
Abstract
Background
Seroepidemiological studies have reported associations between exposure to sexually transmitted organisms and prostate cancer risk. This study sought DNA evidence of candidate organisms in archival prostate cancer tissues with the aim of assessing if a subset of these cancers show any association with common genital infections.
Methods
221 archival paraffin-embedded tissue blocks representing 128 histopathologically confirmed prostate cancers comprising 52 “aggressive” (Gleason score ≥ 7) and 76 “non-aggressive” (Gleason score ≤ 6) TURP or radical prostatectomy specimens were examined, as well as unaffected adjacent tissue when available. Representative tissue sections were subjected to DNA extraction, quality tested and screened by PCR for HSV-1, HSV-2, XMRV, BKV, HPV, Chlamydia trachomatis, Ureaplasma parvum, Ureaplasma urealyticum, Mycoplasma genitalium, and Trichomonas vaginalis.
Results
195 of 221 DNA samples representing 49 “aggressive” and 66 “non-aggressive” prostate cancer cases were suitable for analysis after DNA quality assessment. Overall, 12.2% (6/49) aggressive and 7.6% (5/66) non-aggressive cases were positive for any of the candidate organisms. Mycoplasma genitalium DNA was detected in 4/66 non-aggressive, 5/49 aggressive cancers and in one cancer-unaffected adjacent tissue block of an aggressive case. Ureaplasma urealyticum DNA was detected in 0/66 non-aggressive and 1/49 aggressive cancers and HSV DNA in 1/66 non-aggressive and 0/49 aggressive cancers. This study did not detect BKV, XMRV, T. vaginalis, U. parvum, C. trachomatis or HPV DNA.
Conclusions
The low prevalence of detectable microbial DNA makes it unlikely that persistent infection by the selected candidate microorganisms contribute to prostate cancer risk, regardless of tumour phenotype.
Similar content being viewed by others

Background
The infection hypothesis for prostate cancer was first proposed in the mid-twentieth century [1]. Subsequently, many studies have sought associations between sexually transmitted infections (STIs) and prostate cancer risk but no clear association with a pathogen has been established. A meta-analysis of 29 case–control studies (1966–2003) reported associations between prostate cancer risk and any STI (OR 1.48 95% CI 1.26-1.73), gonorrhoea (OR 1.35 95% CI 1.05-1.83), and HPV (OR 1.39 95% CI 1.12-2.06) [2]. Recently, large prospective sero-epidemiological studies examining the associations between seropositivity to infectious agents and prostate cancer [3, 4] have reported only modest associations between positive serology and prostate cancer.
There is also growing evidence of associations between prostate cancer risk and variants in genes involved in the response to infection and inflammation. Common genetic variants of genes functionally linked to inflammation and immunity such as COX-2 [5], RNASEL [6] and TLR4 [7] have been associated with prostate cancer risk suggesting that infection and host response to infection may be involved in its development.
Case–control studies nested within large prospective seroepidemiological cohort studies have reported only modest associations between evidence of exposure to common STIs and prostate cancer risk (T. vaginalis OR 1.43 95% CI 1.00-2.03) [3] or no association (HPV-33 OR 1.14, 95% CI 0.76-1.72; C. trachomatis OR 1.13, 95% CI 0.65-1.96) [4]. It is likely that these studies would have been limited by the biases inherent in the measures of exposure applied. Serological methods to measure past infection by organisms such as C. trachomatis, N. gonorrhoea and HPV may underestimate actual exposure due to poor sensitivity. Kirnbauer et al. [8] demonstrated that only 59% of those positive for HPV16 DNA at the cervix produced a measureable serological response. The low sensitivity of serological assays may be due to the waning of antibody titres over time. In addition, the time to seroconversion may be lengthy and those infected may not seroconvert at all [9].
It has also been suggested that these studies may have been prone to misclassification bias, due to the widespread use of prostate specific antigen (PSA) testing as a screening device for prostate cancer within the study period. This may have led to the inclusion of subclinical slow-growing prostatic neoplasms that diminished their ability to detect meaningful associations between measures of exposure and clinically significant phenotypes. Therefore, in the current environment with respect to PSA screening, studies should incorporate subgroup analysis into their design in order to discriminate factors that may influence the aetiology or progression of clinically relevant tumours from indolent phenotypes [10].
We examined archival tissue from aggressive and non-aggressive prostate cancer phenotypes and used semi-quantitative molecular methods to seek evidence of infection by common sexually transmitted or other organisms at the tissue level.
We hypothesised that the prevalence of DNA from C. trachomatis, U. urealyticum, U. parvum, T. vaginalis, M. genitalium, herpes simplex virus (HSV) 1 and 2, BK virus, Xenotropic murine leukemia virus-related virus (XMRV), and human papillomavirus (HPV), was the same across tumour phenotypes (non-aggressive and aggressive prostate cancer). We screened samples against a panel of sexually transmitted and other infectious organisms to determine prevalence according to tumour phenotype.
Methods
Cases were drawn from three existing prostate cancer research projects, (1) the Melbourne Collaborative Cohort Study (MCCS) [11], a population-based prospective cohort study, recruited over the period 1990–1994, (2) the Risk Factors for Prostate Cancer Study (RFPCS) [12], a population-based case control study and (3) the Early Onset Prostate Cancer Study (EOPCS) [13], a population based case series of males diagnosed with prostate cancer aged ≤56 years of age. Approval for use of the samples arising from these studies was given by the Human Research Ethics Committee of Cancer Council Victoria.
Specimens were selected on the basis of Gleason score [14] determined by review of diagnostic haemotoxylin and eosin stained slides by a single pathologist (JP). Aggressive and non-aggressive tumours were compared. Aggressive tumours were defined as Gleason score ≥7, poorly-differentiated, including tumours staged at T4, N + (lymph node positive), or M + (distant metastases) regardless of their Gleason score or grade of differentiation. Non-aggressive tumours were defined as well-differentiated with a Gleason score ≤6.
We used archival prostate tissues resected from men that had undergone either radical prostatectomy (RP) or transurethral resection of the prostate (TURP) within the period 1992–2005. A total of 221 formalin-fixed paraffin-embedded tissue blocks (including unaffected adjacent tissue when available) representing 128 histopathologically confirmed prostate cancers comprising TURP and RP specimens were examined.
We processed formalin-fixed, paraffin-embedded radical prostatectomy and TURP specimens using the sandwich sectioning method [15]. To minimize cross-contamination between the samples, gloves and the microtome blade were changed and the microtome washed with histolene, bleach, and 80% ethanol between each sample. Formalin-fixed paraffin-embedded breast tissue was sectioned between every four prostate tissue blocks to ensure no carry-over of DNA. The outer three-micrometer sections were stained with haematoxylin and eosin and validated by a single pathologist to confirm the presence of cancer and the initial histological diagnosis (AL). The four inner seven-micrometer sections remained unstained and were utilised for DNA extraction and molecular assays.
Sections selected for DNA extraction were deparaffinised with histolene and absolute ethanol and the tissue pellet air-dried. Digestion of the tissue was achieved by resuspending the pellet in 160 μL Tissue Lysis Buffer (Roche, Australia) and 40 μL proteinase K (Roche, Australia) and incubating overnight in a heat block at 37°C. A 200 μL volume of lysate was extracted using the MagNA Pure LC instrument and MagNA Pure LC DNA Isolation Kit I (Roche, Australia) with an elution volume of 100 μL as per the manufacturer’s protocol.
Integrity of the DNA extracted from prostate tissue was ascertained by amplification of a 268 bp region of the human beta-globin gene as previously described [16].
We qualitatively screened samples for Chlamydia trachomatis by the COBAS® TaqMan® CT Test, v2.0 (Roche, Australia). Amplification and detection of HPV on all samples was carried out using the PapType High-Risk (HR) HPV Detection and Genotyping kit (Genera Biosystems, Melbourne, Victoria, Australia) [17]. In addition, 49 aggressive cases were screened by DNA ELISA kit HPV SPF10, version 1 (Labo Bio-medical Products BV, Rijswijk, The Netherlands) according to the manufacturer’s instructions. Published primers, probes and Real-Time PCR protocols for Ureaplasma urealyticum[18], Ureaplasma parvum[18], Mycoplasma genitalium[19], Trichomonas vaginalis[20, 21], Xenotropic Murine Retrovirus [22], BK virus [23] AND HSV [24] were applied to the screening of samples with minor modifications (Table 1). Assays to detect T. vaginalis and HSV 1 and 2 were performed on the LightCycler Carousel (Roche, Australia) and all other assays on the LightCycler 480 (Roche, Australia).
Results and discussion
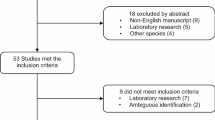
Of the 221 samples, 195 (88.2%) produced a 268 bp product of the human beta-globin gene in quality control PCR testing and were deemed suitable for further analysis. Of these, 49 cases were classified as aggressive and 66 cases as non-aggressive. Of the 49 aggressive cases, 13 cases also had an adjacent normal tissue block. Of the 66 non-aggressive cases, 38 had both a tumour and normal block available.
Table 2 shows the prevalence of M. genitalium, U. urealyticum, and HSV (7.8%, <1% and <1% respectively) and that no difference in prevalence between aggressive and non-aggressive phenotypes was observed. Herpes simplex virus (indeterminate type) DNA was detected in 1/66 non-aggressive prostate cancer tissues and in none of 49 aggressive prostate cancer tissues. Mycoplasma genitalium DNA was detected in 4/66 (6.0%) non-aggressive, 5/49 (10.2%) aggressive and in one cancer-unaffected tissue block of an aggressive case. Ureasplasma urealyticum DNA was detected in none of the non-aggressive and 1/49 (2.0%) aggressive prostate cancer cases. Ureaplasma parvum, T. vaginalis, C. trachomatis, BKV, XMRV or HPV DNA was not detected in any prostate cancer tissue screened in this study.
Our negative findings with respect to the presence of viral DNA in formalin-fixed prostate cancer tissues are consistent with those of Bergh et al. [25] who screened 352 formalin-fixed paraffin embedded tissues of benign prostatic hyperplasia cases for evidence of HSV 1 and 2, BKV or HPV infection and detected no viral DNA. In addition, Martinez-Fierro and colleagues [26] reported a low and insignificant prevalence of XMRV and BKV DNA in fresh frozen prostate material but reported a positive association between prostate cancer and HPV prevalence (OR 3.98, 95% CI 1.17-13.56, p = 0.027), in contrast to our study that did not detect HPV DNA in any prostate sample.
One of the weaknesses of our study is the limited statistical power to detect moderate differences in the prevalence of infectious organisms due to the low prevalence we observed in all our samples. For example, for M. genitalia, the most prevalent organism in our samples, the statistical power to detect a four-fold higher prevalence in tumour tissue samples than in normal tissue samples (i.e. 8% vs 2%) at a 0.05 level of statistical significance was lower than 50%.
Conclusions
The methods we employed for this study were direct and robust with respect to sensitivity and specificity for the target organisms. We chose primers that generated small amplimers (≤268 bp) to account for fragmentation of the DNA extracted from formalin-fixed paraffin embedded tissues. We conclude that it is unlikely that the microorganisms which were included in the candidate panel contributed to the development of prostate cancer in our Australian sample of prostate cancers due to the low prevalence or complete absence of detectable microbial DNA in the tissue samples. Our study hypothesis and aims assumed persistent infection with the candidate organisms allowing for molecular detection in the FFPE material. We cannot exclude the possibility of an initial infection leading to oncogenic sequelae followed by clearance either by natural immunity or administration of antibiotics.
Abbreviations
- BKV:
-
BK virus
- DNA:
-
Deoxyribonucleic acid
- EOPCS:
-
Early onset prostate cancer study
- FFPE:
-
Formalin-fixed paraffin-embedded
- HPV:
-
Human papillomavirus
- HSV-1:
-
Herpes simplex virus 1
- HSV-2:
-
Herpes simplex virus 2
- MCCS:
-
Melbourne Collaborative Cohort Study
- PCR:
-
Polymerase chain reaction
- PSA:
-
Prostate specific antigen
- qPCR:
-
Quantitative polymerase chain reaction
- RP:
-
Radical prostatectomy
- RFPCS:
-
Risk factors for prostate cancer study
- STI:
-
Sexually transmitted infection
- TURP:
-
Transurethral resection of the prostate
- XMRV:
-
Xenotropic murine leukemia virus-related virus.
References
Ravich A, Ravich RA: Prophylaxis of cancer of the prostate, penis, and cervix by circumcision. N Y State J Med. 1951, 51: 1519-1520.
Taylor ML, Mainous AG, Wells BJ: Prostate cancer and sexually transmitted diseases: a meta-analysis. Fam Med. 2005, 37: 506-512.
Sutcliffe S, Giovannucci E, Alderete JF, Chang T-H, Gaydos CA, Zenilman JM, De Marzo AM, Willett WC, Platz EA: Plasma antibodies against Trichomonas vaginalis and subsequent risk of prostate cancer. Cancer Epidemiol Biomarkers Prev. 2006, 15: 939-945.
Sutcliffe S, Giovannucci E, Gaydos CA, Viscidi RP, Jenkins FJ, Zenilman JM, Jacobson LP, De Marzo AM, Willett WC, Platz EA: Plasma antibodies against Chlamydia trachomatis, human papillomavirus, and human herpesvirus type 8 in relation to prostate cancer: a prospective study. Cancer Epidemiol Biomarkers Prev. 2007, 16: 1573-1580.
Panguluri RCK, Long LO, Chen W, Wang S, Coulibaly A, Ukoli F, Jackson A, Weinrich S, Ahaghotu C, Isaacs W, Kittles RA: COX-2 gene promoter haplotypes and prostate cancer risk. Carcinogenesis. 2004, 25: 961-966.
Meyer MS, Penney KL, Stark JR, Schumacher FR, Sesso HD, Loda M, Fiorentino M, Finn S, Flavin RJ, Kurth T, Price AL, Giovannucci EL, Fall K, Stampfer MJ, Ma J, Mucci LA: Genetic variation in RNASEL associated with prostate cancer risk and progression. Carcinogenesis. 2010, 31: 1597-1603.
Chen Y-C, Giovannucci E, Lazarus R, Kraft P, Ketkar S, Hunter DJ: Sequence variants of Toll-like receptor 4 and susceptibility to prostate cancer. Cancer Res. 2005, 65: 11771-11778.
Kirnbauer R, Hubbert NL, Wheeler CM, Becker TM, Lowy DR, Schiller JT: A virus-like particle enzyme-linked immunosorbent assay detects serum antibodies in a majority of women infected with human papillomavirus type 16. J Natl Cancer Inst. 1994, 86: 494-499.
Dillner J: The serological response to papillomaviruses. Semin Cancer Biol. 1999, 9: 423-430.
Platz EA, De Marzo AM, Giovannucci E: Prostate cancer association studies: pitfalls and solutions to cancer misclassification in the PSA era. J Cell Biochem. 2004, 91: 553-571.
Giles GG, English DR: The Melbourne collaborative cohort study. IARC Sci Publ. 2002, 156: 69-70.
Giles GG, Severi G, McCredie MR, English DR, Johnson W, Hopper JL, Boyle P: Smoking and prostate cancer: findings from an Australian case–control study. Ann Oncol. 2001, 12: 761-765.
MacInnis RJ, Severi G, Baglietto L, Dowty JG, Jenkins MA, Southey MC, Hopper JL, Giles GG: Population-based estimate of prostate cancer risk for carriers of the HOXB13 missense mutation G84E. PLoS One. 2013, 8: e54727-
Gleason DF: Histologic grading of prostate cancer: a perspective. Hum Pathol. 1992, 23: 273-279.
Garland SM, Hernandez-Avila M, Wheeler CM, Perez G, Harper DM, Leodolter S, Tang GWK, Ferris DG, Steben M, Bryan J, Taddeo FJ, Railkar R, Esser MT, Sings HL, Nelson M, Boslego J, Sattler C, Barr E, Koutsky LA, Females United to Unilaterally Reduce Endo/Ectocervical Disease (FUTURE) I Investigators: Quadrivalent vaccine against human papillomavirus to prevent anogenital diseases. N Engl J Med. 2007, 356: 1928-1943.
Resnick RM, Cornelissen MT, Wright DK, Eichinger GH, Fox HS, Scheggetter J, Manos MM: Detection and typing of human papillomavirus in archival cervical cancer specimens by DNA amplification with consensus primers. J Natl Cancer Inst. 1990, 82: 1477-1484.
Tabrizi SN, Stevens MP, Khan ZA, Chow C, Devitt MA, Garland SM: Comparison of PapType to Digene Hybrid Capture 2, Roche linear array, and Amplicor for detection of high-risk human papillomavirus genotypes in women with previous abnormal pap smears. J Clin Microbiol. 2012, 50: 2796-2798.
Mallard K, Schopfer K, Bodmer T: Development of real-time PCR for the differential detection and quantification of Ureaplasma urealyticum and Ureaplasma parvum. J Microbiol Methods. 2005, 60: 13-19.
Jensen JS, Björnelius E, Dohn B, Lidbrink P: Use of TaqMan 5′ nuclease real-time PCR for quantitative detection of Mycoplasma genitalium DNA in males with and without urethritis who were attendees at a sexually transmitted disease clinic. J Clin Microbiol. 2004, 42: 683-692.
Riley DE, Roberts MC, Takayama T, Krieger JN: Development of a polymerase chain reaction-based diagnosis of Trichomonas vaginalis. J Clin Microbiol. 1992, 30: 465-472.
Tabrizi SN, Paterson B, Fairley CK, Bowden FJ, Garland SM: A self-administered technique for the detection of sexually transmitted diseases in remote communities. J Infect Dis. 1997, 176: 289-292.
Schlaberg R, Choe DJ, Brown KR, Thaker HM, Singh IR: XMRV is present in malignant prostatic epithelium and is associated with prostate cancer, especially high-grade tumors. Proc Natl Acad Sci U S A. 2009, 106: 16351-16356.
Hirsch HH, Mohaupt M, Klimkait T: Prospective monitoring of BK virus load after discontinuing sirolimus treatment in a renal transplant patient with BK virus nephropathy. J Infect Dis. 2001, 184: 1494-1495. author reply 1495–6
Powell KF, Anderson NE, Frith RW, Croxson MC: Non-invasive diagnosis of herpes simplex encephalitis. Lancet. 1990, 335: 357-358.
Bergh J, Marklund I, Gustavsson C, Wiklund F, Grönberg H, Allard A, Alexeyev O, Elgh F: No link between viral findings in the prostate and subsequent cancer development. Br J Cancer. 2007, 96: 137-139.
Martinez-Fierro ML, Leach RJ, Gomez-Guerra LS, Garza-Guajardo R, Johnson-Pais T, Beuten J, Morales-Rodriguez IB, Hernandez-Ordoñez MA, Calderon-Cardenas G, Ortiz-Lopez R, Rivas-Estilla AM, Ancer-Rodriguez J, Rojas-Martinez A: Identification of viral infections in the prostate and evaluation of their association with cancer. BMC Cancer. 2010, 10: 326-
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/14/579/prepub
Acknowledgements
This work was supported by the National Health and Medical Research Council (project 504702) and the Prostate Cancer Foundation of Australia (projects YIG19 and PG2709). Technical assistance was provided by the Molecular Microbiology Laboratory, Royal Women’s Hospital. Biospecimen retrieval was coordinated by Sonia Terre’Blanche and Charmaine Smith of the Cancer Epidemiology Centre, Cancer Council Victoria.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare no competing interests.
Authors’ contributions
GGG, GS, DB and SG conceived, designed and successfully sought funding for the study. GGG was the principal investigator of the prostate study resources utilized. MS and ST coordinated, designed and supervised the molecular studies. MY carried out the laboratory-based work and drafted the manuscript. JP and AL provided expert pathology review. All authors read and approved the manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Yow, M.A., Tabrizi, S.N., Severi, G. et al. Detection of infectious organisms in archival prostate cancer tissues. BMC Cancer 14, 579 (2014). https://doi.org/10.1186/1471-2407-14-579
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-14-579