
Abstract
Background
Breast cancer is rarely diagnosed in very young women (35years old or younger), and it often presents with distinct clinical-pathological features related to a more aggressive phenotype and worse prognosis when diagnosed at this early age. A pending question is whether breast cancer in very young women arises from the deregulation of different underlying mechanisms, something that will make this disease an entity differentiated from breast cancer diagnosed in older patients.
Methods
We performed a comprehensive study of miRNA expression using miRNA Affymetrix2.0 array on paraffin-embedded tumour tissue of 42 breast cancer patients 35 years old or younger, 17 patients between 45 and 65 years old and 29 older than 65 years. Data were statistically analyzed by t-test and a hierarchical clustering via average linkage method was conducted. Results were validated by qRT-PCR. Putative targeted pathways were obtained using DIANA miRPath online software.
Results
The results show a differential and unique miRNA expression profile of 121 miRNAs (p-value <0.05), 96 of those with a FDR-value <0.05. Hierarchical clustering grouped the samples according to their age, but not by subtype nor by tumour characteristics. We were able to validate by qRT-PCR differences in the expression of 6 miRNAs: miR-1228*, miR-3196, miR-1275, miR-92b, miR-139 and miR-1207. Moreover, all of the miRNAs maintained the expression trend. The validated miRNAs pointed out pathways related to cell motility, invasion and proliferation.
Conclusions
The study suggests that breast cancer in very young women appears as a distinct molecular signature. To our knowledge, this is the first time that a validated microRNA profile, distinctive to breast cancer in very young women, has been presented. The miRNA signature may be relevant to open an important field of research in order to elucidate the underlying mechanism in this particular disease, which in a more clinical setting, could potentially help to identify therapeutic targets in this particular set of patients.
Similar content being viewed by others

Background
Breast cancer (BC) is the most common cancer in women worldwide. It is estimated that breast cancer will account for up to 29% of all new cases of cancer in women in the USA in 2013 [1]. Although the median age at onset is 61 years, approximately one in forty women diagnosed with early breast cancer is very young, constituting 5 to 7% of all cancer deaths in these women [2, 3].
Breast cancer in very young women is typically more aggressive than in their older counterparts, in part owing to the over-representation of high-grade, triple-negative tumours in the former patients. Indeed, very young patients diagnosed with breast cancer usually present larger and poorly differentiated tumours, lymph node invasion, HER2 overexpression and an absence of hormone receptor (HR) expression [4, 5]. It is still a matter of controversy, however, as to whether breast cancer in very young patients (BCVY) is a biologically unique entity that should be considered separately or if its behaviour and outcome is solely due to the higher frequency of more aggressive subtypes. There are contradictory studies on the identification of young age as an independent prognostic factor [5–7].
MicroRNAs (miRNAs) are post-transcriptional regulators that bind to complementary sequences on target messenger RNA transcripts (mRNAs), usually resulting in translational repression or target degradation and gene silencing [8–10]. Deregulation of many of the miRNA’s expression has been linked to various types of disease [11–13]; (http://www.mir2disease.org/). The expression of several miRNAs has been found to be deregulated in some types of cancer [14, 15]. High levels of some miRNA have been linked to stem cell promotion [16–18], while others exhibit a reduced expression of many, promoting loss of differentiation [19, 20]. Both are common traits in tumour development, but many other unknown underlying mechanisms could be affected [21]. Moreover, miRNA expression profiling has been previously shown to be a useful tool for classifying different cancer risk stratification, outcome prediction and classification of histological subtypes [22, 23]. Regarding breast cancer, several studies have identified molecular markers, such as miR-21, miR-9, let-7, miR-205, miR-200 family, miR-126 and miR-335, which correlate to tumours of high metastatic and proliferative capacity, larger size and thus poorer prognosis [18, 24–29].
As previously stated, very little is known about the biology of BCVY, and published studies have focused mainly on hereditary tumours and genomic traits [30, 31]. Nevertheless, the data from two previous published studies point to the possibility that BCVY could have a distinct molecular identity [32, 33].
We performed a study with the aim to identify whether BCVY had a different miRNA profile compared with older age. A secondary objective was to identify those miRNA that are typically up- or downregulated in the tumours from very young patients, which could reveal the ongoing mechanisms of the development of the tumour and of its aggressiveness at an early age.
Methods
Sample selection
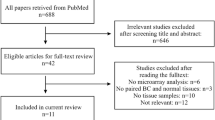
Tissue samples were obtained from patients undergoing surgery for breast cancer at the Hospital Clinico Universitario of Valencia from among the Spanish population. As shown in Figure 1, we selected two groups of women according to their age, one group of women 35 years old or younger (BCVY), and the other of those older than 65 years of age. After excluding those samples with known BRCA1/2 mutation, we got a total of 148 women 35 years old or younger and 3140 patients over 65 years of age with an available biopsy sample before any treatment. We screened for BRCA1/2 status according to the ESMO Guidelines for Hereditary Breast Cancer that follow standard criteria for high-risk patients and from those, we excluded the positive cases from our study as well as those with known or suspected family history of breast cancer. All the participants, with one exception, were Caucasian women of European origin and gave written, informed consent by signing an approved Biobank document allowing research use of the remaining diagnostic tumour material. The study was approved by the Institutional Health Incliva-Hospital Clinico Ethics Committee.

Flow diagram representing the guidelines followed in the selection of the patients suitable for the present study.
After evaluating the quantity and quality of the material, a total of 33 breast cancer patients were included in this study: 21 women diagnosed before the age of 35 categorized as the BCVY group, and 12 breast cancer patients older than 65 years old and matched by tumour subtype (Table 1). We also collected tissue samples from three young cancer-free women undergoing mammary reduction surgery, with ages 28, 34 and 40 years old, and BMI within normal values (between 20 and 24).
For the validation set, we included a total of 55 new independent samples from HCUV, selected with the same aforementioned criteria, 21 of which were BCVY patients, 17 were older than 65 years, and 17 individuals were post-menopausal women between 45 and 65 years old. We included this new age group in order to better establish the cut-off age of the miRNA profile. Overall, the present study was performed on a total of 88 breast cancer patients.
All of the tissue samples were biopsied from primary tumour and locked in FFPE (Formalin-fixed paraffin-embedded). Three-μm thick paraffin sections were stained with haematoxylin and eosin (H&E) in order to obtain a histological control of all samples. All tissue samples contained >30% tumour material. Tumour grade was assessed based on Bloom-Richardson scoring system. Images of the sample’s tissues can be seen in Additional file 1.
To obtain the molecular characteristics of the tumour, presence of molecular markers ER, PR, HER2 and Ki67 was evaluated following the ACO/CAP guidelines. ER and PR status were obtained by IHC staining and HER2 Immunohistochemical staining on TMA sections was performed by the EnVision method with a heat-induced antigen retrieval step. Staining results were assessed by a pathologist. ER and PR were scored based on two-stage scoring system: positive (1) for >10% of ER/PR positive cells and negative (0) for less than 10%, as described previously. Proliferation was assessed measuring percentage of Ki-67 expression. HER2 was called positive either by detection of ERBB2 gene amplification by FISH analysis and/or 3+ staining by DAKO system on HercepTestTM. Where duplicate cores gave discordant results, the higher score was used. Breast cancer tumours were classified into four subtypes based on IHC-model (Tang P. et al., 2009) as: luminal A (ER + and/or PR+, HER2−, Ki67 < 14%); luminal B (ER + and/or PR+, HER2-, Ki67 > 14%);, Triple Negative (ER−, PR−, HER2−) and HER2 overexpressed/amplified (ER−, PR−, HER2+), plus an additional group Luminal/HER2 (ER + and/or PR+, HER2+).
RNA isolation
Total RNA was isolated using RecoverAll Total Nucleic Acid Isolation Kit (Applied Biosystems by Life Technologies, Carlsbad, California, USA) following the manufacturer’s protocol. RNA concentration was measured using a NanoDrop ND 2000 UV–vis Spectrophotometer (Thermo Fisher Scientific Inc., Wilmington DE, USA).
RNA integrity determined by RIN (RNA integrity number) value was assessed with the Agilent 2100 Bioanalyzer using the RNA 6000 Nano Assay (Agilent Technologies Inc., Santa Clara, CA, USA).
miRNA microarray
Microarray expression profiling was performed using GeneChip® miRNA 2.0 Array (Affymetrix, Santa Clara, CA, USA), containing a total of 15,644 probes, in 11 replicates, including 1100 human mature miRNA, their precursors, 2334 human snoRNA (small nucleolar RNA) and scaRNA (small Cajal body-specific RNA) annotated in the miRBase v.15 database. Hybridisation and scanning were performed according to the Affymetrix standard protocol, using Affymetrix Expression Console software. The microarray dataset is publicly available at GEO database (GSE48088) http://www.ncbi.nlm.nih.gov/geo/info/linking.html.
Array data processing and analysis
Data passed quality controls implemented in Expression Console software, build 1.2.0.20, and QC Tool (Affymetrix, Santa Clara, CA, USA). All data were normalised by the DABG-RMA, detected above background (DABG) Robust Multichip Average (RMA) method. We selected 1100 human miRNAs, and set as the threshold for low expression the highest common intensity value of miRNA from vegetable organisms (not supposed to be expressed in humans) included in the microarray. After filtering the data, in order to determine the differences in expression pattern between tumours from BCVY patients and from older ones, we carried out differential expression analyses with the POMELO II tool (http://asterias.bioinfo.cnio.es). We performed a t-test by permutation testing, and p-values were adjusted for multiple comparisons by Benjamini & Hochberg False Discovery Rate (FDR). Those miRNAs with a FDR p-value <0.05 were considered statistically significant. Average linkage hierarchical clustering was performed to obtain clusters of data sets, using Gene Cluster and Treeview software (http://www.eisenlab.org/eisen/).
In order to evaluate whether the miRNA profile detected was a result of age differences or if it was influenced by confounding factors such as tumour size, histological grade and other features characteristic of more aggressive tumours, we performed a t-test analysis on the filtered miRNA expression data after correcting by the influence of nodal status, histological grade, percentage of ki67 expression and tumour size. Data were adjusted obtaining the correlation between miRNAs’ expression and tumour characteristics via linear regression with R Studio (http://www.rstudio.com/), using the remaining residuals for the correction.
Validation by qRT-PCR
Quantitative real time-PCR (qRT-PCR) of selected miRNAs was performed on breast tumour tissue samples in independent series of women younger than 35 years old, women between 45 and 65 years old, and women older than 65 years using TaqMan microRNA Assays (Applied Biosystems by Life Technologies, Carlsbad, California, USA). Normalisation was done with RNU43 snoRNA. The data were managed using the Applied Biosystems software RQ Manager v1.2.1. Relative expression was calculated by using the comparative Ct method and obtaining the fold-change value (ΔΔCt). Data analyses were performed via GraphPad Prism v6.00. The ANOVA test was performed, and Tukey’s test was used to correct for multiple comparisons (p-value threshold of 0.05). Brown-Forsythe test was used to assess the homogeneity of the variances in the different samples groups. Finally, the association of the validated miRNAs was adjusted by breast cancer subtypes using RStudio (http://www.rstudio.com/).
Pathway enrichment analysis and candidate gene searching
DIANA miRPath pathway enrichment analysis was used to gain insight into global molecular networks and canonical pathways related to differentially expressed miRNAs (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=mirpath/index). The software performs an enrichment analysis of multiple miRNA target genes comparing each set of miRNA targets to all known KEGG pathways. Those pathways showing a FDR p-value <0.05 were considered significantly enriched between classes under comparison. We also searched for candidate genes using Target-scan online software (http://www.targetscan.org/) and previously published data.
Results
miRNA expression profiling in primary breast tumours from discovery set
After initial pre-processing of 1100 miRNA expression data, we discarded miRNAs with uniformly low expression, reducing the number of miRNA to a total of 251 hsa-miRNAs. Finally we detected 119 miRNAs differentially expressed between the two groups (p-value < 0.05) and after adjusting for FDR, 96 miRNAs remained significant (Additional file 2). In addition, all of these miRNA’s expression remained significantly different even after correcting by possible confounding factors (tumour size, nodal status, ki67% and histological grade), suggesting that young age is an independent factor (Additional file 3).Mammary tissue from three cancer-free women was included as healthy control group. When unsupervised hierarchical clustering was performed two clearly different two clearly different groups of samples (Figure 2). One group included all except one of the BC tissue samples from very young women. The other group contained all the samples from both older women and normal tissue. We were able to identify two major nodes of differentially regulated miRNA in the BCVY group. As shown in Figure 2, the 52 miRNAs overexpressed in the BCVY group can be seen clustered in the top node, 115, (correlation 0.383) while the lower node, 119, shows the 67 miRNAs found to be repressed in this BCVY set of tumours (correlation 0.102).

Heatmap representing an unsupervised cluster performed according to a hierarchical clustering method centered on the median, using differently expressed human miRNA expression values from an Affymetrix array assay. The miRNAs plotted are those which gave significant FDR p-values after performing a t-test according to the age group of the samples. Correlation values (0–1) of different sub-nodes are shown in the figure according to data obtained from Cluster and Treeview software. The two most correlated, overexpressed-sub-nodes are highlighted in red (from top to bottom, sub-node 92 and sub-node 29); highlighted in green are the most correlated repressed sub-nodes (from top to bottom 102, 79, 52, 54 and 73). The selected nodes for validation are shown in pink (node 15 and node 63).
From the overexpressed node (node 115) we were able to identify two differentiated sub-nodes: node 92 (correlation value 0.63) and node 84 (correlation value 0.66), that appears to be more homogenous inside each group of patients. Within node 84 we selected sub-node 15 (correlation 0.923) including miRNAs with the highest significant expression difference between Young vs Old women-Normal. When scanning the repressed node 119, we identified six interesting sub-nodes, covering most of the node: sub-node 102 (correlation 0.582), sub-node 79 (correlation 0.667), sub-node 76 (correlation 0.678), sub-node 63 (correlation 0.72), sub-node 54 (correlation 0.75) and sub-node 52 (correlation 0.75). While sub-nodes 54 and 52 reached the highest correlation value, we selected sub-node 63 for the further study, since the expression of the whole cluster in cancer-free tissue samples was remarkably high, and also because the BC patients showed levels uniformly higher than the BCVY ones. MicroRNAs from the selected sub-nodes are listed in Figure 2, and their expression fold change and p-values are shown in Additional file 4.
Regarding the hsa-miRNA-star (hsa-miR-92*, hsa-miR-149* and hsa-miR-1228*), when the main form of both miRNA species was also differentially expressed (hsa-miR-92* and hsa-miR-149*), we selected the main form of each miRNA (hsa-miR-92-3p and hsa-miR-149-5p) in order to get more reliable results.
Pathway enrichment and selection of candidate miRNAs for validation step
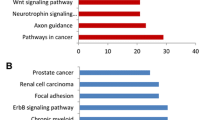
With the 20 selected miRNAs, composing the two most striking sub-nodes, we performed in silico analyses and database searches to determine which ones might be more biologically relevant in the breast cancer context, and eligible for validation. Considering that the miRNAs target from small to a very large number of mRNA transcripts, common deregulation of a set of miRNA could have a potential impact on various biological pathways. To asses which pathways could be affected by the deregulation, we used DIANA miRPath v2.0. When studying miRNAs from the selected sub-nodes, KEGG pathway enrichment analysis revealed several pathways overrepresented with a FDR p-value <0.05, including adhesion and mobility related pathways (extracellular matrix-receptor interactions, glycosaminoglycan biosynthesis, adherens junction, focal adhesion, cell adhesion molecules, regulation of actin cytoskeleton), biological proliferation-or differentiation-related pathways relevant in cancer development or progression (MAPK signalling, endocytosis, axon guidance), circadian rhythm and several cardiomyopathies . Within the genes involved in the pathways related with the selected miRNAs, we found many integrins, laminins, collagen, MAPkinases, semaforins, interleukins, claudins and calmodulins, among others. More information and FDR p-values associated with each pathway are shown in Additional file 5.
We have finally selected 12 miRNAs (7 from the sub-node 15 and 5 from sub-node 63) as putative candidate targets to perform further studies according to their significance, previous published studies, and implications in biological pathways: hsa-miR-1275, hsa-miR-1207-5p, hsa-miR-149*, hsa-miR-762, hsa-miR-3196, hsa-miR-1228*, hsa-miR-92*, hsa-miR-139-5p, hsa-miR-132, hsa-miR-379, hsa-miR-409-3p, and hsa-miR-433. Supplementary information is available in Additional file 6.
qRT-PCR validation in an independent set of samples
We performed a validation of the 12 selected miRNAs using qRT-PCR, on an independent set of samples with similar characteristics as those used in the Discovery phase. However, in the validation set, we classified the 55 samples into three groups (21 tumours from women younger than 35 years, 17 tumours from women aged between 45 and 65 and finally 17 tumours from women older than 65) (see the clinical characteristics for the entire sample set in Table 1).
We did not find evidence suggesting than women older than 65 had any differential characteristics with the 45 to 65 year age group in terms of miRNA expression (Figure 3). Six of the twelve selected miRNAs were confirmed to have a differential expression across groups: miR-1228* (p-value 4.73×10−5), miR-3196 (p-value 1.98×10−4), miR-1275 (p-value 2.86×10−3), miR-92b (p-value 0.012), miR-139-5p (p-value 0.018), and miR-1207 (p-value 0.047). After Tukey’s correction for multiple comparisons, we obtained p-values between each group, confirming that the differences were higher between BCVY samples and the other two older groups (Figure 3). The first four values agree with an upregulation in BCVY, while the last two, miR-92b and miR-139-5p, were indeed downregulated in young women group (see Figure 3 and Additional file 2). Afterward, a t-test analysis was used to compare the BCVY group with the two older patient groups, treating the last two as only one group. The results obtained were the following: miR-1228 (p-value 7.13×10−6), miR-3196 (p-value 3.23×10−5), miR-1275 (p-value 7.77×10−4), miR-92b (p-value 2.94×10−3), miR-139-5p (p-value 6.57×10−3), and miR-1207 (p-value 0.523). After adjusting for tumour subtype, four of them remained statistically significant: miR-1228 (p-value 0.0032), miR-1275 (p-value 0.0010), miR-3196 (p-value 0.0095), miR-139 (p-value 0.0172). It is worth mentioning that all of the other miRNAs tested did indeed lose expression, as was expected; however, no significant value could be obtained due to a lack of detection sensitivity.

Relative expression of the validated miRNAs which have significantly different expression in BCVY tumour samples. A: miRNAs with increased expression in women ≤35 years old (BCVY). B: miRNAs with decreased expression in BCVY. Measures of the expression were quantified using the qRT-PCR technique and calculated using the ΔΔCt method. Boxes represent the sample distribution with the mean, vertical lines mark the 10th percentile, and outliers are represented as dots. P-values are calculated via One-Way ANOVA, and Tukey’s method was implemented as a correction for multiple comparisons. The top p-value represents differences between those ≤35 (BCVY) and >65; the bottom-left represents differences between those >65 and those 45–65 (middle group, between 45 and 65 years old); and bottom-right, the differences between those 45–65 and those ≤35 (BCVY).
Putative functional implication of the validated miRNAs
In order to establish a possible underlying biological implication with the validated deregulated miRNAs (hsa-miR-1228*, hsa-miR-3196, hsa-miR-1275, hsa-miR-1207-5p, hsa-miR-92b, hsa-miR-139-5p), we used online software and miRNA databases to perform an exhaustive search for enrichment of candidate pathways and genes. Moreover, we text-mined all published studies related to the six validated miRNAs with the terms cancer and/or putatively target genes. We found several enriched pathways related to cell motility and invasion: adherens junction (FDR p-value 1.03×10−4), glycosaminoglycan biosynthesis (FDR p-value 2.23×10−3), cell adhesion molecules (CAMs) (FDR p-value 0.035) and several related genes such as integrins, claudins and cadherins. We also found enriched pathways involved in proliferation and apoptosis: calcium signalling (FDR p-value 0.035) and Wnt signalling (FDR p-value 0.041) (Table 2).
Discussion
Our data lead us to think of a differentiated miRNA signature in the group of patients diagnosed at ≤35 years. We have detected a wide range of differentially regulated miRNAs in BCVY when compared to tumours diagnosed in older women and healthy breast tissue. The fact that the older women’s miRNA profile was highly similar to the healthy breast tissue profile (obtained from women under 40 years old) highlights the distinctiveness of the miRNA profile being characteristic of BCVY patients. These differences were maintained after validation, in an independent series of samples using a different technique, in which controls between 45 and 65 years were included, suggesting that our findings are consistent and not a result of the false positives characteristic of high throughput technologies. Although from a limited sample size, the in silico searches done, pointed out to pathways closely related to high cell proliferation, motility and invasion, which support the fact that tumours from young woman have an aggressive behaviour and are prone to metastasis. Other studies in breast cancer showing miRNA profiles characterized by bad prognosis, revealed similar signalling deregulated pathways [34].
Recently, miRNA profiling has arisen as a major approach study technique, that aims to gain more insight into tumour biology, and widespread deregulated miRNA has been demonstrated in various tumour types [35]. It has been shown that specific miRNA, such as miR-195 and let-7a, may play a role as a potential diagnostic tool and a new biomarker for both detecting non-invasive, early breast cancer as well as monitoring treatment effectiveness [36, 37]. Furthermore, other studies have revealed miRNA expression to be specific for each breast tissue type, but it seems not to exist a specific miRNA profile distinctive of BC subtypes, which may point to both differences in the biology of tumours, as well as to patterns of response to treatment [38–44]. As suggested in previous studies, our results reinforce the applicability of the study of miRNA expression in FFPE samples, since the small size of the miRNA preserves them from degradation [45]. In addition, the use of Taq-Man-assay for microRNA expression profiling and validation in FFPE breast tissue samples is reported to offer excellent inter-experimental reproducibility and biological accuracy [41, 46].
Several studies confirm that BC in very young patients is a more aggressive disease given the high frequency of adverse prognosis factors or more aggressive subtypes at this age [4]. In most studies published the cut-off age for defining young women is 45 years. However, results from a large Korean breast cancer study with 9885 pre-menopausal patients detects a sharp increase in risk of death from breast cancer in women younger than 35 years old, establishing this age as the more reasonable cut-off defining young age-onset breast cancer [47]. Following this recommendation, we have determined 35 years old as the threshold for the young group, since we considered that, despite this cut-off makes harder patient’s recruitment within the young age group, it identifies a more homogeneous population.
Whether breast cancer in the young is a distinct biological entity or just a reflection of a higher percentage of cases with aggressive phenotypes (that are otherwise indistinctive from aggressive tumours in older patients) remains a matter of controversy and an important outstanding question with possible therapeutic implications [6].
In order to minimize clinical-pathological putative confounding effects, we have performed several comparisons in the expression of these miRNA in terms of tumour size, axillary node involvement, histological grade and ki67, with the results showing no significant differences. This absence of significant differences, having considered the most relevant adverse prognostic factors, suggests that young age could be indeed a factor independent of subtype, size, nodal involvement, or proliferative characteristics that merits further study.
Moreover, no distinct therapy is considered for young or very young patients, but in many cases these patients are over-treated as a consequence of their young age. Thus identification of new prognostic or diagnostic platforms and identification of specific targets is needed for this subgroup [48].
Data about genomic profile in young breast cancer patients are scarce in the literature; however, Colak and cols. have recently published an age-specific gene expression signature in breast cancer [33]. Although, the study focused on women younger than 45, it also included 6 samples from those younger than 35 (BCVY). The study pointed to genes barely implicated in the pathways found in our study. That notwithstanding, the authors proposed that there is a different molecular profile characterizing breast cancer in very young women.
A recent retrospective study analysed data from 20 data sets and assessed the role of these genetic signatures in predicting the prognosis in breast cancer in young women. This study confirmed that BC patients aged 40 or less were diagnosed more frequently with oestrogen receptors and HER2 negative tumours. Furthermore, although proliferation-related gene signatures were not associated with age in this study, in two independent cohorts BC in younger patients was associated with immature epithelial cell and growth factor signalling pathways, leading to the conclusion that BC in young women seems to be a distinct entity beyond the intrinsic breast cancer subtype classification [32].
The data obtained with our miRNA profiling supports the evidence proposed by Yau and cols that could be detected an age-dependent signature in BC with a number of samples from young women very similar to ours. Although the conclusions are raised on gene expression analyses the signaling pathways detected including cell cycle, mammary gland development and extracellular matrix (ECM) are also highlighted in our study [49].
In our study, the most significantly set of deregulated miRNAs in BCVY pointed out to pathways related to apoptosis, cell motility, proliferation, mitotic regulatory processes and the PI3K and IGFR transduction that confer tumours high metastatic capacity, increasing progression and invasion (Table 3). Furthermore, many of the pathways involved include integrins and laminins as target genes, described as proliferation genes portraits of breast tumours [50].
If we take into account the set of validated miRNAs (upregulated miR-1228*, miR-3196, miR-1275, miR-1207 and downregulated miR-139-5p and miR-92b) we were able to detect in the literature their implications in escaping of apoptosis and therefore, contributing to migration and invasion [15, 51–69]. Downregulation of one particular miRNA, miR-139, might promote gastric tumour progression and metastasis via upregulating CXCR4, Bao and cols suggest that miR-139 might be suppressed by upstream HER2 signaling, a strongly deregulated pathway in breast cancer [52].
We cannot fully reject an influence of demographics or epidemiological factors inherent to young women in the tumour profile obtained. Bearing this in mind, we believe that the inclusion of healthy young breast tissue in the analysis might have minimized these putative confounding epidemiologic aspects.
Oestrogen levels are an age-dependent factor increasing the risk of breast cancer, which can themselves modulate the expression of several miRNAs. Moreover, miRNAs can as well regulate oestrogen receptor levels [70, 71]. None of the miRNAs described in the literature related to oestrogen appears to be deregulated in the BCVY profile presented. The different profile obtained between BCVY and normal breast samples from young women (with similar oestrogen levels) supports the idea that the intrinsic levels of oestrogen are not responsible of the miRNA deregulation.
Early pregnancies and lactation are considered protector factors in BC and they could bias the comparison between young and older woman. In our series, older women in general have early age at first birth and still have developed breast cancer, while in the young women we have representation of both circumstances and all of them fall in the same miRNA profile, highlighting young age as the main leading factor of the profiling.
Mammary density is the strongest risk factor for non-familial breast cancer among women, apart from older age. Young women present denser breast tissue in mammographies, which makes more difficult the diagnosis. In addition, differences in normal breast microenvironment seem to exert an influence on the behavior of breast cancer cells in premenopausal women [72]. A recent molecular profiling study performed on adjacent tissue of breast tumours, classifies extratumoral stromal microenvironments into two primary gene expression phenotypes (Active and Inactive). The Active subtype has high expression of genes involved in activation of fibrosis, cellular movement, increased TWIST expression, and positive expression of TGF-b signatures. Inactive phenotype is overrepresented by samples from young women and according to Perez and cols, means higher levels of cell adhesion and cell–cell contact genes [73, 74]. The miRNA profile presented in our study pointed out to pathways also related with cell motility and adhesion, however the upregulation of implicated miRNAs indicates a downregulation of the targeted genes and therefore, stroma that correlates with young and denser breast tissue is not the driver in our deregulated miRNAs signature.
Finally, BMI (Body Mass Index) has been recently related to BC risk and survival. In older women higher BMI results in elevated risk of developing BC, while in young women is a protector factor [75]. In our study the mean value of BMI in BCVY is within normal ranges, which makes difficult any speculation about its role in BCVY.
In our study all patients but one are confirmed to be of European Caucasian ethnicity. This implies that our miRNA profile might not be representative of other genetic backgrounds such as African descent, where exists a higher proportion of breast cancer in young women. On the other side, the study has been performed in a very homogeneous population which reinforces the conclusions obtained and we encourage the replication of our findings in other ethnicity breast cancer collections.
Conclusions
Our results suggest that breast cancer in young patients appears to be a different biological entity. Previously published works and in silico enrichment analyses of the miRNAs deregulated in young women suggest that targeted genes involved in proliferation pathways, cell adhesion, apoptosis, extracellular matrix and cell motility, deregulation of these pathways may lead to more aggressive, proliferative and metastatic tumour phenotypes. A more detailed analysis of those miRNA significantly up- or downregulated could guide both to the establishment of a different hypothesis about the potential molecular mechanisms involved in the carcinogenesis and to identify more specific therapeutic targets in this particular set of patients.
Abbreviations
- ANOVA:
-
Analysis of variances
- BC:
-
Breast cancer
- BCVY:
-
Breast cancer in very young women
- BMI:
-
Body mass index
- BRCA1/2:
-
Breast cancer gene 1/2
- cDNA:
-
Complementary DNA
- Ct:
-
Mean cycle above threshold
- DABG-RMA:
-
Detected above background-robust multichip average
- FDR:
-
False discovery rate
- FFPE:
-
Formalin fixed-paraffin embedded
- H&E:
-
Hematoxilin and eosin
- HCUV:
-
Valencian Clinic-University Hospital
- HER2:
-
Human epidermal growth factor receptor 2
- HR:
-
Hormone receptor
- Hsa:
-
Homo sapiens
- IGFR:
-
Insulin-like growth factor receptor
- MAPK:
-
Mitogen-activated protein kinase
- miR:
-
microRNA
- miRNA:
-
microRNA
- mRNA:
-
messenger RNA
- PCR:
-
Polymerase chain reaction
- PI3K:
-
Phosphoinositol-3-kinase
- qRT-PCR:
-
Quantitative real time-PCR
- RIN:
-
RNA integrity number
- RNA:
-
Ribonucleic acid
- RNU43:
-
Ribonucleoprotein
- scaRNA:
-
Small cajal-body RNA
- snoRNA:
-
Small nucleolar RNA
- Wnt:
-
Wingless-type MMTV integration site family.
References
Siegel R, Naishadham D, Jemal A: Cancer statistics, 2013. CA Cancer J Clin. 2013, 63 (1): 11-30.
Cardoso F, Loibl S, Pagani O, Graziottin A, Panizza P, Martincich L, Gentilini O, Peccatori F, Fourquet A, Delaloge S, Marotti L, Penault-Llorca F, Kotti-Kitromilidou AM, Rodger A, Harbeck N: The European Society of Breast Cancer Specialists recommendations for the management of young women with breast cancer. Eur J Cancer. 2012, 48 (18): 3355-3377.
Kothari AS, Fentiman IS: Breast cancer in young women. Int J Clin Pract. 2002, 56 (3): 184-187.
Anders CK, Fan C, Parker JS, Carey LA, Blackwell KL, Klauber-DeMore N, Perou CM: Breast Carcinomas Arising at a Young Age: Unique Biology or a Surrogate for Aggressive Intrinsic Subtypes?. J Clin Oncol. 2011, 29 (1): e18-e20.
Anders CK, Hsu DS, Broadwater G, Acharya CR, Foekens JA, Zhang Y, Wang Y, Marcom PK, Marks JR, Febbo PG, Nevins JR, Potti A, Blackwell KL: Young age at diagnosis correlates with worse prognosis and defines a subset of breast cancers with shared patterns of gene expression. J Clin Oncol. 2008, 26 (20): 3324-3330.
Colleoni M, Anders CK: Debate: the biology of breast cancer in young women is unique. Oncologist. 2013, 18 (4): e13-e15.
Anders CK, Acharya CR, Hsu DS, Broadwater G, Garman K, Foekens JA, Zhang Y, Wang Y, Marcom K, Marks JR, Mukherjee S, Nevins JR, Blackwell KL, Potti A: Age-specific differences in oncogenic pathway deregulation seen in human breast tumors. PLoS One. 2008, 3 (1): e1373-
Bartels CL, Tsongalis GJ: MicroRNAs: novel biomarkers for human cancer. Clin Chem. 2009, 55 (4): 623-631.
Chen K, Rajewsky N: The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet. 2007, 8 (2): 93-103.
Kusenda B, Mraz M, Mayer J, Pospisilova S: MicroRNA biogenesis, functionality and cancer relevance. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2006, 150 (2): 205-215.
Hammond SM: RNAi, microRNAs, and human disease. Cancer Chemother Pharmacol. 2007, 58 (Suppl 1): s63-s68.
Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Hoydal M, Autore C, Russo MA, Dorn GW, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G: MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007, 13 (5): 613-618.
Beveridge NJ, Gardiner E, Carroll AP, Tooney PA, Cairns MJ: Schizophrenia is associated with an increase in cortical microRNA biogenesis. Mol Psychiatry. 2009, 15 (12): 1176-1189.
Hammond SM: MicroRNAs as oncogenes. Curr Opin Genet Dev. 2006, 16 (1): 4-9.
He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM: A microRNA polycistron as a potential human oncogene. Nature. 2005, 435 (7043): 828-833.
Gunaratne PH: Embryonic stem cell microRNAs: defining factors in induced pluripotent (iPS) and cancer (CSC) stem cells?. Curr Stem Cell Res Ther. 2009, 4 (3): 168-177.
Nimmo RA, Slack FJ: An elegant miRror: microRNAs in stem cells, developmental timing and cancer. Chromosoma. 2009, 118 (4): 405-418.
Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, Diehn M, Liu H, Panula SP, Chiao E, Dirbas FM, Somlo G, Pera RA, Lao K, Clarke MF: Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009, 138 (3): 592-603.
Gurtan AM, Ravi A, Rahl PB, Bosson AD, Jnbaptiste CK, Bhutkar A, Whittaker CA, Young RA, Sharp PA: Let-7 represses Nr6a1 and a mid-gestation developmental program in adult fibroblasts. Genes Dev. 2013, 27 (8): 941-954.
Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ: RAS is regulated by the let-7 microRNA family. Cell. 2005, 120 (5): 635-647.
Gurtan AM, Sharp PA: The Role of miRNAs in Regulating Gene Expression Networks. J Mol Biol. 2013, 425 (19): 3582-3600.
Di Leva G, Croce CM: miRNA profiling of cancer. Curr Opin Genet Dev. 2013, 23 (1): 3-11.
Jay C, Nemunaitis J, Chen P, Fulgham P: Tong AW: miRNA profiling for diagnosis and prognosis of human cancer. DNA Cell Biol. 2007, 26 (5): 293-300.
Gravgaard KH, Lyng MB, Laenkholm AV, Sokilde R, Nielsen BS, Litman T, Ditzel HJ: The miRNA-200 family and miRNA-9 exhibit differential expression in primary versus corresponding metastatic tissue in breast cancer. Breast Cancer Res Treat. 2012, 134 (1): 207-217.
Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ: The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008, 10 (5): 593-601.
Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Menard S, Palazzo JP, Rosenberg A, Musiani P, Volinia S, Nenci I, Calin GA, Querzoli P, Negrini M, Croce CM: MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005, 65 (16): 7065-7070.
Lee YM, Lee JY, Ho CC, Hong QS, Yu SL, Tzeng CR, Yang PC, Chen HW: miRNA-34b as a tumor suppressor in estrogen-dependent growth of breast cancer cells. Breast Cancer Res. 2011, 13 (6): R116-
Sun X, Qin S, Fan C, Xu C, Du N, Ren H: Let-7: a regulator of the ERalpha signaling pathway in human breast tumors and breast cancer stem cells. Oncol Rep. 2013, 29 (5): 2079-2087.
Tavazoie SF, Alarcon C, Oskarsson T, Padua D, Wang Q, Bos PD, Gerald WL, Massague J: Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008, 451 (7175): 147-152.
Pastrello C, Polesel J, Della Puppa L, Viel A, Maestro R: Association between hsa-mir-146a genotype and tumor age-of-onset in BRCA1/BRCA2-negative familial breast and ovarian cancer patients. Carcinogenesis. 2010, 31 (12): 2124-2126.
Tanic M, Yanowsky K, Rodriguez-Antona C, Andres R, Marquez-Rodas I, Osorio A, Benitez J, Martinez-Delgado B: Deregulated miRNAs in hereditary breast cancer revealed a role for miR-30c in regulating KRAS oncogene. PLoS One. 2012, 7 (6): e38847-
Azim HA, Michiels S, Bedard PL, Singhal SK, Criscitiello C, Ignatiadis M, Haibe-Kains B, Piccart MJ, Sotiriou C, Loi S: Elucidating prognosis and biology of breast cancer arising in young women using gene expression profiling. Clin Cancer Res. 2012, 18 (5): 1341-1351.
Colak D, Nofal A, Albakheet A, Nirmal M, Jeprel H, Eldali A, Al-Tweigeri T, Tulbah A, Ajarim D, Malik OA, Inan MS, Kaya N, Park BH, Bin Amer SM: Age-specific gene expression signatures for breast tumors and cross-species conserved potential cancer progression markers in young women. PLoS One. 2013, 8 (5): e63204-
de Rinaldis E, Gazinska P, Mera A, Modrusan Z, Fedorowicz GM, Burford B, Gillett C, Marra P, Grigoriadis A, Dornan D, Holmberg L, Pinder S, Tutt A: Integrated genomic analysis of triple-negative breast cancers reveals novel microRNAs associated with clinical and molecular phenotypes and sheds light on the pathways they control. BMC Genomics. 2013, 14 (1): 643-
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR: MicroRNA expression profiles classify human cancers. Nature. 2005, 435 (7043): 834-838.
Heneghan HM, Miller N, Kelly R, Newell J, Kerin MJ: Systemic miRNA-195 differentiates breast cancer from other malignancies and is a potential biomarker for detecting noninvasive and early stage disease. Oncologist. 2010, 15 (7): 673-682.
Heneghan HM, Miller N, Kerin MJ: Circulating miRNA signatures: promising prognostic tools for cancer. J Clin Oncol. 2010, 28 (29): e573-e574. author reply e575-576
Enerly E, Steinfeld I, Kleivi K, Leivonen SK, Aure MR, Russnes HG, Ronneberg JA, Johnsen H, Navon R, Rodland E, Makela R, Naume B, Perala M, Kallioniemi O, Kristensen VN, Yakhini Z, Borresen-Dale AL: miRNA-mRNA integrated analysis reveals roles for miRNAs in primary breast tumors. PLoS One. 2011, 6 (2): e16915-
Farazi TA, Horlings HM, Ten Hoeve JJ, Mihailovic A, Halfwerk H, Morozov P, Brown M, Hafner M, Reyal F, van Kouwenhove M, Kreike B, Sie D, Hovestadt V, Wessels LF, van de Vijver MJ, Tuschl T: MicroRNA sequence and expression analysis in breast tumors by deep sequencing. Cancer Res. 2011, 71 (13): 4443-4453.
Rothe F, Ignatiadis M, Chaboteaux C, Haibe-Kains B, Kheddoumi N, Majjaj S, Badran B, Fayyad-Kazan H, Desmedt C, Harris AL, Piccart M, Sotiriou C: Global microRNA expression profiling identifies MiR-210 associated with tumor proliferation, invasion and poor clinical outcome in breast cancer. PLoS One. 2011, 6 (6): e20980-
Bockmeyer CL, Christgen M, Muller M, Fischer S, Ahrens P, Langer F, Kreipe H, Lehmann U: MicroRNA profiles of healthy basal and luminal mammary epithelial cells are distinct and reflected in different breast cancer subtypes. Breast Cancer Res Treat. 2011, 130 (3): 735-745.
Cascione L, Gasparini P, Lovat F, Carasi S, Pulvirenti A, Ferro A, Alder H, He G, Vecchione A, Croce CM, Shapiro CL, Huebner K: Integrated microRNA and mRNA signatures associated with survival in triple negative breast cancer. PLoS One. 2013, 8 (2): e55910-
Dvinge H, Git A, Graf S, Salmon-Divon M, Curtis C, Sottoriva A, Zhao Y, Hirst M, Armisen J, Miska EA, Chin SF, Provenzano E, Turashvili G, Green A, Ellis I, Aparicio S, Caldas C: The shaping and functional consequences of the microRNA landscape in breast cancer. Nature. 2013, 497 (7449): 378-382.
Majumder S, Jacob ST: Emerging role of microRNAs in drug-resistant breast cancer. Gene Expr. 2012, 15 (3): 141-151.
Liu A, Xu X: MicroRNA isolation from formalin-fixed, paraffin-embedded tissues. Methods Mol Biol. 2011, 724: 259-267.
Hui AB, Shi W, Boutros PC, Miller N, Pintilie M, Fyles T, McCready D, Wong D, Gerster K, Waldron L, Jurisica I, Penn LZ, Liu FF: Robust global micro-RNA profiling with formalin-fixed paraffin-embedded breast cancer tissues. Lab Invest. 2009, 89 (5): 597-606.
Han W, Kang SY: Relationship between age at diagnosis and outcome of premenopausal breast cancer: age less than 35 years is a reasonable cut-off for defining young age-onset breast cancer. Breast Cancer Res Treat. 2010, 119 (1): 193-200.
Goldhirsch A, Gelber RD, Yothers G, Gray RJ, Green S, Bryant J, Gelber S, Castiglione-Gertsch M, Coates AS: Adjuvant therapy for very young women with breast cancer: need for tailored treatments. J Natl Cancer Inst Monogr. 2001, 30: 44-51.
Yau C, Fedele V, Roydasgupta R, Fridlyand J, Hubbard A, Gray JW, Chew K, Dairkee SH, Moore DH, Schittulli F, Tommasi S, Paradiso A, Albertson DG, Benz CC: Aging impacts transcriptomes but not genomes of hormone-dependent breast cancers. Breast Cancer Res. 2007, 9 (5): R59-
Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D: Molecular portraits of human breast tumours. Nature. 2000, 406 (6797): 747-752.
Awsare NS, Martin TA, Haynes MD, Matthews PN, Jiang WG: Claudin-11 decreases the invasiveness of bladder cancer cells. Oncol Rep. 2011, 25 (6): 1503-1509.
Bao W, Fu HJ, Xie QS, Wang L, Zhang R, Guo ZY, Zhao J, Meng YL, Ren XL, Wang T, Li Q, Jin BQ, Yao LB, Wang RA, Fan DM, Chen SY, Jia LT, Yang AG: HER2 interacts with CD44 to up-regulate CXCR4 via epigenetic silencing of microRNA-139 in gastric cancer cells. Gastroenterology. 2011, 141 (6): 2076-2087. e2076
Beauchemin D, Lacombe C, Van Themsche C: PAX2 is activated by estradiol in breast cancer cells of the luminal subgroup selectively, to confer a low invasive phenotype. Mol Cancer. 2011, 10: 148-
Castano Z, Marsh T, Tadipatri R, Kuznetsov HS, Al-Shahrour F, Paktinat M, Greene-Colozzi A, Nilsson B, Richardson AL, McAllister SS: Stromal EGF and IGF1 Together Modulate Plasticity of Disseminated Triple Negative Breast Tumors. Cancer Discov. 2013, 3 (8): 922-935.
Fan Q, He M, Deng X, Wu WK, Zhao L, Tang J, Wen G, Sun X, Liu Y: Derepression of c-Fos caused by MicroRNA-139 down-regulation contributes to the metastasis of human hepatocellular carcinoma. Cell Biochem Funct. 2012, 31 (4): 319-324.
Guo H, Hu X, Ge S, Qian G, Zhang J: Regulation of RAP1B by miR-139 suppresses human colorectal carcinoma cell proliferation. Int J Biochem Cell Biol. 2012, 44 (9): 1465-1472.
Katsushima K, Shinjo K, Natsume A, Ohka F, Fujii M, Osada H, Sekido Y, Kondo Y: Contribution of microRNA-1275 to Claudin11 protein suppression via a polycomb-mediated silencing mechanism in human glioma stem-like cells. J Biol Chem. 2012, 287 (33): 27396-27406.
Kovacevic Z, Fu D, Richardson DR: The iron-regulated metastasis suppressor, Ndrg-1: identification of novel molecular targets. Biochim Biophys Acta. 2008, 1783 (10): 1981-1992.
Leidner RS, Li L, Thompson CL: Dampening enthusiasm for circulating microRNA in breast cancer. PLoS One. 2013, 8 (3): e57841-
Lian ZQ, Wang Q, Li WP, Zhang AQ, Wu L: Screening of significantly hypermethylated genes in breast cancer using microarray-based methylated-CpG island recovery assay and identification of their expression levels. Int J Oncol. 2012, 41 (2): 629-638.
Milde-Langosch K, Karn T, Muller V, Witzel I, Rody A, Schmidt M, Wirtz RM: Validity of the proliferation markers Ki67, TOP2A, and RacGAP1 in molecular subgroups of breast cancer. Breast Cancer Res Treat. 2013, 137 (1): 57-67.
Ozata DM, Caramuta S, Velazquez-Fernandez D, Akcakaya P, Xie H, Hoog A, Zedenius J, Backdahl M, Larsson C, Lui WO: The role of microRNA deregulation in the pathogenesis of adrenocortical carcinoma. Endocr Relat Cancer. 2011, 18 (6): 643-655.
Qian YW, Chen Y, Yang W, Fu J, Cao J, Ren YB, Zhu JJ, Su B, Luo T, Zhao XF, Dai RY, Li JJ, Sun W, Wu MC, Feng GS, Wang HY: p28(GANK) prevents degradation of Oct4 and promotes expansion of tumor-initiating cells in hepatocarcinogenesis. Gastroenterology. 2012, 142 (7): 1547-1558. e1514
Romanuik TL, Ueda T, Le N, Haile S, Yong TM, Thomson T, Vessella RL, Sadar MD: Novel biomarkers for prostate cancer including noncoding transcripts. Am J Pathol. 2009, 175 (6): 2264-2276.
Shen K, Liang Q, Xu K, Cui D, Jiang L, Yin P, Lu Y, Li Q, Liu J: MiR-139 inhibits invasion and metastasis of colorectal cancer by targeting the type I insulin-like growth factor receptor. Biochem Pharmacol. 2012, 84 (3): 320-330.
Tajnik M, Strazisar M, Volavsek M, Bostjancic E, Glavac D: BBC3 is down-regulated with increased tumor size independently of p53 expression in head and neck cancer. Cancer Biomark. 2012, 11 (5): 197-208.
Webb PG, Spillman MA, Baumgartner HK: Claudins play a role in normal and tumor cell motility. BMC Cell Biol. 2013, 14: 19-
Wong FY, Liem N, Xie C, Yan FL, Wong WC, Wang L, Yong WP: Combination therapy with gossypol reveals synergism against gemcitabine resistance in cancer cells with high BCL-2 expression. PLoS One. 2012, 7 (12): e50786-
Yan B, Zhao JL: miR-1228 prevents cellular apoptosis through targeting of MOAP1 protein. Apoptosis. 2012, 17 (7): 717-724.
Leivonen SK, Makela R, Ostling P, Kohonen P, Haapa-Paananen S, Kleivi K, Enerly E, Aakula A, Hellstrom K, Sahlberg N, Kristensen VN, Borresen-Dale AL, Saviranta P, Perala M, Kallioniemi O: Protein lysate microarray analysis to identify microRNAs regulating estrogen receptor signaling in breast cancer cell lines. Oncogene. 2009, 28 (44): 3926-3936.
Queiros AM, Eschen C, Fliegner D, Kararigas G, Dworatzek E, Westphal C, Sanchez Ruderisch H, Regitz-Zagrosek V: Sex- and estrogen-dependent regulation of a miRNA network in the healthy and hypertrophied heart. Int J Cardiol. 2013, 169 (5): 331-338.
Fleming JM, Miller TC, Quinones M, Xiao Z, Xu X, Meyer MJ, Ginsburg E, Veenstra TD, Vonderhaar BK: The normal breast microenvironment of premenopausal women differentially influences the behavior of breast cancer cells in vitro and in vivo. BMC Med. 2010, 8: 27-
Roman-Perez E, Casbas-Hernandez P, Pirone JR, Rein J, Carey LA, Lubet RA, Mani SA, Amos KD, Troester MA: Gene expression in extratumoral microenvironment predicts clinical outcome in breast cancer patients. Breast Cancer Res. 2012, 14 (2): R51-
Sun X, Gierach GL, Sandhu R, Williams T, Midkiff BR, Lissowska J, Wesolowska E, Boyd NF, Johnson NB, Figueroa JD, Sherman ME, Troester MA: Relationship of mammographic density and gene expression: analysis of normal breast tissue surrounding breast cancer. Clin Cancer Res. 2013, 19 (18): 4972-4982.
Anderson WF, Jatoi I, Sherman ME: Qualitative age interactions in breast cancer studies: mind the gap. J Clin Oncol. 2009, 27 (32): 5308-5311.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/14/529/prepub
Acknowledgements
MPC is funded by the Generalitat Valenciana VALi + d, ACIF/2011/270. MTM is funded by”Rio Hortega Project” (CM12/00264). GR is a FIS “Miquel Servet” Researcher. AB holds a Translational Research Grant awarded by the Spanish Society of Medical Oncology (SEOM). This project was carried out thanks to Fundación LeCadó – proyecto Flor de Vida and co-funded by FIS project PI13/00606 and FEDER. We would like to give thanks to all the patients and volunteers for their participation and also to the INCLIVA Biobank, integrated into the Spanish Hospital Biobanks Network (ReTBioH) and supported by the Instituto de Salud Carlos III/FEDER (grant number: RD09/0076/00132). We also wish to thank several private Breast cancer associations that funded this study and the Unit for Multigenic Analysis from the Central Unit for Medical Research (UCIM/INCLIVA) for the performance of the Affymetrix microRNA profiles.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MPC processed the material and extracted RNA, prepare samples for Affymetrix array, performed qRT-PCR, acquired and analyzed all the data and performed statistical analyses, carried out databases and literature searching and contributed in the drafting of the manuscript; MTM performed the selection of patients suitable for the study, helped processing the samples, gave intellectual support, performed literature searching and contributed writing the manuscript; JAPF conceived the study, participated in the patients selection, contributed in the interpretation of the results and in the manuscript writing; LPC processed samples, optimized the used technique and acquired data, also provide useful information and intellectual content to the manuscript drafting; ET, PE and JC helped in the sample processing, interpretation of the results and revised final manuscript; BMD helped in the data analyses, interpretation of the results and in drafting the manuscript; EA, OB and JFL performed the pathological analyses, characterized the tumour tissue samples and performed immunohistochemical techniques, also provide the FFPE material and provide intellectual content and information for the manuscript drafting; AB has been involved in revising the manuscript critically for important intellectual content; AL performed the conceptual design of the study, supervision of the whole study, revised the manuscript and provide intellectual content, also participated in the acquisition of funding; GR conceived and supervised the whole study, contributed in the interpretation of the data, gave intellectual support, contributed in the manuscript writing and helped in the acquisition of funding. All authors have given final approval of the version to be published.
Electronic supplementary material
12885_2013_4714_MOESM1_ESM.pdf
Additional file 1: Haematoxylin and eosin staining images from tissue samples used in the present study. Sections from FFPE tissue blocks stained with haematoxylin and eosin. A, B: correspond to samples from normal healthy mammary tissue. C, D: represent the mammary tumour from young women. E, F: show breast tumour tissue obtained from older women. (PDF 320 KB)
12885_2013_4714_MOESM2_ESM.pdf
Additional file 2: List and p-values of miRNAs significantly differences in expression between BCVY and BC. Significantly different expressed miRNAs among two sample groups: young women in breast cancer (BCVY, younger than 35 years old) and older than 65 years women with breast cancer (BC). P-values and FDR p-values (corrected for Benjamini and Hochberg’s False Discovery Rate for multiple comparisons) were obtained performing a t-test with 200000 permutations. Nomenclature of miRNAs belong to miRBase v.15. (PDF 253 KB)
12885_2013_4714_MOESM3_ESM.pdf
Additional file 3: List and p-values of miRNAs significantly differences in expression between BCVY and BC. Significantly different expressed miRNAs among two sample groups: young women in breast cancer (BCVY, younger than 35 years old) and older than 65 years women with breast cancer (BC), headed by Age FDR (from Additional file 1: Table S1), the association of miRNA was further analyzed correcting for tumour characteristics (grade, tumour size, Ki67 and nodal status) to assess whether there are any confounding factor. FDR p-values are corrected for Benjamini and Hochberg’s False Discovery Rate for multiple comparisons and were obtained performing a t-test with 200000 permutations. Nomenclature of miRNAs belong to miRBase v.15. (PDF 347 KB)
12885_2013_4714_MOESM4_ESM.pdf
Additional file 4: MicroRNA selected for this study from miRNAs significantly associated with young women’s breast cancer in discovery set and its validation. Clusters were constructed using Average hierarchical clustering method. Statistics of the discovery set were calculated via t-test. FDR stands for False Discovery rate multitesting corrected p-value. Only miRNAs remarked in bold were validated on an independent set of patients. Fold change of the validation set were calculated using ΔΔCt method and p-values were estimated via ANOVA test comparing the age groups and adjusted for multiple testing using Tukey’s method. Bold denotes significant associated results in validation set. (PDF 210 KB)
12885_2013_4714_MOESM5_ESM.pdf
Additional file 5: Putative pathways enriched with the miRNAs included in the sub-node 15 and 63. Code between brackets refers to the pathway code on KEGG, hsa means homo sapiens. FDR refers to p-value adjusted by False Discovery Rate. 20 miRNAs were included in a pathway enrichment analysis and obtained the genes union results. (PDF 300 KB)
12885_2013_4714_MOESM6_ESM.pdf
Additional file 6: Information about miRNAs selected for validation. Information of the miRNAs selected for validation, including 3p or 5p form (3’ or 5’), MIMAT ID, family and sequence of the mature miRNA. “*” refers to the minor form of the miRNA, according to miRBase v.15 nomenclature. (PDF 192 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Peña-Chilet, M., Martínez, M.T., Pérez-Fidalgo, J.A. et al. MicroRNA profile in very young women with breast cancer. BMC Cancer 14, 529 (2014). https://doi.org/10.1186/1471-2407-14-529
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-14-529