.A), which uses an avidin-biotin complex method and Fuchsin as the chromogen. Phospho-Akt, Ser473 (#9277 L, Cell Signalling, MA. USA) was used at a dilution of 1:50 and incubated with sections for 18 hours at 4°C. Positive (prostate cancer tissue) and negative controls were included in each staining procedure. Samples were assessed as described previously.
Results
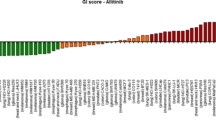
Gefitinib showed low inhibition (IndexSUM >300) across the range of concentrations tested in the ATP-TCA, with little evidence of increasing inhibition with increasing drug concentration. 7% (6/86) of tumours exhibited considerable inhibition (>50% inhibition at 100% TDC), but most showed a more modest response resulting in a low maximum percentage inhibition (Figure 1). The estimated median IC50 and IC90 value for single agent gefitinib in all tumours tested was 3.98 microM (<0.1–69.9 microM) and 6.45 microM (2.4–125.9 microM) respectively. The median IC50 for individual tumour types tested is shown in Table 2.

Median effect of gefitinib on tumour-derived cells compared with a sensitive and non-sensitive colorectal tumour. Error bars show 25th and 75th inter-quartile range.
There was heterogeneity in the degree of inhibition observed when tumours were tested against single agent gefitinib (Figure 2). To compare between tumours, an IndexSUM of <300 corresponding to 50% inhibition across the range of concentrations tested was used to compare results. On this basis, single agent gefitinib was effective against 5% (4/86) of samples, comprising 1 colorectal tumour, 1 ovarian tumour, 1 uveal melanoma and 1 unknown primary carcinoma. In 88% (76/86) of samples there was sufficient material to test gefitinib in combination with different cytotoxics.

Frequency histogram showing heterogeneity of the IndexSUM for gefitinib alone in all tumours tested (n = 86).
Table 3 shows the median results for single-agent cytotoxics tested compared to results when tested in combination with gefitinib. In samples tested with gefitinib in combination with cisplatin (n = 6) only 33% (2/6) showed increased sensitivity (i.e. a decrease in their IndexSUM), compared to when cisplatin was used alone. The remaining 67% (4/6) showed increased resistance (i.e. an increase in their IndexSUM). This compares with gefitinib in combination with oxaliplatin (n = 10) where 90% (9/10) of samples showed an increase in sensitivity with the combination, with 1 sample showing a >50% decrease in the IndexSUM. When gefitinib was combined with gemcitabine (n = 2), both samples showed an increase in their sensitivity.
Of the tumours tested with treosulfan + gefitinib, 38% (13/34) were of ovarian origin. Of these, 62% (8/13) showed potentiation, with 1 sample showing a >50% decrease in IndexSUM. 31% (4/13) showed increased resistance with the combination in comparison with treosulfan alone and 1 sample showed no change (Figure 3). Of the remaining samples tested with gefitinib + treosulfan, 57% (12/21) showed an increase in sensitivity, 38% (8/21) showed an increase in their resistance and one sample showed no change. Figures 4a and 4b show differential effects of gefitinib in combination with treosulfan in cells derived from a skin melanoma sample (Figure 4a) and an ovarian carcinoma sample (Figure 4b). When gefitinib was tested in combination with treosulfan + gemcitabine (n = 24), 54% (13/24) showed an increase in sensitivity, with 3 samples showing a >50% decrease in their IndexSUM and 46% (11/24) showed an increase in resistance.

The effect of gefitinib on tumour-derived cells from recurrent ovarian cancer (n = 13). Legend Some tumours show increased sensitivity (lower IndexSUM), while others show enhanced resistance (higher IndexSUM)

Tumour growth inhibition by gefitinib vs. treosulfan + gefitinib in 2 different tumour types. Legend Figure 4a shows the positive effect of combining treosulfan plus gefitinib in cells derived from a skin melanoma compared with figure 4b in which the combination of treosulfan plus gefitinib has a negative effect in cells derived from an ovarian tumour.
In summary, the addition of gefitinib appeared to potentiate the effect of the cytotoxic agent or combination in 59% (45/76) of tumours tested; of these 11% (5/45) had a >50% decrease in their IndexSUM. In 38% of tumours (29/76), the combination of gefitinib + cytotoxic caused the IndexSUM to increase thereby increasing resistance. In the remaining 3% (2/76) there was no change observed.
Immunostaining for EGFR was positive in 32% (10/31) of samples comprising of 1 breast carcinomas, 5 colon carcinomas, 1 ovarian carcinoma, 1 sarcoma, 1 skin melanoma and 1 carcinoma of unknown primary site. Immunostaining for pAkt was positive in 81% (25/31) of samples comprising of 3 breast carcinomas, 10 colon carcinomas, 2 oesophageal carcinomas, 2 ovarian carcinomas, 4 skin melanomas, 3 uveal melanomas and 1 carcinoma of unknown primary site. Of the positive samples, 8 were positive for both antibodies (comprising 1 breast carcinoma, 4 colon carcinomas, 1 ovarian carcinoma, 1 skin melanoma and 1 carcinoma of unknown primary site). In 74% (23/31) of samples that were stained for EGFR and pAkt, there was an IC50, IC90 and IndexSUM value available for comparison. In all cases tested there was no relationship with gefitinib activity and EGFR or pAkt staining.
Discussion
This is the first study in which a TKI has been successfully tested in the ATP-TCA. ATP-TCA has potential to assist drug development for TKIs and possibly to direct therapy for individual patients. It represents one possible answer to the need for predictive oncology testing of these agents, and could be performed alongside clinical trials to obtain correlation data with outcome in patients treated with gefitinib. However, it is difficult to ascertain whether these were specific or non-specific effects of gefitinib and whether similar outcomes would be seen in the clinical setting. This would need to be determined before using this test for routine screening. Gefitinib showed activity in the assay and even though cytotoxic effects were not expected, in some cases the diminution in ATP levels suggests that these may occur. In general, flat concentration – activity curves were observed which are consistent with a cytostatic rather than a cytotoxic effect. Gefitinib alone showed activity in lung, ovarian and colon carcinomas. These results were consistent with previous findings in cell lines [24].
When gefitinib was tested in combination with a limited number of cytotoxic drugs, increases and decreases in the activity of the cytotoxic agent were observed. For example, gefitinib in combination with cisplatin caused 67% of samples to have a decrease in the activity of the cytotoxic. This compares with gefitinib in combination with a second platinum-containing agent, oxaliplatin, where 91% of samples showed an increase in the activity of the cytotoxic. However, it should be noted that oxaliplatin was virtually ineffective against the cells tested and this is therefore likely to reflect the effect of the gefitinib alone (Figure 5). Decreased activity of cytotoxic agents when these were combined with gefitinib was seen in 4 samples with cisplatin, 13 with treosulfan, 1 with oxaliplatin and 11 with treosulfan + gemcitabine. This could be detrimental to patients. It is similar to the effect of tamoxifen treatment on the success of breast cancer chemotherapy [25].

Tumour growth inhibition by gefitinib vs. gefitinib + oxaliplatin in a NSCLC. Legend The combination of oxaliplatin + gefitinib has a positive effect compared with single agent oxaliplatin which is inactive.
Although there was heterogeneity in the response of tumours to single agent gefitinib, there was no relationship between immunostaining for EGFR and gefitinib activity, consistent with other published studies [26]. Sirotnak et al. [9] showed that gefitinib caused growth inhibition in human tumour xenografts that was not dependent on high levels of EGFR expression. However, EGFR activation leads to activation of at least three separate second messenger cascades. While the ras/raf pathway may mediate the proliferative effects, survival signals are thought be mediated by the PI3/Akt pathway. As cells have to die in the ATP-TCA to register increased inhibition, sensitivity might be related to the degree of activation of the Akt pathway by other mechanisms. Sensitivity to gefitinib and other non-TKI EGFR inhibitors might therefore be related to pathway activation assessed by detection of pAkt, rather than the levels of EGFR expression. However, this study has not found any such relationship and, when EGFR staining was compared to pAkt staining there was no correlation between EGFR levels to pAkt activity. A similar observation was made by Campiglio et al., [27] whose data suggested that neither MAPK nor pAkt were reliable markers of gefitinib activity. It should be noted that many receptors lead to Akt activation and that constitutive activation of the PI3/Akt pathway may be the result of PTEN inactivation.
Of the 4 samples that had an IndexSUM of <300 and the 6 samples that demonstrated >50% inhibition at 100% TDC when tested with single agent gefitinib, 2 samples (a uveal melanoma and an unknown primary) had material available for immunohistochemical staining with EGFR and pAkt. The uveal melanoma was negative for EGFR and positive for pAkt compared to the unknown primary, which was positive for both EGFR and pAkt. However, there were samples with similar IHC results that did not show sensitivity to gefitinib. As the EGFR (HER1) dimerizes with the other HER molecules and mediates greater activity as a heterodimer, it is likely that the expression of these molecules is also important to the activity of gefitinib [12]. Sensitivity to gefitinib is therefore likely to be the end result of a complex series of interactions within the cell.
Conclusion
In this study we have found that gefitinib in combination with different cytotoxic agents (cisplatin; gemcitabine; oxaliplatin; treosulfan and treosulfan + gemcitabine) is a double-edged sword: their effect on growth rate may make some tumours more resistant to concomitant cytotoxic chemotherapy, while their effect on cytokine-mediated cell survival (anti-apoptotic) mechanisms may potentiate sensitivity to the same drugs in tumours from other individuals.
References
Woodburn JR: The Epidermal Growth Factor Receptor and Its Inhibition in Cancer Therapy. Pharmacology & Therapeutics. 1999, 82: 241-250. 10.1016/S0163-7258(98)00045-X.
Wakeling AE, Guy SP, Woodburn JR, Ashton SE, Curry BJ, Barker AJ, Gibson KH: ZD1839 (Iressa): An Orally Active Inhibitor of Epidermal Growth Factor Signaling with Potential for Cancer Therapy. Cancer Research. 2002, 62: 5749-5754.
Ranson M, Hammond LA, Ferry D, Kris M, Tullo A, Murray PI, Miller V, Averbuch S, Ochs J, Morris C, Feyereislova A, Swaisland H, Rowinsky EK: ZD1839, a selective oral epidermal growth factor receptor-tyrosine kinase inhibitor, is well tolerated and active in patients with solid, malignant tumors: results of a phase I trial. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology. 2002, 20: 2240-2250.
Fukuoka M, Yano S, Giaccone G, Tamura T, Nakagawa K, Douillard JY, Nishiwaki Y, Vansteenkiste J, Kudoh S, Rischin D, Eek R, Horai T, Noda K, Takata I, Smit E, Averbuch S, Macleod A, Feyereislova A, Dong RP, Baselga J: Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer. Journal of Clinical Oncology. 2003, 21: 2237-2246. 10.1200/JCO.2003.10.038.
Kris MG, Natale RB, Herbst RS, Lynch TJ, Prager D, Belani CP, Schiller JH, Kelly K, Spiridonidis H, Sandler A, Albain KS, Cella D, Wolf MK, Averbuch SD, Ochs JJ, Kay AC: Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA. 2003, 290: 2149-2158. 10.1001/jama.290.16.2149.
Giaccone G, Herbst RS, Manegold C, Scagliotti G, Rosell R, Miller V, Natale RB, Schiller JH, Von Pawel J, Pluzanska A, Gatzemeier U, Grous J, Ochs JS, Averbuch SD, Wolf MK, Rennie P, Fandi A, Johnson DH: Gefitinib in combination with gemcitabine and cisplatin in advanced non-small-cell lung cancer: a phase III trial – INTACT 1. J Clin Oncol. 2004, 22: 777-784. 10.1200/JCO.2004.08.001.
Herbst RS, Giaccone G, Schiller JH, Natale RB, Miller V, Manegold C, Scagliotti G, Rosell R, Oliff I, Reeves JA, Wolf MK, Krebs AD, Averbuch SD, Ochs JS, Grous J, Fandi A, Johnson DH: Gefitinib in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: a phase III trial – INTACT 2. J Clin Oncol. 2004, 22: 785-794. 10.1200/JCO.2004.07.215.
Meye A, Fiedler U, Kunert K: Growth Inhibitory effects of ZD1839 ('Iressa') on human bladder cancer cell lines. Proc Am Assoc Cancer Research. 2001, 43: A3901-
Sirotnak FM, Zakowski MF, Miller VA, Scher HI, Kris MG: Efficacy of Cytotoxic Agents against Human Tumor Xenografts Is Markedly Enhanced By Coadministration of ZD1839 (Iressa), an Inhibitor of EGFR Tyrosine Kinase. Clin Cancer Res. 2000, 6: 4885-4892.
Bailey R, Kris M, Wolf M, Kay A, Averbuch S, Askaa J, Janas M, Schmidt K, Fukuoka M: Tumor EGFR membrane staining is not clinically relevant for predicting response in patients receiving gefitinib ('Iressa', ZD1839) monotherapy for pretreated advanced non-small-cell lung cancer: IDEAL 1 and 2 [abstract]. Proc Am Assoc Cancer Res. 2003, 44: LB-170.
Arteaga CL: Epidermal growth factor receptor dependence in human tumors: more than just expression?. The Oncologist. 2002, 7: 31-39.
Kolch W: Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem J. 2000, 351: 289-305. 10.1042/0264-6021:3510289.
Myers MP, Pass I, Batty IH, Van der Kaay J, Stolarov JP, Hemmings BA, Wigler MH, Downes CP, Tonks NK: The lipid phosphatase activity of PTEN is critical for its tumor suppressor function. Proc Natl Acad Sci U S A. 1998, 95: 13513-13518. 10.1073/pnas.95.23.13513.
Andreotti PE, Cree IA, Kurbacher CM, Hartmann DM, Linder D, Harel G, Gleiberman I, Caruso PA, Ricks SH, Untch M, Sartori C, Bruckner H: Chemosensitivity testing of human tumors using a microplate adenosine triphosphate luminescence assay: clinical correlation for cisplatin resistance of ovarian carcinoma. Cancer Research. 1995, 55: 5276-5282.
Cree IA, Kurbacher CM, Untch M, Sutherland LA, Hunter EM, Subedi AM, James EA, Dewar JA, Preece PE, Andreotti PE, Bruckner HW: Correlation of the clinical response to chemotherapy in breast cancer with ex vivo chemosensitivity. Anti-Cancer Drugs. 1996, 7: 630-635.
Kurbacher CM, Cree IA, Bruckner HW, Brenne U, Kurbacher JA, Muller K, Ackermann T, Gilster TJ, Wilhelm LM, Engel H, Mallmann PK, Andreotti PE: Use of an ex vivo ATP luminescence assay to direct chemotherapy for recurrent ovarian cancer. Anti-Cancer Drugs. 1998, 9: 51-57.
Sharma S, Neale MH, Di Nicolantonio F, Knight LA, Whitehouse PA, Mercer SJ, Higgins BR, Lamont A, Osborne R, Hindley AC, Kurbacher CM, Cree IA: Outcome of ATP-based tumor chemosensitivity assay directed chemotherapy in heavily pre-treated recurrent ovarian carcinoma. BMC Cancer. 2003, 3: 19-10.1186/1471-2407-3-19.
Kurbacher CM, Untch M, Cree IA: A randomised trial of chemotherapy directed by a tumor chemosensitivity assay versus physician's choice in patients with recurrent platinum-resistant ovarian adenocarcinoma. The Lancet. 1997
Neale MH, Myatt N, Cree IA, Kurbacher CM, Foss AJ, Hungerford JL, Plowman PN: Combination chemotherapy for choroidal melanoma: ex vivo sensitivity to treosulfan with gemcitabine or cytosine arabinoside. British Journal of Cancer. 1999, 79: 1487-1493. 10.1038/sj.bjc.6690237.
Di Nicolantonio F, Neale MH, Knight LA, Lamont A, Skailes GE, Osborne RJ, Allerton R, Kurbacher CM, Cree IA: Use of an ATP-based chemosensitivity assay to design new combinations of high-concentration doxorubicin with other drugs for recurrent ovarian cancer. Anti-Cancer Drugs. 2002, 13: 625-630. 10.1097/00001813-200207000-00009.
Hunter EM, Sutherland LA, Cree IA, Dewar JA, Preece PE, Wood RA, Linder D, Andreotti PE: Heterogeneity of chemosensitivity in human breast carcinoma: use of an adenosine triphosphate (ATP) chemiluminescence assay. European Journal of Surgical Oncology: the Journal of the European Society of Surgical Oncology and the British Association of Surgical Oncology. 1993, 19: 242-249.
Konecny G, Crohns C, Pegram M, Felber M, Lude S, Kurbacher C, Cree IA, Hepp H, Untch M: Correlation of Drug Response with the ATP Tumor chemosensitivity Assay in Primary FIGO Stage III Ovarian Cancer. Gynecologic Oncology. 2000, 77: 258-263. 10.1006/gyno.2000.5728.
Neale MH, Myatt NE, Khoury GG, Weaver P, Lamont A, Hungerford JL, Kurbacher CM, Hall P, Corrie PG, Cree IA: Comparison of the ex vivo chemosensitivity of uveal and cutaneous melanoma. Melanoma Research. 2001, 11: 601-609. 10.1097/00008390-200112000-00006.
Ranson M: ZD1839 (Iressa(TM)): For More Than Just Non-Small Cell Lung Cancer. The Oncologist. 2002, 7: 16-24.
Gelber RD, Cole BF, Goldhirsch A, Rose C, Fisher B, Osborne CK, Boccardo F, Gray R, Gordon NH, Bengtsson NO, Sevelda P: Adjuvant chemotherapy plus tamoxifen compared with tamoxifen alone for postmenopausal breast cancer: meta-analysis of quality-adjusted survival. The Lancet. 1996, 347: 1066-1071. 10.1016/S0140-6736(96)90277-9.
Cappuzzo F, Gregorc V, Rossi E, Cancellieri A, Magrini E, Paties CT, Ceresoli G, Lombardo L, Bartolini S, Calandri C, de Rosa M, Villa E, Crino L: Gefitinib in Pretreated Non-Small-Cell Lung Cancer (NSCLC): Analysis of Efficacy and Correlation With HER2 and Epidermal Growth Factor Receptor Expression in Locally Advanced or Metastatic NSCLC. J Clin Oncol. 2003, 21: 2658-2663. 10.1200/JCO.2003.01.039.
Campiglio M, Locatelli A, Olgiati C, Normanno N, Somenzi G, Vigano L, Fumagalli M, Menard S, Gianni L: Inhibition of proliferation and induction of apoptosis in breast cancer cells by the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor ZD1839 ('Iressa') is independent of EGFR expression level. J Cell Physiol. 2004, 198: 259-268. 10.1002/jcp.10411.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/4/83/prepub
Acknowledgements
We are grateful to AstraZeneca for providing the supply of gefitinib used in this study. We wish to thank the patients, oncologists and surgeons who provided tumour tissue for this study.
This study was funded by Cantech Ltd.
'Iressa' is a trademark of the AstraZeneca group of companies
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
IAC is director of Cantech Ltd.
Authors' contributions
LAK drafted the manuscript and carried out ATP-TCA assays. FDN, PW, SM, SS and SG also carried out ATP-TCA assays. PJ carried out the immunohistochemical work and IAC conceived the study and participated in its co-ordination.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Knight, L.A., Di Nicolantonio, F., Whitehouse, P. et al. The in vitroeffect of gefitinib ('Iressa') alone and in combination with cytotoxic chemotherapy on human solid tumours. BMC Cancer 4, 83 (2004). https://doi.org/10.1186/1471-2407-4-83
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-4-83