
Abstract
Background
About 10-15% of adult gastrointestinal stromal tumors (GIST) and the vast majority of pediatric GIST do not harbour KIT or platelet-derived growth factor receptor alpha (PDGFRA) mutations (J Clin Oncol 22:3813–3825, 2004; Hematol Oncol Clin North Am 23:15–34, 2009). The molecular biology of these GIST, originally defined as KIT/PDGFRA wild-type (WT), is complex due to the existence of different subgroups with distinct molecular hallmarks, including defects in the succinate dehydrogenase (SDH) complex and mutations of neurofibromatosis type 1 (NF1), BRAF, or KRAS genes (RAS-pathway or RAS-P).
In this extremely heterogeneous landscape, the clinical profile and molecular abnormalities of the small subgroup of WT GIST suitably referred to as quadruple wild-type GIST (quadruple WT or KIT WT/PDGFRA WT/SDH WT/RAS-P WT) remains undefined. The aim of this study is to investigate the genomic profile of KIT WT/PDGFRA WT/SDH WT/RAS-P WT GIST, by using a massively parallel sequencing and microarray approach, and compare it with the genomic profile of other GIST subtypes.
Methods
We performed a whole genome analysis using a massively parallel sequencing approach on a total of 16 GIST cases (2 KIT WT/PDGFRA WT/SDH WT and SDHB IHC+/SDHA IHC+, 2 KIT WT/PDGFRA WT/SDHA mut and SDHB IHC-/SDHA IHC- and 12 cases of KIT mut or PDGFRA mut GIST). To confirm and extend the results, whole-genome gene expression analysis by microarray was performed on 9 out 16 patients analyzed by RNAseq and an additional 20 GIST patients (1 KIT WT/PDGFRA WT SDHA mut GIST and 19 KIT mut or PDGFRA mut GIST). The most impressive data were validated by quantitave PCR and Western Blot analysis.
Results
We found that both cases of quadruple WT GIST had a genomic profile profoundly different from both either KIT/PDGFRA mutated or SDHA-mutated GIST. In particular, the quadruple WT GIST tumors are characterized by the overexpression of molecular markers (CALCRL and COL22A1) and of specific oncogenes including tyrosine and cyclin- dependent kinases (NTRK2 and CDK6) and one member of the ETS-transcription factor family (ERG).
Conclusion
We report for the first time an integrated genomic picture of KIT WT/PDGFRA WT/SDH WT/RAS-P WT GIST, using massively parallel sequencing and gene expression analyses, and found that quadruple WT GIST have an expression signature that is distinct from SDH-mutant GIST as well as GIST harbouring mutations in KIT or PDGFRA. Our findings suggest that quadruple WT GIST represent another unique group within the family of gastrointestintal stromal tumors.
Similar content being viewed by others


Background
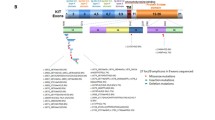
About 10-15% of adult gastrointestinal stromal tumors (GIST) and the vast majority of pediatric GIST do not harbour KIT or platelet-derived growth factor receptor alpha (PDGFRA) mutations [1, 2]. These GIST were originally defined as KIT/PDGFRA wild-type (KIT WT/PDGFRA WT) and generally are less sensitive to tyrosine-kinase inhibitors [3–5]. Their molecular biology is heterogeneous as evidence by the existence of different subgroups with distinct molecular abnormalities (Figure 1). KIT WT/PDGFRA WT GIST can be divided into two main groups according to the succinate dehydrogenase subunit B (SDHB) immunohistochemical status (IHC): SDHB positive (SDHB IHC+), or type 1 GIST which, includes neurofibromatosis type 1 (NF1)-mutated GIST and some sporadic KIT WT/PDGFRA WT GIST. The second group of KIT WT/PDGFRA WT, called as type 2 GIST, is characterized by a lack of SDHB protein expression (SDHB IHC-). In some cases SDHB IHC- is due to germline and/or de novo mutations of any of the four SDH subunits (SDHA mut) [6–8]. The SDHB IHC- includes additional subgroups that can be distinguished on the basis of the SDHA IHC status, which strictly correlates with the presence of SDHA-inactivating mutations (SDHA mut) [9–16]. In particular, SDHB IHC-/SDHA IHC- GIST include a subgroup of young adult women patients with a well defined clinical and biological profile, generally characterized by the gastric primary tumour localization, a predominantly mixed epithelioid and spindle cell morphology, diffuse IHC positivity for KIT and discovered on gastrointestinal stromal tumours 1 (DOG1), frequent lymph node metastases, and an indolent course of disease even if metastasis is present [17]. Moreover, they are characterized by overexpression of the insulin growth factor 1 receptor (IGF1R) [18–21]. On the contrary, SDHB IHC-, but SDHA IHC+ subgroup include 1) cases of syndromic GIST arising from the Carney-Stratakis Syndrome (CSS), that are characterized by SDHB, SDHC or SDHD inactivating mutations (SDHB mut, SDHC mut, or SDHD mut); and 2) cases of Carney Triad (CT), that lack SDHx-mutations [6, 22–24]. More rarely, SDHB IHC-/SDHA IHC+ subgroup may include sporadic KIT WT/PDGFRA WT GIST characterized by SDHB, −C or D mutations (most of them germline, and in few cases by SDHA mutations), arising mainly from the stomach, with a lesser female prevalence, but histologically similar to SDHA IHC- GIST [15].

The complexity of KIT WT / PDGFRA WT GIST molecular biology. KIT WT/PDGFRA WT GIST could be firstly divided two main group according to the SDHB immunohistochemical status: SDHB IHC+ (including NF1-mutated GIST and sporadic KIT WT/PDGFRA WT GIST with or without KRAS/BRAF mutations) and SDHB IHC- or SDH-deficient GIST. The latter could be further divided according to the SDHA immunohistochemical status: SDHB IHC-/SDHA IHC- GIST (pediatric type or young adult GIST characterized by germline or somatic inactivating SDHA mutations) and SDHB IHC/SDHA IHC+ GIST (including Carney-Stratakis Syndrome-related GIST, characterized by germline or somatic inactivating SDHB, −C, −D mutations, Carney Triad-related GIST that lack SDHx mutations, and sporadic KIT WT/PDGFRA WT GIST, characterized by germline or somatic inactivating SDHB, −C, −D mutations and SDHA mutations, reported in only three cases [15]. In red the subset of KIT WT/PDGFRA WT GIST referred to as quadruple WT GIST (KIT WT/PDGFRA WT/SDH WT/RAS-P WT), that represent the subject of this study.
The SDHB IHC+ subgroup includes cases of NF1-mutated GIST, that are commonly intestinal, multifocal and have an IGF1R negative staining, and also sporadic KIT WT/PDGFRA WT GIST, arising in the adult from any part of gastrointestinal tract [15, 21, 25]. In about 15% of cases of sporadic KIT WT/PDGFRA WT GIST there may be an activating mutation in BRAF or, more rarely, RAS[26–28]. Taken together, cases of BRAF, RAS, or NF1 mutant GIST can be referred to as RAS-pathway (RAS-P) mutant GIST (RAS-P mut).
In this extremely heterogeneous landscape, the clinical profile and molecular abnormalities of the small subgroup of WT GIST suitably referred to as quadruple wild-type GIST (quadruple WT or KIT WT/PDGFRA WT/SDH WT/RAS-P WT) remains undefined [29]. The aim of this study is to investigate the genomic profile of KIT WT/PDGFRA WT/SDH WT/RAS-P WT GIST, by using a massively parallel sequencing and microarray approach, and compare it with the genomic profile of other GIST subtypes.
Results and discussion
Whole-Transcriptome Paired-End RNA Sequencing and copy number analysis
Whole-Transcriptome Paired-End RNA Sequencing was performed on a total of 16 GIST samples, of which 2 were KIT WT/PDGFRA WT without SDH-inactivating mutations and SDHB IHC+/SDHA IHC+ (GIST_133 and GIST_127), 2 were KIT WT/PDGFRA WT/SDHA mut and SDHB IHC-/SDHA IHC- (GIST_7 and GIST_10), and 12 were KIT mut or PDGFRA mut. The principal component analysis showed that both GIST_133 and GIST_127 (KIT WT/PDGFRA WT/SDH WT and SDHB IHC+/SDHA IHC+) are characterized by a gene expression profile profoundly different from both GIST_7 and GIST_10 (KIT WT/PDGFRA WT/SDHA mut and SDHB IHC/SDHA IHC), while clustering in proximity of a subset of KIT mut or PDGFRA mut GIST (Figure 2A).

Principal Component Analysis (PCA) performed on samples analyzed with RNA-seq (Figure 2 A) and microarray (Figure 2 B). In both cases the patients with SDHA mutations are arranged in a strongly separated cluster (yellow points), as were the KIT WT/PDGFRA WT/SDH WT/RAS-P WT samples (red point) although closer to KIT or PDGFRA mutated (respectively blue and green point).
To investigate the presence of novel mutations or small ins/del in the whole coding regions of KIT and PDGFRA we analyzed whole transcriptome sequencing data for single nucleotide variant (SNV) and found no private or cryptic mutations. Moreover, no NF-1, BRAF, RAS mutations were found by whole transcriptome sequencing. Therefore, the GIST from these two patients were KIT WT/PDGFRA WT/SDH WT/RAS-P WT, or quadruple WT GIST. Analysis of deleterious mutations from whole transcriptome sequencing did not identify any known oncogenic event or shared alteration in the two patients (Additional file 1: Table S1). Copy number analysis was performed on the two KIT WT/PDGFRA WT/SDH WT/RAS-P WT GIST: GIST_133 showed no genomic imbalances, while GIST_127 harbors several macroscopic cytogenetic alterations, including loss of chromosome arms 14q and 22q frequently observed in KIT/PDGFRA mutated GIST.
Gene expression analysis
To confirm and extend the results, whole-genome gene expression analysis by microarray was performed on 9 out 16 patients analyzed by RNAseq and an additional 20 GIST patients (1 KIT WT/PDGFRA WT SDHA mut GIST and 19 KIT mut or PDGFRA mut GIST). The principal component analysis confirmed that both KIT WT/PDGFRA WT/SDH WT/RAS-P WT GIST have a genetic profile significantly different from all three KIT WT/PDGFRA WT/SDHA mut GIST, and cluster in close proximity to some KIT mut GIST samples (Figure 2B). Supervised gene expression analysis revealed the presence of specific genetic signatures characterizing the different molecular subgroups of GIST (Figure 3); the SDHA mut group showed a gene signature mainly characterized by the over-expression of IGF1R (p value 2.7X10−11) and of neural markers (LHX2, KIRREL3) [30], whereas as expected, all PDGFRA mut GIST were clearly separated from KIT mut GIST, especially for the expression of PDGFRA.

Representation of top-scoring genes significantly over-expressed in the four GIST classes, ( KIT WT / PDGFRA WT / SDH WT / RAS-P WT , SDHA mut , KIT mut and PDGFRA mut ), when each of them is compared with all other cases together.
The quadruple WT (KIT WT/PDGFRA WT/SDH WT/RAS-P WT) samples were characterized by a distinct gene expression profile (Figure 4), with 65 genes over-expressed or under-expressed (p value < 0.005) compared with all the other GIST molecular subgroups. GSEA analysis of the transcriptional profile of quadruple WT tumors showed enrichment of Polycomb target genes with respect to SDHA mut GIST, in particular of the classes of PRC2 targets (p value 0.043) and H3K27-bound genes (p value 0.021). The function of the upregulated genes was related to cell cycle progression and MAPK signaling, ad exemplified by increased expression of SKP2, CDK6, FGF4, NTRK2). The quadruple WT GIST tumors are characterized by the overexpression of molecular markers (CALCRL and COL22A1) and of specific oncogenes including tyrosine and cyclin- dependent kinases (NTRK2 and CDK6) and one member of the ETS-transcription factor family (ERG). Overexpression of CALCRL, COL22A1, NTRK2 (TrkB) and of the ETS-transcription factor ERG was confirmed by quantitative PCR, showing that only the KIT WT/PDGFRA WT/SDH WT/RAS-P WT GIST subgroup expressed these molecular markers and possible therapeutic targets (Figure 5). NTRK2 protein expression level was also evaluated by Western Blot analysis and its overexpression in quadruple WT GIST was confirmed (Additional file 2: Figure S1). No mutations, gene fusions or amplifications were identified in NTRK2 and ERG.

Unsupervised hierarchical clustering representation of differential expressed genes (P-value < 0.005) in KIT WT / PDGFRA WT / SDH WT / RAS-P WT GIST with respect to the other GIST classes ( SDHx mut , KIT mut and PDGFRA mut ).

Quantitative PCR estimation of ERG , NTRK2 , CALCRL and COL22A1 expression in GIST. Relative expression of ERG (upper left panel), NTRK2 (upper right panel), CALCRL (lower left panel) and COL22A1 (lower right panel) mRNA in the two KIT WT/PDGFRA WT/SDH WT/RAS-P WT GIST in respect to the others molecular subgroups (4 SDHx mut, 19 KIT mut and 10 PDGFRA mut GIST). Significance was estimated by the Student T-test: *p-value < 0.05; **p-value < 0.01; ***p-value < 0.001.
Discussion
The pathogenesis and underlying biology of KIT WT/PDGFRA WT with intact SDH complex (SDHx WT) and non-mutated RAS-pathway members (RAS-P WT) suitably referred to as quadruple WT GIST remains undefined. In the present study, we performed a whole genome analysis using a massively parallel sequencing approach on a total of 16 GIST cases that included 2 KIT WT/PDGFRA WT/SDH WT and SDHB IHC+/SDHA IHC+,, 2 KIT WT/PDGFRA WT/SDHA mut and SDHB IHC-/SDHA IHC- and 12 cases of KIT mut or PDGFRA mut GIST. Notably, we found that both cases of quadruple WT GIST had a transcriptome profile profoundly different from both KIT/PDGFRA mutated and SDHA-mutated GIST, suggesting a different molecular background underlying quadruple WT GIST. Since both cases of KIT WT/PDGFRA WT/SDH WT lacked mutations of BRAF, RAS family members or NF1, the GIST of these two patients was classified KIT WT/PDGFRA WT/SDH WT/RAS-P WT or quadruple WT GIST. We further validated our data using genome wide gene expression analysis, performed on 9 cases from a previous series that was expanded to include an additional 20 GIST cases (1 KIT WT/PDGFRA WT/SDHA mut GIST and 19 KIT mut or PDGFRA mut GIST). This larger analysis confirmed the unique gene expression signature of the two quadruple WT GIST compared to KIT mutant, PDGFRA mutant or SDHA-mutant GIST. Interestingly, the gene signatures of the quadruple WT GIST, which both arose in the small intestine, clustered in close proximity to a single KIT mut GIST sample (GIST_13). This case was a small intestine GIST of intermediate risk of relapse radically resected from a 46 year old; it harbored an exon 11 KIT point mutation (KIT exon 11 V559D). Our current sample size does not allow us to draw definitive conclusions, but we hypothesize that the intestinal origin of all three tumors may have influenced the gene signature. However, several other cases of small intestinal origin did not cluster near the cases of quadruple WT GIST. The influence of the tissue of origin on the gene signature is consistent with the recent data by Beadling et al., who described five cluster groups among 136 GIST patients (53 KIT mut, 12 PDGFRA mut, 65 adult KIT WT/PDGFRA WT and 7 pediatric KIT WT/PDGFRA WT) defined by the expression patterns of 14 target genes, that were in some cases paralleled by the location of the primary tumour [31].
Using a supervised analysis, we found four gene cluster subgroups based on KIT/PDGFRA/SDH-mutational status. Due to the rarity of RAS-P mutated GIST, we did not have any cases suitable for these genomic studies. Consistent with previous reports, KIT WT/PDGFRA WT/SDHA mut GIST over-expressed IGF1R, further confirming the potential role of this receptor as a target or diagnostic marker for this specific molecular subgroup [18–21]. Moreover, as already described, the gene signature of KIT WT/PDGFRA WT/SDHA mut GIST was largely characterized by the expression of neural-commitment transcription markers, in support of the theory that this subgroup may have a different cellular origin or may derive from interstitial cells of Cajal (ICCs) during a different differentiation step, such as from precursors of ICCs [30]. Notably, both quadruple WT GIST had a distinct gene expression signature that was separated from the KIT WT/PDGFRA WT/SDHA mut GIST. Amongst the differentially expressed genes, it is interesting to note the over-expression of CALCRL, a G protein-coupled receptor that acts as a receptor for adrenomedullin and calcitonin gene-related peptide (CGRP), and is strongly expressed by in several vascular tumours and types of gliomas [32–35]. Also of interest, we found over-expression of COL22A1, a member of the collagen protein family which specifically localizes to tissue junctions and acts as a cell adhesion ligand for skin epithelial cells and fibroblasts [36]. Taken together, these findings may suggest the potential role of CALCRL and COL22A1 as diagnostic markers for the identification of this GIST subgroup. This would need to be validated in a larger series of GIST.
We found that both quadruple WT GIST, in comparison with the other samples, strongly expressed several oncogenes, including ERG and NTRK2 (TrkB). This was confirmed by quantitative PCR. ERG is a well-known member of the erythroblast transformation-specific (ETS) family of transcription factors, which function as transcriptional regulators [37]. ETS proteins are regulated by the mitogenic (RAS/MAPK) signalling transduction pathway, and play an important role in cell differentiation, proliferation, apoptosis and tissue remodelling [38]. There is evidence for an oncogenic role of ERG and the other ETS transcription factors in many human cancers, including sarcomas, prostate cancer, and acute myeloid leukemia, in most cases via chromosomal translocations [39–41]. More recently, it has been shown that the IHC detection of ERG may be a useful marker for vascular tumors, prostate carcinoma and ERG-rearranged Ewing sarcoma [42–44]. Over-expression of NTRK2 (TrkB) in quadruple WT GIST is also of interest, as NTRK2 helps regulated neuronal cell function, including synaptic plasticity, differentiation, growth, survival, and motility [45]. It has also been shown that Trks regulate important processes in non-neuronal cells, contributing to the pathogenesis of several kinds of cancer, such as medullary thyroid carcinoma, prostate cancer, non-small cell lung cancer, head and neck squamous cell carcinoma and pancreatic cancer, in addition to tumors of neural origin [46–51]. Given the relevant biological role played by Trks in cancer, different small molecule inhibitors have been developed and evaluated both in mono-therapy and in combination with chemotherapy in phase 1 and 2 clinical trials [52–58].
To our knowledge, the over-expression of ERG and TrkB in GIST has not been previously reported. However, it is well known that ETV1, another member of ETS family, is highly expressed in GIST and certain subsets of ICC. ETV1 expression plays an important role in regulating the growth of KIT mutant GIST cell lines [59]. On the basis of our results, the overexpression of ERG and TrkB seems to be a unique feature of the quadruple WT GIST, suggesting that it could play a relevant role in the pathogenesis of this subset of GIST. To translate these observations into clinical practice, the over-expression of both molecules could be investigated as diagnostic markers of quadruple WT GIST.
Conclusions
In conclusion, we report for the first time an integrated genomic picture of the quadruple WT GIST, using massively parallel sequencing and gene expression analyses, and have identified a unique subset of GIST among the family of the KIT/PDGFRA WT GIST [60]. The frequency of this GIST subset amongst the family of GIST will need to be defined in future studies as well as any unique clinical-pathological features of this GIST subset, including response to conventional GIST medical therapy. In addition, ongoing studies of ICC developmental biology may help identify the “normal” precursor cells that give rise to this unique GIST subgroup.
Methods
This study was approved by the institutional review board of Azienda Ospedaliero-Universitaria Policlinico S.Orsola-Malpighi, Bologna, Italy (approval number 113/2008/U/Tess). All patients provided written informed consent.
Patients and tumor samples
Fresh tissue specimens of GIST from 36 patients were collected during the surgical operation, snap-frozen in liquid nitrogen and stored at −80°C until analysis. Patient’s characteristics are listed in Table 1.
Whole-Transcriptome Paired-End RNA Sequencing was performed on 16 GIST, including 2 KIT WT/PDGFRA WT GIST patients without SDH-inactivating mutations (GIST_133 and GIST_127), 2 KIT WT/PDGFRA WT GIST patients harbouring SDHA-mutations (GIST_7 and GIST_10), and 12 KIT or PDGFRA mutated GIST patients (7 harboured exon 11 KIT mutations and 5 harboured exon 18 PDGFRA mutations).
Whole-genome gene expression analysis was performed on 9 of the above 16 GIST and extended to include an additional 20 GIST: 1 KIT WT/PDGFRA WT/SDHA mut GIST and 19 KIT or PDGFRA mutated GIST, of which 13 harboured KIT mutations (12 in exon 11 and 2 in exon 9), and 5 harboured PDGFRA mutations (2 in exon 12, 1 in exon 14 and 2 in exon 18).
SDH status
SDH protein expression status was evaluated by both immunohistochemistry (IHC) of SDHB and SDH subunits sequencing. IHC was performed on 4-μm sections of FFPE GIST tumor samples. Rabbit polyclonal anti-SDHB (HPA002868, Sigma-Aldrich, St Louis, MO, USA, 1:800) antibody was used. The sections were deparaffinized, rehydrated, and subjected to the appropriate antigen retrieval treatment (SDHB: microwave heating in citrate buffer pH 6.0 at 100 1C for 40 min). After cooling at room temperature, the activity of endogenous peroxidises was inhibited using methanol/H2O2 (0.5% v/v) for 20 min. The sections were then washed in phosphate-buffered saline (PBS, pH 7.2–7.4) and incubated with the specific primary antibody overnight at room temperature. After that, the sections were washed in PBS and treated using the Novolink Polymer Detection System (Novocastra, Newcastle upon Tyne, UK) according to the manufacturer’s instructions. Liver tissues (for SDHB) were used as positive controls. These tissues showed strong granular staining in the cytoplasm and mitochondria with both of the antibodies.
SDHA gene exons [1–15], SDHB gene exons [1–8], SDHC (exon 1–6) and SDHD (exon 1–4) were sequenced on fresh-frozen tumor specimens of KIT WT/PDGFRA WT GIST patients by Sanger Sequencing method. DNA was extracted by the QIAmp DNA Mini kit (Qiagen, Milan, Italy) in accordance with manufacturer’s directions. Each exon was amplified with Polymerase Chain Reaction (PCR) amplification using specific primer pairs designed with Primer Express 3.0 Software (Applied Biosystem) to amplify exons but not SDHA pseudo-genes located on chromosomes 3 and 5. Then, PCR products were purified with the Qiaquick PCR purification kit (Qiagen, Milan, Italy) and sequenced on both strands using the Big Dye Terminator v1.1 Cycle Sequencing kit (Applied Biosystems). Sanger sequencing was performed on ABI 3730 Genetic Analyzer (Applied Biosystems).
Whole-transcriptome paired-end RNA sequencing
Total RNA was extracted from tumor specimens with RNeasy Mini Kit (Qiagen, Milan, Italy), then cDNA libraries were synthesized from 250 ng total RNA with TruSeq RNA Sample Prep Kit v2 (Illumina, San Diego, CA) according to the manufacturer’s recommendations. Sequencing by synthesis was performed on HiScanSQ sequencer (Illumina) at 75 bp in paired-end mode. Whole-transcriptome sequencing yielded an average of 61 million mapped reads/patient, thus reaching an average coverage of 44X. Two SDHA mut tumor specimens were previously analyzed by whole transcriptome sequencing at the Genome Sciences Centre (Vancouver, Canada) [9].
Bioinformatic analysis
After demultiplexing and FASTQ generation (both steps performed with Casava1.8, an application software specifically developed by Illumina), the paired-end reads quality were analyzed with the function fastx_quality_stats (part of FASTX Toolkit available at http://hannonlab.cshl.edu/fastx_toolkit/index.html). Based on these results we decided to trim each read of each sample at 74 bp in order to maximize sequence quality. The paired-end reads were mapped with the pipeline TopHat/Bowtie [61] on human reference genome HG19, collected from UCSC Genome Browser (http://www.genome.ucsc.edu/). After the alignment procedure the BAM file obtained was manipulated with Samtools [62] in order to remove the optical/PCR duplicate, to sort and to index it.
The analysis of gene expression was performed in two steps: 1) the function htseq-count (Python package HTseq) [63] was adopted to count the number of reads mapped on known genes, included in the Ensembl release 72 annotation features (http://www.ensembl.org); 2) the differential expressed genes were evaluated using the R-Bioconductor package edger [64]. DeFuse, ChimeraScan and FusionMap packages were used to detect chimeric transcripts from RNA-seq data.
Gene expression analysis
RNA was extracted using RNeasy Mini Kit (Qiagen), quality-controlled and labeled as directed by the Affymetrix expression technical manual before hybridization to U133Plus 2.0 arrays. Gene expression data were quantified by the RMA algorithm, filtered and analyzed with supervised techniques by Limma modified t-test for the detection of differentially expressed genes. Differential expressed genes hierarchical clustering and Principal Component Analysis (PCA) were performed with Multiple Array Viewer (MEV available at http://www.tm4.org/mev.html). The same software was used to represent the data in the Figure 3 and Figure 4. Gene expression data of KIT/PDGFRA-mutated and SDHA-mutated samples were previously reported [30].
Copy number analysis
Genomic DNA was labelled and hybridized to SNP array Genome Wide SNP 6.0 (Affymetrix) following manufacturer’s instructions. Quality control was performed by Contrast QC and MAPD calculation. Copy number analysis was performed by Genotyping Console and visualized with Chromosome Analysis Suite (ChAS) Software (Affymetrix). Hidden Markov Model algorithm was used to detect amplified and deleted segments with stringent parameters. To control for hyperfragmentation adjacent segments separated by < 50 probes were combined into one single segment, and only segments > 100 probes were considered.
Quantitative PCR (qPCR)
Total RNA was reverse transcribed using Transcriptor First Strand cDNA synthesis kit (Roche Applied Science, Monza, Italy) with oligo-dT primers, according to the manufacturer’s guidelines. Gene-specific primers were designed with Primer Express 3.0 Software (Applied Biosystems) and qPCR was performed using FastStart Sybr Green (Roche) on the LightCycler 480 apparatus (Roche). DDCt method was used to quantify gene product levels relative to the GAPDH and ATP5B housekeeping genes. Significance was estimated by the Student’s t test: * p-value < 0.05; ** p-value < 0.01, *** p-value < 0.01.
Western blot
Protein expression of NTRK2 was evaluated on 2 KIT WT/PDGFRA WT/SDH WT/RAS-P WT GIST and 8 KIT or PDGFRA or SDH mutant GISTs, of which fresh-frozen tissues were available. Tissue were disrupted in RIPA buffer (Sigma-Aldrich) supplemented with proteases inhibitors and lysed for 1 h with gentle agitation at 4°C. Lysates were centrifuged at 13,000 × g for 15 min at 4°C and supernatants were stored at −80°C. Protein concentrations were determined with the BCA protein assay (Pierce, Rockford, IL). Twenty micrograms of protein were resolved on a 8% SDS-PAGE gel and transferred onto polyvinylidene difluoride (PVDF) membranes. Nonspecific binding sites were blocked by incubation in blocking buffer (PBS containing 0.1% Tween-20 with 5% w/v milk) for 1 h at room temperature. Membranes were incubated overnight at 4°C, with the following primary antibodies: rabbit polyclonal TRKB antibody (ab18987 Abcam 1:500), and rabbit polyclonal β-Tubulin antibody (sc-9104 Santa Cruz Biotechnology, Santa Cruz, CA, 1:500). Then, membranes were washed and incubated with peroxidase conjugate secondary antibodies for 1 h at room temperature. Antigens were revealed using Enhanced Chemiluminescence Reaction (ECL Select, Amersham Pharmacia Biotech, Les Ulis, France).
Nomenclature
KIT WT No mutations of KIT
PDGFRA WT No mutations of PDGFRA
SDH WT No abnormalities of SDHA/B/C/D protein expression and/or gene mutation
SDHA IHC – No expression of SDHA protein
SDHA IHC + Normal expression of SDHA protein
SDHB IHC – No expression of SDHB protein
SDHB IHC + Normal expression of SDHB protein
SDHA mut Mutation of SDHA protein (homozygous or compound heterozygote)
SDHB mut Mutation of SDHB protein (homozygous or compound heterozygote)
SDHC mut Mutation of SDHC protein (homozygous or compound heterozygote)
SDHD mut – Mutation of SDHD protein (homozygous or compound heterozygote)
Abbreviations
- CGRP :
-
Calcitonin gene-related peptide
- CSS:
-
Carney-Stratakis Syndrome
- CT:
-
Carney Triad
- DOG1 :
-
Discovered on gastrointestinal stromal tumours 1
- ETS :
-
Erythroblast transformation-specific
- GIST:
-
Gastrointestinal stromal tumors
- ICCs:
-
Cells of Cajal
- IGF1R :
-
Insulin growth factor 1 receptor
- IHC:
-
Immunohistochemistry
- NF1 :
-
Neurofibromatosis type 1
- PDGFRA :
-
Platelet-derived growth factor receptor alpha
- RAS-P:
-
RAS-pathway
- SDH :
-
Succinate dehydrogenase
- SNV:
-
Single nucleotide variant
- WT:
-
Wild-type.
References
Corless CL, Fletcher JA, Heinrich MC: Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004, 22: 3813-3825. 10.1200/JCO.2004.05.140.
Janeway KA, Pappo AS: Pediatric gastrointestinal stromal tumor. Hematol Oncol Clin North Am. 2009, 23: 15-34. 10.1016/j.hoc.2008.11.005.
Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, McGreevey LS, Chen CJ, Van den Abbeele AD, Druker BJ, Kiese B, Eisenberg B, Roberts PJ, Singer S, Fletcher CD, Silberman S, Dimitrijevic S, Fletcher JA: Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003, 21: 4342-4349. 10.1200/JCO.2003.04.190.
Debiec-Rychter M, Sciot R, Le Cesne A, Schlemmer M, Hohenberger P, van Oosterom AT, Blay JY, Leyvraz S, Stul M, Casali PG, Zalcberg J, Verweij J, Van Glabbeke M, Hagemeijer A, Judson I, EORTC Soft Tissue and Bone Sarcoma Group; Italian Sarcoma Group; Australasian GastroIntestinal Trials Group: KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006, 42: 1093-1103. 10.1016/j.ejca.2006.01.030.
Heinrich MC, Owzar K, Corless CL, Hollis D, Borden EC, Fletcher CD, Ryan CW, von Mehren M, Blanke CD, Rankin C, Benjamin RS, Bramwell VH, Demetri GD, Bertagnolli MM, Fletcher JA: Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol. 2008, 26: 5360-5367. 10.1200/JCO.2008.17.4284.
Gill AJ, Chou A, Vilain R, Clarkson A, Lui M, Jin R, Tobias V, Samra J, Goldstein D, Smith C, Sioson L, Parker N, Smith RC, Sywak M, Sidhu SB, Wyatt JM, Robinson BG, Eckstein RP, Benn DE, Clifton-Bligh RJ: Immunohistochemistry for SDHB divides gastrointestinal stromal tumors (GISTs) into 2 distinct types. Am J Surg Pathol. 2010, 34: 636-644.
Miettinen M, Wang ZF, Sarlomo-Rikala M, Osuch C, Rutkowski P, Lasota J: Succinate dehydrogenase-deficient GISTs: a clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric GISTs with predilection to young age. Am J Surg Pathol. 2011, 35: 1712-1721. 10.1097/PAS.0b013e3182260752.
Janeway KA, Kim SY, Lodish M, Nosé V, Rustin P, Gaal J, Dahia PL, Liegl B, Ball ER, Raygada M, Lai AH, Kelly L, Hornick JL, O’Sullivan M, de Krijger RR, Dinjens WN, Demetri GD, Antonescu CR, Fletcher JA, Helman L, Stratakis CA, NIH Pediatric and Wild-Type GIST Clinic: Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A. 2011, 108: 314-318. 10.1073/pnas.1009199108.
Pantaleo MA, Astolfi A, Indio V, Moore R, Thiessen N, Heinrich MC, Gnocchi C, Santini D, Catena F, Formica S, Martelli PL, Casadio R, Pession A, Biasco G: SDHA loss-of-function mutations in KIT-PDGFRA wild-type gastrointestinal stromal tumors identified by massively parallel sequencing. J Natl Cancer Inst. 2011, 103: 983-987. 10.1093/jnci/djr130.
Pantaleo MA, Nannini M, Astolfi A, Biasco G, GIST Study Group Bologna: A distinct pediatric-type gastrointestinal stromal tumor in adults: potential role of succinate dehydrogenase subunit A mutations. Am J Surg Pathol. 2011, 35: 1750-1752. 10.1097/PAS.0b013e318230a523.
Italiano A, Chen CL, Sung YS, Singer S, DeMatteo RP, LaQuaglia MP, Besmer P, Socci N, Antonescu CR: SDHA loss of function mutations in a subset of young adult wild-type gastrointestinal stromal tumors. BMC Cancer. 2012, 12: 408-10.1186/1471-2407-12-408.
Wagner AJ, Remillard SP, Zhang YX, Doyle LA, George S, Hornick JL: Loss of expression of SDHA predicts SDHA mutations in gastrointestinal stromal tumors. Mod Pathol. 2013, 26: 289-294. 10.1038/modpathol.2012.153.
Dwight T, Benn DE, Clarkson A, Vilain R, Lipton L, Robinson BG, Clifton-Bligh RJ, Gill AJ: Loss of SDHA expression identifies SDHA mutations in succinate dehydrogenase-deficient gastrointestinal stromal tumors. Am J Surg Pathol. 2013, 37: 226-233. 10.1097/PAS.0b013e3182671155.
Oudijk L, Gaal J, Korpershoek E, van Nederveen FH, Kelly L, Schiavon G, Verweij J, Mathijssen RH, den Bakker MA, Oldenburg RA, van Loon RL, O’Sullivan MJ, de Krijger RR, Dinjens WN: SDHA mutations in adult and pediatric wild-type gastrointestinal stromal tumors. Mod Pathol. 2013, 26: 456-463. 10.1038/modpathol.2012.186.
Miettinen M, Killian JK, Wang ZF, Lasota J, Lau C, Jones L, Walker R, Pineda M, Zhu YJ, Kim SY, Helman L, Meltzer P: Immunohistochemical loss of succinate dehydrogenase subunit A (SDHA) in gastrointestinal stromal tumors (GISTs) signals SDHA germline mutation. Am J Surg Pathol. 2013, 37: 234-240. 10.1097/PAS.0b013e3182671178.
Pantaleo MA, Astolfi A, Urbini M, Nannini M, Paterini P, Indio V, Saponara M, Formica S, Ceccarelli C, Casadio R, Rossi G, Bertolini F, Santini D, Pirini MG, Fiorentino M, Basso U, Biasco G, on behalf of GIST Study Group, University of Bologna, Bologna, Italy: Analysis of all subunits, SDHA, SDHB, SDHC, SDHD, of the succinate dehydrogenase complex in KIT/PDGFRA wild-type GIST. Eur J Hum Genet. 2013, Epub online before print
Rege TA, Wagner AJ, Corless CL, Heinrich MC, Hornick JL: “Pediatric-type” gastrointestinal stromal tumors in adults: distinctive histology predicts genotype and clinical behavior. Am J Surg Pathol. 2011, 35: 495-504. 10.1097/PAS.0b013e31820e5f7d.
Chou A, Chen J, Clarkson A, Samra JS, Clifton-Bligh RJ, Hugh TJ, Gill AJ: Succinate dehydrogenase-deficient GISTs are characterized by IGF1R overexpression. Mod Pathol. 2012, 25: 1307-1313. 10.1038/modpathol.2012.77.
Nannini M, Astolfi A, Paterini P, Urbini M, Santini D, Catena F, Indio V, Casadio R, Pinna AD, Biasco G, Pantaleo MA: Expression of IGF-1 receptor in KIT/PDGF receptor-α wild-type gastrointestinal stromal tumors with succinate dehydrogenase complex dysfunction. Future Oncol. 2013, 9: 121-126. 10.2217/fon.12.170.
Belinsky MG, Rink L, Flieder DB, Jahromi MS, Schiffman JD, Godwin AK, Mehren M: Overexpression of insulin-like growth factor 1 receptor and frequent mutational inactivation of SDHA in wild-type SDHB-negative gastrointestinal stromal tumors. Genes Chromosomes Cancer. 2013, 52: 214-224. 10.1002/gcc.22023.
Lasota J, Wang Z, Kim SY, Helman L, Miettinen M: Expression of the receptor for type I insulin-like growth factor (IGF1R) in gastrointestinal stromal tumors: an immunohistochemical study of 1078 cases with diagnostic and therapeutic implications. Am J Surg Pathol. 2013, 37: 114-119. 10.1097/PAS.0b013e3182613c86.
McWhinney SR, Pasini B, Stratakis CA: Familial gastrointestinal stromal tumors and germ-line mutations. N Engl J Med. 2007, 357: 1054-1056. 10.1056/NEJMc071191.
Pasini B, McWhinney SR, Bei T, Matyakhina L, Stergiopoulos S, Muchow M, Boikos SA, Ferrando B, Pacak K, Assie G, Baudin E, Chompret A, Ellison JW, Briere JJ, Rustin P, Gimenez-Roqueplo AP, Eng C, Carney JA, Stratakis CA: Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet. 2008, 16: 79-88. 10.1038/sj.ejhg.5201904.
Zhang L, Smyrk TC, Young WF, Stratakis CA, Carney JA: Gastric stromal tumors in Carney triad are different clinically, pathologically, and behaviourally from sporadic gastric gastrointestinal stromal tumors: findings in 104 cases. Am J Surg Pathol. 2010, 34: 53-64. 10.1097/PAS.0b013e3181c20f4f.
Bajor J: Gastrointestinal stromal tumors in patients with type 1 neurofibromatosis. Clin Exp Med J. 2009, 3: 247-254. 10.1556/CEMED.3.2009.28478.
Agaram NP, Wong GC, Guo T, Maki RG, Singer S, Dematteo RP, Besmer P, Antonescu CR: Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer. 2008, 47: 853-859. 10.1002/gcc.20589.
Hostein I, Faur N, Primois C, Boury F, Denard J, Emile JF, Bringuier PP, Scoazec JY, Coindre JM: BRAF mutation status in gastrointestinal stromal tumors. Am J Clin Pathol. 2010, 133: 141-148. 10.1309/AJCPPCKGA2QGBJ1R.
Daniels M, Lurkin I, Pauli R, Erbstösser E, Hildebrandt U, Hellwig K, Zschille U, Lüders P, Krüger G, Knolle J, Stengel B, Prall F, Hertel K, Lobeck H, Popp B, Theissig F, Wünsch P, Zwarthoff E, Agaimy A, Schneider-Stock R: Spectrum of KIT/PDGFRA/BRAF mutations and Phosphatidylinositol-3-Kinase pathway gene alterations in gastrointestinal stromal tumors (GIST). Cancer Lett. 2011, 312: 43-54. 10.1016/j.canlet.2011.07.029.
Pantaleo MA, Nannin M, Corless CL, Heinrich MC: Quadruple wild-type (WT) GIST: defining the subset of GISTs that lack abnormalities of KIT, PDGFRA, SDH, and the RAS signalling pathway. Cancer Med. 2014, in press
Pantaleo MA, Astolfi A, Nannini M, Ceccarelli C, Formica S, Santini D, Heinrich MC, Corless C, Dei Tos AP, Paterini P, Catena F, Maleddu A, Saponara M, Di Battista M, Biasco G: Differential expression of neural markers in KIT and PDGFRA wild-type gastrointestinal stromal tumours. Histopathology. 2011, 59: 1071-1080. 10.1111/j.1365-2559.2011.04071.x.
Beadling C, Patterson J, Justusson E, Nelson D, Pantaleo MA, Hornick JL, Chacón M, Corless CL, Heinrich MC: Gene expression of the IGF pathway family distinguishes subsets of gastrointestinal stromal tumors wild type for KIT and PDGFRA. Cancer Med. 2013, 2: 21-31. 10.1002/cam4.57.
Muff R, Born W, Fischer JA: Calcitonin, calcitonin gene-related peptide, adrenomedullin and amylin: homologous peptides, separate receptors and overlapping biological actions. Eur J Endocrinol. 1995, 133: 17-20. 10.1530/eje.0.1330017.
Hagner S, Stahl U, Grimm T, Stürzl M, Lang RE: Expression of calcitonin receptor-like receptor in human vascular tumours. J Clin Pathol. 2006, 59: 1104-1107. 10.1136/jcp.2005.026930.
Mennel HD, Hallier-Neelsen M, Hagner S, Benes L: Two novel cell specific receptor proteins, CRLR and CD 117 in human glial tumors. Clin Neuropathol. 2006, 25: 107-114.
Benes L, Kappus C, McGregor GP, Bertalanffy H, Mennel HD, Hagner S: The immunohistochemical expression of calcitonin receptor-like receptor (CRLR) in human gliomas. J Clin Pathol. 2004, 57: 172-176. 10.1136/jcp.2003.12997.
Koch M, Schulze J, Hansen U, Ashwodt T, Keene DR, Brunken WJ, Burgeson RE, Bruckner P, Bruckner-Tuderman L: A novel marker of tissue junctions, collagen XXII. J Biol Chem. 2004, 279: 22514-22521. 10.1074/jbc.M400536200.
Reddy ES, Rao VN, Papas TS: The erg gene: a human gene related to the ets oncogene. Proc Natl Acad Sci U S A. 1987, 84: 6131-6135. 10.1073/pnas.84.17.6131.
Hart AH, Corrick CM, Tymms MJ, Hertzog PJ, Kola I: Human ERG is a proto-oncogene with mitogenic and transforming activity. Oncogene. 1995, 10: 1423-1430.
Sorensen PH, Lessnick SL, Lopez-Terrada D, Liu XF, Triche TJ, Denny CT: A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat Genet. 1994, 6: 146-151. 10.1038/ng0294-146.
Sorensen PH, Lessnick SL, Lopez-Terrada D, Liu XF, Triche TJ, Denny CT: Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005, 310: 644-648. 10.1126/science.1117679.
Martens JH: Acute myeloid leukemia: a central role for the ETS factor ERG. Int J Biochem Cell Biol. 2011, 43: 1413-1416. 10.1016/j.biocel.2011.05.014.
Miettinen M, Wang ZF, Paetau A, Tan SH, Dobi A, Srivastava S, Sesterhenn I: ERG transcription factor as an immunohistochemical marker for vascular endothelial tumors and prostatic carcinoma. Am J Surg Pathol. 2011, 35: 432-441. 10.1097/PAS.0b013e318206b67b.
Wang WL, Patel NR, Caragea M, Hogendoorn PC, López-Terrada D, Hornick JL, Lazar AJ: Expression of ERG, an Ets family transcription factor, identifies ERG-rearranged Ewing sarcoma. Mod Pathol. 2012, 25: 1378-1383. 10.1038/modpathol.2012.97.
Miettinen M, Wang Z, Sarlomo-Rikala M, Abdullaev Z, Pack SD, Fetsch JF: ERG expression in epithelioid sarcoma: a diagnostic pitfall. Am J Surg Pathol. 2013, 37: 1580-1585. 10.1097/PAS.0b013e31828de23a.
Thiele CJ, Li Z, McKee AE: On Trk–the TrkB signal transduction pathway is an increasingly important target in cancer biology. Clin Cancer Res. 2009, 15: 5962-5967. 10.1158/1078-0432.CCR-08-0651.
Brodeur GM, Nakagawara A, Yamashiro DJ, Ikegaki N, Liu XG, Azar CG, Lee CP, Evans AE: Expression of TrkA, TrkB and TrkC in human neuroblastomas. J Neurooncol. 1997, 31: 49-55. 10.1023/A:1005729329526.
McGregor LM, McCune BK, Graff JR, McDowell PR, Romans KE, Yancopoulos GD, Ball DW, Baylin SB, Nelkin BD: Roles of trk family neurotrophin receptors in medullary thyroid carcinoma development and progression. Proc Natl Acad Sci U S A. 1999, 96: 4540-4545. 10.1073/pnas.96.8.4540.
Satoh F, Mimata H, Nomura T, Fujita Y, Shin T, Sakamoto S, Hamada Y, Nomura Y: Autocrine expression of neurotrophins and their receptors in prostate cancer. Int J Urol. 2001, 8: S28-S34. 10.1046/j.1442-2042.2001.00331.x.
Harada T, Yatabe Y, Takeshita M, Koga T, Yano T, Wang Y, Giaccone G: Role and relevance of TrkB mutations and expression in non-small cell lung cancer. Clin Cancer Res. 2011, 17: 2638-2645. 10.1158/1078-0432.CCR-10-3034.
Kupferman ME, Jiffar T, El-Naggar A, Yilmaz T, Zhou G, Xie T, Feng L, Wang J, Holsinger FC, Yu D, Myers JN: TrkB induces EMT and has a key role in invasion of head and neck squamous cell carcinoma. Oncogene. 2010, 29: 2047-2059. 10.1038/onc.2009.486.
Sclabas GM, Fujioka S, Schmidt C, Li Z, Frederick WA, Yang W, Yokoi K, Evans DB, Abbruzzese JL, Hess KR, Zhang W, Fidler IJ, Chiao PJ: Overexpression of tropomysin-related kinase B in metastatic human pancreatic cancer cells. Clin Cancer Res. 2005, 11: 440-449.
Camoratto AM, Jani JP, Angeles TS, Maroney AC, Sanders CY, Murakata C, Neff NT, Vaught JL, Isaacs JT, Dionne CA: CEP-751 inhibits TRK receptor tyrosine kinase activity in vitro exhibits anti-tumor activity. Int J Cancer. 1997, 72: 673-679. 10.1002/(SICI)1097-0215(19970807)72:4<673::AID-IJC20>3.0.CO;2-B.
Miknyoczki SJ, Dionne CA, Klein-Szanto AJ, Ruggeri BA: The novel Trk receptor tyrosine kinase inhibitor CEP-701 (KT-5555) exhibits antitumor efficacy against human pancreatic carcinoma (Panc1) xenograft growth and in vivo invasiveness. Ann N YAcad Sci. 1999, 880: 252-262. 10.1111/j.1749-6632.1999.tb09530.x.
Evans AE, Kisselbach KD, Yamashiro DJ, Ikegaki N, Camoratto AM, Dionne CA, Brodeur GM: Antitumor activity of CEP-751 (KT-6587) on human neuroblastoma and medulloblastoma xenografts. Clin Cancer Res. 1999, 5: 3594-3602.
Strock CJ, Park JI, Rosen M, Dionne C, Ruggeri B, Jones-Bolin S, Denmeade SR, Ball DW, Nelkin BD: CEP-701 and CEP-751 inhibit constitutively activated RET tyrosine kinase activity and block medullary thyroid carcinoma cell growth. Cancer Res. 2003, 63: 5559-5563.
Marshall JL, Kindler H, Deeken J, Bhargava P, Vogelzang NJ, Rizvi N, Luhtala T, Boylan S, Dordal M, Robertson P, Hawkins MJ, Ratain MJ: Phase I trial of orallyadministeredCEP-701, anovelneurotrophin receptor-linked tyrosine kinase inhibitor. Invest New Drugs. 2005, 23: 31-37.
Wang T, Lamb ML, Scott DA, Wang H, Block MH, Lyne PD, Lee JW, Davies AM, Zhang HJ, Zhu Y, Gu F, Han Y, Wang B, Mohr PJ, Kaus RJ, Josey JA, Hoffmann E, Thress K, Macintyre T, Wang H, Omer CA, Yu D: Identification of 4-aminopyrazolylpyrimidines as potent inhibitors of Trk kinases. J Med Chem. 2008, 51: 4672-4684. 10.1021/jm800343j.
Chan E, Mulkerin D, Rothenberg M, Holen KD, Lockhart AC, Thomas J, Berlin J: A phase I trial of CEP-701 + gemcitabine in patients with advanced adenocarcinoma of the pancreas. Invest New Drugs. 2008, 26: 241-247. 10.1007/s10637-008-9118-3.
Chi P, Chen Y, Zhang L, Guo X, Wongvipat J, Shamu T, Fletcher JA, Dewell S, Maki RG, Zheng D, Antonescu CR, Allis CD, Sawyers CL: ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature. 2010, 467: 849-853. 10.1038/nature09409.
Nannini M, Biasco G, Astolfi A, Pantaleo MA: An overview on molecular biology of KIT/PDGFRA wild type (WT) gastrointestinal stromal tumours (GIST). J Med Genet. 2013, 50: 653-661. 10.1136/jmedgenet-2013-101695.
Trapnell C, Pachter L, Salzberg SL: TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009, 9: 1105-1111.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup: The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009, 25: 2078-2079. 10.1093/bioinformatics/btp352.
Anders S, Huber W: Differential expression analysis for sequence count data. Genome Biol. 2010, 11: R106-10.1186/gb-2010-11-10-r106.
Robinson MD, McCarthy DJ, Smyth GK: edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010, 26: 139-140. 10.1093/bioinformatics/btp616.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/14/685/prepub
Acknowledgments
All staff of Bologna GIST Study Group: Annalisa Altimari, Claudio Ceccarelli, Paolo Castellucci, Fausto Catena, Monica Di Battista, Massimo Del Gaudio, Valerio Di Scioscio, Stefano Fanti, Michelangelo Fiorentino, Pietro Fusaroli, Lidia Gatto, Franco W. Grigioni, Elisa Gruppioni, Alessandra Maleddu, Maria Caterina Pallotti, Antonio Daniele Pinna, Paola Tommasetti, Maurizio Zompatori.
Funding
The present work was done with a financial contribution by Novartis Oncology, Italy, and with funds by My First Grant 2013, AIRC 2013.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MN: have made substantial contributions to conception and design of the study, interpretation of data and drafted the manuscript; AA: carried out the molecular genetic studies, the sequence alignment and have been involved in drafting the manuscript. MU: carried out the molecular genetic studies, the sequence alignment and have been involved in drafting the manuscript. VI: carried out the bioinformatic analysis and interpretation of data and have been involved in drafting the manuscript. DS: carried out the pathological analysis and the collection of samples. MCH: have been involved in revising the manuscript critically for important intellectual content and have given final approval of the version to be published. CLC: have been involved in revising the manuscript critically for important intellectual content and have given final approval of the version to be published. MS, AM and CL have helped to draft and revised the manuscript. GE: carried out the surgical collection of samples. GB: have been involved in revising the manuscript critically for important intellectual content and have given final approval of the version to be published. MAP: have made substantial contributions to conception and design of the study, interpretation of data and drafted the manuscript; All authors read and approved the final manuscript.
Electronic supplementary material
12885_2014_4877_MOESM1_ESM.xlsx
Additional file 1: Table S1: NTRK2 protein overexpression in KIT WT/PDGFRA WT/SDH WT/RAS-P WT GIST. Western blot immunostaining of NTRK2 was perfomed on proteins extracted from two quadruple WT GIST and from eight PDGFRA or KIT or SDH mutated GIST. HL-60 cell line protein extract was used as positive control. (XLSX 18 KB)
12885_2014_4877_MOESM2_ESM.docx
Additional file 2: Figure S1: NTRK2 protein overexpression in KIT WT/PDGFRA WT/SDH WT/RAS-P WT GIST. Western blot immunostaining of NTRK2 was perfomed on proteins extracted from two quadruple WT GIST and from eight PDGFRA or KIT or SDH mutated GIST. HL-60 cell line protein extract was used as positive control. (DOCX 159 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Nannini, M., Astolfi, A., Urbini, M. et al. Integrated genomic study of quadruple-WT GIST (KIT/PDGFRA/SDH/RAS pathway wild-type GIST). BMC Cancer 14, 685 (2014). https://doi.org/10.1186/1471-2407-14-685
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-14-685