
Abstract
Background
Therapeutic decisions in cancer are generally guided by molecular biomarkers or, for some newer therapeutics, primary tumor genotype. However, because biomarkers or genotypes may change as new metastases emerge, circulating tumor cells (CTCs) from blood are being investigated for a role in guiding real-time drug selection during disease progression, expecting that CTCs will comprehensively represent the full spectrum of genomic changes in metastases. However, information is limited regarding mutational heterogeneity among CTCs and metastases in breast cancer as discerned by single cell analysis. The presence of disseminated tumor cells (DTCs) in bone marrow also carry prognostic significance in breast cancer, but with variability between CTC and DTC detection. Here we analyze a series of single tumor cells, CTCs, and DTCs for PIK3CA mutations and report CTC and corresponding metastatic genotypes.
Methods
We used the MagSweeper, an immunomagnetic separation device, to capture live single tumor cells from breast cancer patients’ primary and metastatic tissues, blood, and bone marrow. Single cells were screened for mutations in exons 9 and 20 of the PIK3CA gene. Captured DTCs grown in cell culture were also sequenced for PIK3CA mutations.
Results
Among 242 individual tumor cells isolated from 17 patients and tested for mutations, 48 mutated tumor cells were identified in three patients. Single cell analyses revealed mutational heterogeneity among CTCs and tumor cells in tissues. In a patient followed serially, there was mutational discordance between CTCs, DTCs, and metastases, and among CTCs isolated at different time points. DTCs from this patient propagated in vitro contained a PIK3CA mutation, which was maintained despite morphological changes during 21 days of cell culture.
Conclusions
Single cell analysis of CTCs can demonstrate genotypic heterogeneity, changes over time, and discordance from DTCs and distant metastases. We present a cautionary case showing that CTCs from any single blood draw do not always reflect metastatic genotype, and that CTC and DTC analyses may provide independent clinical information. Isolated DTCs remain viable and can be propagated in culture while maintaining their original mutational status, potentially serving as a future resource for investigating new drug therapies.
Similar content being viewed by others

Background
Clinical use of some newer or investigational drug therapies in cancer requires that primary tumors be assayed for specific mutations associated with response or lack of response [1–5]. However, not only are primary tumors known to be mutationally heterogeneous [6–8], new mutations may become apparent in recurrent tumors, emerging during disease progression [9–11]. Yet sequential biopsy and evaluation of molecular biomarkers and mutations in metastases is not routinely done, even during clinical trials [11], largely due to the multiplicity and internal location of many metastases (such as liver, lung, and/or brain metastases in breast cancer), and potential morbidity associated with sequential biopsy. An appealing alternative is a “liquid biopsy” with CTC capture and characterization [12, 13]. As they are easily accessible by simple blood draw, CTCs can be sequentially sampled at multiple time points during the course of disease for biomarker or genotype determination. Moreover, it is hoped, but not ascertained, that CTCs represent mixtures of tumor cells that reflect the full spectrum of molecular phenotypes and genotypes present in multiple metastases.
Following a slightly different tack, some groups are also investigating biomarker and genetic characteristics of DTCs from bone marrow [14–16], which are postulated to serve as a reservoir for active and dormant tumor cells [12, 17]. However, genetic analyses of mutations in CTCs and DTCs are still in an early discovery stage, having been done on few patients, so clinical significance and utility is postulated but remains unproven. Moreover, it is not known whether CTC analysis can replace DTC analysis for there is, as yet, incomplete understanding of the relationship between these two populations [18].
In the present study, we used a previously described magnetic separation technology that isolates live single cells [19–21] for mutation analysis of single cells from different compartments in metastatic breast cancer patients and also demonstrate growth in culture of patient DTCs from bone marrow. For single CTC/DTC/tumor mutation analysis, we have chosen to interrogate exons 9 and 20 of the PIK3CA gene, one of the most frequently mutated genes in breast cancer [22–25]. We demonstrate that this mutation can be detected in single tumor cells isolated from breast cancer patient primary tumor, blood, bone marrow, and metastases, and track mutational status of CTCs over time in a metastatic breast cancer case example and in cultured DTCs from this patient. While we have previously shown that individual CTCs in breast cancer, even from the same blood draw, are transcriptionally heterogeneous [21], here we investigate mutational heterogeneity and concordance among CTCs, DTCs, and single tumor cells from primary tumors and metastases. In particular, for CTCs to be ultimately used to guide drug selection, we hypothesized that CTCs should indeed contain the mutational changes found in metastases. However, our results were surprising and we present here a case that provides a cautionary note that CTCs from any one blood draw alone may not always represent the mutational status of tumor cells in bone marrow or distant metastases.
Methods
Ethics statement
This study protocol was approved by Stanford’s Human Subjects Research and Institutional Review Board (Protocol 5630). Written informed consent was explained and signed by all participating patients prior to sample collection.
Tumor cell isolation, staining, and culture
Single cell suspensions used for MagSweeper tumor cell isolation were prepared from primary and metastatic tissue from breast cancer patients. Tumor chunks were finely minced, gently pulled to release single cells or small cell clusters, filtered through a 70 micron mesh followed by centrifugation of the filtrate at 1900 g. The supernatant was discarded and the pellet was resuspended in 1x trypsin (Invitrogen/Life Technologies, Carlsbad, CA, USA) for 5–10 minutes. DMEM culture media with 10% FBS (Gibco/Life Technologies, Carlsbad, CA, USA) was added to stop the trypsin reaction. The target single tumor cells were labeled with EpCAM-conjugated microbeads and isolated by the MagSweeper as previously described [19, 21]. Individual tumor cells were aspirated under direct microscopic visualization (Axio Observer A1, Zeiss, Thornwood, NY, USA). Authenticated MCF7 and BT474 human breast cancer cell lines (ATCC, Manassas, VA, USA) were grown in DMEM and trypsinized to release single cells, which were then isolated by the MagSweeper and manually aspirated as single cells.
For immunostaining assays, EpCAM-captured cells were treated with DNase I Solution (StemCell Technologies, Vancouver, BC, Canada) to remove the DNA-linker on the magnetic microbeads, and placed on slides. Tumor cells were defined by immunostain assay [26–28] as cells that stained positive for purified anti-cytokeratin (CK+) CAM 5.2 (BD Biosciences, San Jose, CA, USA) and DAPI nuclear stain (DAPI+) (VECTASHIELD Mounting Medium with DAPI, Vector Laboratories, Burlingame, CA, USA), and negative for CD45 (CD45-) (CD45 Ab-1/Bra55, NeoMarkers Lab Vision, Thermo Fisher Scientific, Kalamazoo, MI, USA). White blood cells (WBCs) from patients were collected using the MagSweeper and CD45 Dynabeads (Invitrogen, Carlsbad, CA, USA) and similarly immunostained.
Bone marrow aspirates obtained from the clinic were filtered through a 70 micron mesh to eliminate debris; the passed through solution was adjusted to 10 ml by adding DMEM. DTCs from the prepared bone marrow sample solution were isolated by the MagSweeper and hundreds of DTCs were directly cultured in DMEM with 10% FBS and 50 μg/ml of penicillin-streptomycin (Gibco/Life Technologies, Carlsbad, CA, USA). The cells were then identified by immunostaining assays. The DTCs were cultured for 21 days and single cells were assayed for mutation detection.
Blood sample collection and MagSweeper isolation of CTCs were isolated as previously described [19, 21]. White blood cells (n = 15, from the blood of Patient 12) were collected using anti-CD45 microbeads.
Mutation analysis
Single tumor cells from 17 breast cancer patients were lysed by incubation with a 1:10 dilution of proteinase K (Qiagen, Valencia, CA, USA) in individual PCR tubes for 20 min at 65°C in 10 μl of 1x GeneAmpPCR buffer II (Applied Biosystems/Life Technologies, Carlsbad, CA, USA). In one of the 17 patients, additional tumor cell clusters were tested from slides sectioned from formalin-fixed paraffin-embedded (FFPE) blocks of primary tumor and a lung metastasis with location confirmed by hematoxylin and eosin (H&E) stain. For tumor tissue fixed on slides, diluted proteinase K was placed on the targeted tissue section area of the slide. The solution was heated at 65°C for 20 min and collected for DNA mutation analysis. The DNA from single CTC, DTC, or tumor tissue cells was then pre-amplified for exons 9 and 20 of the PIK3CA gene, using Pfu Ultra DNA polymerase (Agilent Technologies, Santa Clara, CA, USA) with one set of primers for each exon (exon 9 forward primer: CTGTGAATCCAGAGGGGA, reverse primer: CAGAGAATCTCCATTTTAGCAC; exon 20 forward primer: GGAATGCCAGAACTACAATCTTTTG, reverse primer: CCTATGCAATCGGTCTTTGC). The reaction was amplified for 30 cycles at 94°C, 55°C, and 72°C for 30 sec per cycle for each temperature. The pre-amplified products were then diluted 1:10 in distilled water (DW) and 5 μl of the diluted products were used for a single 50 μl PCR using the same primers as above for exon 9, and an internal primer pair for exon 20 that decreased non-specific background amplification (forward primer: GTGGAATCCAGAGTGAGC, reverse primer: TTGCATACATTCGAAAGACC) [25]. PCR products were checked by 2% agarose gel against a GeneRuler 50 bp DNA Ladder (Frementas, Glen Burnie, MD, USA) and sequenced by BigDye Terminator v3.1 Cycle Sequencing Kits according to the manufacturer’s recommended protocol (Applied Biosystems/Life Technologies, Carlsbad, CA, USA). The sequenced results were analyzed with Sequencher 4.8 software (Gene Codes Corporation, Ann Arbor, MI, USA). DW was used as a negative template control. Single authenticated MCF7 and BT474 cells were used as amplification and sequencing controls: MCF7 cells contain a G1633A mutation in exon 9 and are wild type in exon 20; BT474 cells contain an exon 1 mutation but are wild type in exons 9 and 20. BT20 cells, which contain the A3140G mutation in exon 20, served as a positive control for sequencing of exon 20 [29]. Fifteen single WBCs isolated from blood samples also served as wild type control for sequencing.
Results
Patient samples and tumor cell identification
From a group of 17 breast cancer patients, single tumor cells were collected using anti-EpCAM microbeads and the MagSweeper from 30 blood samples, a bone marrow biopsy, and six fresh tumor tissues (Table 1, and Table 2). CTCs from blood, DTCs from bone marrow, and tumor cells from fresh primary and metastatic tumors were defined by immunostain assay as cells that were CK+, CD45-, DAPI+. WBCs collected from the blood of one patient (Patient 12) using anti-CD45 microbeads for use as an additional wild type control for sequencing were confirmed by immunostaining to be CK negative/very weak, CD45 positive, and DAPI positive (Figure 1). Overall, 769 individual CTCs from blood, 75 DTCs from bone marrow, and 60 single TCs from fresh primary tumor tissue (chunk and core needle biopsy), lymph node metastasis, and a bone metastasis to the spine were collected. Of these, 185 CTCs, 24 DTCs, 33 single TCs from fresh primary and metastatic tissues (total 242 single cells) were collected for mutational analysis. Additional tumor cell clusters from two FFPE samples of primary tumor and a lung metastasis were also analyzed for PIK3CA mutations (Table 3).

Single cell isolation and identification. Individual tumor cells (TCs) from primary or metastatic tissue, blood, or bone marrow were isolated by the MagSweeper and immunostained (200×). Panel (A) shows examples of CTCs and DTCs (CK+, CD45-, DAPI+); small round circles in two frames of (A) are autofluorescing residual magnetic microbeads; Panel (B) shows an example of TCs from tumor tissue (CK+, CD45-, DAPI+); Panel (C) shows examples of WBCs (CK-/very weak, CD45+, DAPI+). Green = cytokeratin; red = CD45; blue = DAPI nuclear stain.
DTC culture
The MagSweeper facilitates the isolation of viable target cells that can be grown in culture. Pooled bead-captured DTCs (Figure 2A) showed distinct morphological changes when grown over a 21-day cell culture period (Figure 2B and C). At the time of bone marrow biopsy, DTCs had round shapes when examined fresh and after capture by the MagSweeper (Figure 2A). However, onward from day 5, cell shapes started varying, with some changing to elongated or irregular shapes, although immunostaining assays at different stages demonstrated that all cultured cells remained CK+, CD45- and DAPI+, as expected of tumor cells (Figure 2B and C).

Live DTCs captured by the MagSweeper and propagated in vitro. (A) DTCs detected in fresh smear of bone marrow aspirate among a background of red blood cells (RBCs) and white blood cells (WBCs) (left panel); unstained EpCAM-captured DTCs from bone marrow aspirate (right panel); small black circles are EpCAM-conjugated microbeads. (B) EpCAM-captured tumor cells (200×) grown in culture for three weeks: tumor cells proliferated days 1–4, but began changing shape on day 5. (C) Tumor cells identified by immunostaining at different timepoints during cell culture period (a = day 1; b-f = day 21 at 200×); upper panel 1 shows cell morphology by brightfield; middle panel 2 shows immunostain images (all cells were CK+, CD45- and DAPI+); lower panel 3 shows varying nuclear morphology of cells between day 1 day 21. Green = cytokeratin, red = CD45, blue = DAPI nuclear stain.
PIK3CAgene mutations
To first assess our mutation detection method, targeted DNA from single tumor cells or cells from cell lines was successfully pre-amplified by PCR and the expected bands were confirmed for PIK3CA exons 9 (216 bp) and 20 (269 bp) (Figure 3A). Using the pre-amplified PCR products as new DNA templates, exons 9 and 20 were separately amplified for sequencing, amplifying exon 9 using the original primers and exon 20 with internal primers. Single PCR bands for each exon verified the specificity of the amplification prior to sequencing (Figure 3B). A known heterozygous PIK3CA exon 9 mutation, G1633A (E545K), was identified in replicate samples of single MCF7 cells (positive control), single DTCs, and single CTCs, but was not detected in single WBCs (wild type/negative control) or single BT474 cells (which carry a PIK3CA mutation in exon 1, but no mutations in exons 9 or 20, another negative control) [30–33] (Figure 3C). Note that A1634C (E545A) in the chromatogram in Figure 3B sometimes showed a pseudogene that may be co-amplified with these primers [34]. No exon 20 mutation was identified in negative control MCF7 and BT474 cells, any patient tumor cells, or captured WBCs among our samples, although sequencing of BT20 cells (positive control) detected the expected exon 20 mutation at A3140G (H1047R), confirming the accuracy of our sequencing method (Figure 3D).

Single cell PIK3CA mutation detection. (A) Target DNA from two single tumor cells (SC1, SC2) successfully pre-amplified by PCR with the expected bands for PIK3CA exon 9 (216 bp) and exon 20 (269 bp). (B) Second round of amplification using the pre-amplified PCR products from (A) as new DNA templates to separately amplify exon 9 (using original primers, 216 bp) and exon 20 (using internal primers, 192 bp). P1 = PCR product from first round of amplification; P2 = PCR product from second round of amplification. (C) Sanger sequencing results for PIK3CA mutation G1633A on exon 9: two MCF7 single cells shown here carry the G1633A heterozygous mutation; Normal DNA, BT474 single cells, and single WBCs are wild type (G/G); PIK3CA G1633A mutations were detected in single DTCs and CTCs from breast cancer patient 12. The G1633A mutation was distinguishable in the chromatogram from the adjacent A1634C peak from a known pseudogene on chromosome 22 that may be co-amplified with these primers [34], as in sample MCF7.2. (D) Sanger sequencing results for PIK3CA mutation A3140G on exon 20: BT20 cells show the mutation but MCF7 cells, BT474 cells, and CTCs are wildtype for this mutation hotspot.
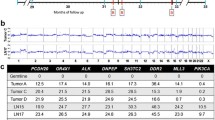
PIK3CA mutation analysis was then performed on the 242 EpCAM-captured single tumor cells (185 CTCs, 24 DTCs, and 33 tumor cells). Three out of 17 (18%) patients showed the PIK3CA exon 9 G1633A mutation in tissue; no exon 20 mutations were detected (Table 1, Table 2 and Table 3). Of these three patients (Patient 12, Patient 14, and Patient 15), 33 individual tumor cells obtained from six fresh clinical samples were sequenced and three showed the exon 9 mutation: 8/8 (100%) single tumor cells from a spinal bone metastasis (Patient 12); 1/5 (20%) single tumor cells from a primary tumor core biopsy (Patient 14); and 3/5 (60%) single tumor cells from a metastatic lymph node (Patient 15). Not unexpectedly, and similar to this patient’s bone metastasis, all 24 (100%) sequenced single DTCs collected from a 3 ml aspirate of Patient 12’s tumor-replaced bone marrow contained the exon 9 mutation (the bone marrow aspirate had been done to determine if tumor overgrowth was causing her pancytopenia, and the DTCs detected were too numerous to count). Interestingly, the FFPE sample of Patient 12’s primary tumor was wild type, but a tissue section of her lung metastasis also showed the PIK3CA exon 9 mutation.
CTCs from Patient 12 were captured periodically over 14 months, during changing treatment of her metastatic breast cancer (Table 1, Table 2, Table 3 and Table 4, Figure 4). Remarkably, in this patient with mutant DTCs in her bone marrow and mutant tumor cells in her spinal bone metastasis and lung metastasis, CTCs in some blood draw samples were discordant and did not show the expected mutation. Notably, of ten blood samples collected from this patient over time, only nine blood samples contained capturable CTCs; of the 128 CTCs sequenced from these nine samples, mutant CTCs were detected in only two blood draws: at one and six weeks after bone marrow biopsy, with mutant cells comprising 50% (10/20) and 29% (2/7) of EpCAM-captured cells, respectively (Table 1, Table 2, Table 3 and Table 4, Figure 4). As expected, all 15 WBCs analyzed from this patient were wild type. Our single cell data thus indicate that not only may there be mutational heterogeneity within a sample, there may be genotypic discordance between CTCs, DTCs and metastases, or between CTC samples isolated at different time points.

PIK3CA G1633A mutation detection in CTCs and tissues over time. One patient, Patient 12, with progressive metastatic breast cancer underwent tissue evaluation and PIK3CA sequencing of a bone (spine) metastasis, lung metastasis, bone marrow biopsy and aspirate (DTCs), and sequential blood draws for one and a half years. Eight out of 10 EpCAM-captured single cells (two cells did not get PCR products for sequencing) in the core needle biopsy of a bone metastases (Bone Bx) from the lumbar spine carried the PIK3CA exon 9 heterozygous mutation G1633A; a lung biopsy (Lung Bx) also showed this mutation; 24 out of 24 EpCAM-captured single cells analyzed from the bone marrow biopsy (BM Bx) carried the heterozygous mutation (EpCAM-captured DTCs retrieved from the bone marrow aspirate). The G1633A mutation was detected only twice in CTCs captured from nine blood samples: one week after bone marrow biopsy, with 10/20 EpCAM-captured cells having the mutation, and six weeks after bone marrow biopsy, with 2/7 EpCAM captured cells having the mutation. Drug treatments are noted (RAD001 = everolimus, an mTOR inhibitor). Note that increasing CTC counts dramatically decreased after treatment with capecitabine and everolimus.
In addition to mutational heterogeneity and discordance, this patient’s clinical pathology showed discordance between different sites when tested for the standard breast cancer biomarkers estrogen receptor (ER), progesterone receptor (PR), and HER2 growth factor receptor: primary tumor and lung were hormone receptor (ER and/or PR) positive, whereas bone marrow and bone were hormone receptor negative (Table 4). No tissue sample was HER2 positive.
Finally, when Patient 12’s DTCs were propagated in vitro, all cultured cells maintained the original PIK3CA exon 9 mutation when again tested on day 21, despite the observed morphological changes described above.
Discussion
Investigations are underway exploring the clinical utility of CTCs and DTCs in monitoring cancer patients undergoing systemic drug therapy [35]. However, a recent prospective randomized phase III clinical trial of patients with metastatic breast cancer (SWOG S0500) showed that early change in therapy based on persistently elevated CTC counts three weeks after starting a drug did not change patient outcome - likely due to poor efficacy of the drugs these metastatic patients received after switching therapy [36]. One upshot of this study is the expectation that, in the future, the measurement of CTC biomarkers or genotype, rather than only CTC enumeration, should offer better prediction of which drugs will be efficacious.
However, as demonstrated here, primary tumors and metastases can be heterogeneous, and different metastases do not always display the same biological markers. It is not clinically feasible to biopsy all metastases in a given patient at any one time point, and certainly not for serial sampling over the course of disease, so it is hoped that sampling CTCs will reflect the spectrum of tumor cells requiring treatment in metastatic disease. Here in a patient with progressive metastatic breast cancer, we compared the PIK3CA mutation status of sequentially sampled CTCs to that of tumor cells from two biopsied metastases and DTCs from bone marrow. Unfortunately, and surprisingly, our data did not support the premise that CTCs in most blood draws were reflective of metastatic genotype. While we did show that different metastases contained discordant biomarkers (Table 4), PIK3CA mutations in CTCs were heterogeneously present in only 2/9 serial blood draws in this patient with multiple distant metastases, two of which (lung and spine) contained tumor cells carrying mutations and whose bone marrow was full of mutant DTCs.
This finding causes pause and suggests that different factors may be at work. One may be that the CTCs analyzed here were captured using the EpCAM cell surface marker. It is postulated that among tumor cells shed from a tissue, many undergo epithelial-mesenchymal transition (EMT), with expression of EMT-associated genes and proteins, and we and others have demonstrated EMT gene and protein expression in CTCs [21, 37–39]. Although CTCs in most EMT studies have been captured with EpCAM antibodies, as EMT progresses, EpCAM expression likely diminishes. Thus, CTCs may consist of populations of EpCAM-expressing and non-EpCAM expressing CTCs. One of the limitations of this study is that because of the technology applied, we studied only EpCAM-expressing CTCs. There may be other non-EpCAM-expressing CTCs present in the blood samples that may have shown a mutant genotype not identified in some of the EpCAM-expressing CTCs. While the mutant tumor cells from metastases in our study were also captured using anti-EpCAM magnetic beads, these cells may potentially have been seeded from EpCAM-negative CTCs that had undergone mesenchymal-epithelial-transition after lodging and growing in the metastatic site, with re-expression of EpCAM on their cell surface. We are now testing different cell surface markers and label-free capture technologies to address this issue, which is particularly important because of recent data suggesting an association between EpCAM-negative CTCs and brain metastases [40].
A second limitation or explanation of our findings is that we do not know the role of drug treatment in suppressing the appearance of mutant tumor cells in the circulation. For example, at the time that 50 of Patient 12’s CTCs showed no mutation, the patient was receiving RAD001 (everolimus), an mTOR inhibitor that may be more active against cells carrying PIK3CA mutations [41]; her CTC count subsequently dropped to zero, perhaps showing response to therapy over time.
A third limitation may be that sequencing only two common hotspots on the PIK3CA gene may miss other genetic variations that could occur during the evolution of progressive metastatic disease, and which may have been shared by the CTCs and metastases. Very recent and exciting work is underway to develop rigorous methods for investigating single cell whole-exome sequencing of CTCs (also captured using MagSweeper technology) [42].
However, given current technology development, our study is important in that it adds a note of caution to using CTCs from only a single blood draw to depict the mutational status in any given patient with metastatic disease for treatment purposes. Treatment decisions based on CTC mutational status should only be done under the auspices of a clinical trial.
While CellSearch™ is the only FDA-approved test for enumeration of CTCs, other CTC capture and characterization technologies, including single cell analysis, are rapidly advancing [43–46]. As sequencing technologies progress and cost decreases, it is anticipated that CTC and DTC genotyping will become more clinically feasible. Genotyping of CTCs or DTCs has generally been performed on pooled samples [47–53]. Studying potentially mixed subpopulations, mutant and wild type, may not be as informative regarding which cells respond to which drugs. However, there are reports describing single cell copy number alterations or mutations in CTCs or DTCs from breast [54–56] or other cancers [12, 57–60] using array comparative genomic hybridization and/or sequencing. Like ours, these studies tend to be small, describing only a few cases with aberrant CTC or DTC DNA, but results are encouraging. Tumor cell genotyping at the single cell level may become important clinically because different tumor cell genotypes may be responsive or resistant to different treatments. Thus, capturing and analyzing single tumor cells using deep sequencing of cancer-related genes may lead to even better clarification for selective drug targeting.
However, the lack of mutational discordance between some of the CTC blood draws in Patient 12 and the known presence of mutations in several metastases, raises questions regarding what tumor cells from metastases are released into the circulation, how they may be preferentially impacted by systemic chemotherapy, and whether bone marrow biopsy would be valuable to augment CTC information prior to switching chemotherapy. In the long run, a prospective clinical trial would be needed to determine whether CTCs alone can optimally guide drug therapy and impact survival, or whether assaying both CTCs and DTCs may be better, or whether this is moot until drugs are developed that will ablate metastatic cancer cells.
On a more positive note, we were able to isolate live DTCs and propagate them in culture. Although cell morphology changed over time, mutation status did not. Reports of culturing DTCs are rare [16, 61], and this is the second to report in vitro conservation of mutational status [16]. As extent of growth of DTCs in culture has been previously associated with poor clinical outcome [16], this lays the groundwork for future in vitro drug testing experiments using novel therapeutics against DTCs isolated in treatment-refractory patients.
Conclusions
Single cell analysis of CTCs can reveal genotypic heterogeneity that may change over time, and can show mutational discordance with DTCs and distant metastases. We present a case suggesting that CTCs may not always reflect the full spectrum of mutations in metastatic disease - or that mutant cells in the circulation may have been more susceptible to the systemic therapy being administered. We postulate that bone marrow may be a more privileged and perhaps chemo-refractory site than the bloodstream, so analyzing both CTCs and DTCs may provide independent clinical information potentially relevant to treatment decisions. Isolated DTCs remain viable and can be propagated in culture while maintaining their original mutational status, and thus may serve as a resource for investigating new drug therapies in the future.
Abbreviations
- CTCs:
-
Circulating tumor cells
- DAPI:
-
4',6-diamidino-2-phenylindole
- DTCs:
-
Disseminated tumor cells
- EDTA:
-
Ethylenediaminetetraacetic acid
- EMT:
-
Epithelial-mesenchymal transition
- EpCAM:
-
Epithelial cell adhesion molecule
- ER:
-
Estrogen receptor
- H&E:
-
Hematoxylin and eosin
- HER2:
-
Human epidermal growth factor receptor 2
- mTOR:
-
Mammalian target of rapamycin
- PCR:
-
Polymerase chain reaction
- PIK3CA:
-
Phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha gene
- PR:
-
Progesterone receptor.
References
Fritsch C, Huang A, Chatenay-Rivauday C, Schnell C, Reddy A, Liu M, Kauffmann A, Guthy D, Erdmann D, De Pover A, Furet P, Gao H, Ferretti S, Wang Y, Trappe J, Brachmann SM, Maira SM, Wilson CJ, Boehm M, Garcia-Echeverria C, Chene P, Wiesmann M, Cozens R, Lehar J, Schlegel R, Caravatti G, Hofmann F, Sellers WR: Characterization of the novel and specific PI3Kalpha inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther. 2014, 13: 1117-1129.
Janku F, Hong DS, Fu S, Piha-Paul SA, Naing A, Falchook GS, Tsimberidou AM, Stepanek VM, Moulder SL, Lee JJ, Luthra R, Zinner RG, Broaddus RR, Wheler JJ, Kurzrock R: Assessing PIK3CA and PTEN in Early-Phase Trials with PI3K/AKT/mTOR Inhibitors. Cell Rep. 2014, 6: 377-387.
Patel SP, Lazar AJ, Papadopoulos NE, Liu P, Infante JR, Glass MR, Vaughn CS, LoRusso PM, Cohen RB, Davies MA, Kim KB: Clinical responses to selumetinib (AZD6244; ARRY-142886)-based combination therapy stratified by gene mutations in patients with metastatic melanoma. Cancer. 2013, 119: 799-805.
Pao W, Girard N: New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011, 12: 175-180.
De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, Santini D, Tonini G, Cappuzzo F, Frattini M, Molinari F, Saletti P, De Dosso S, Martini M, Bardelli A, Siena S, Sartore-Bianchi A, Tabernero J, Macarulla T, Di Fiore F, Gangloff AO, Ciardiello F, Pfeiffer P, et al: Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010, 11: 753-762.
Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C: Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012, 366: 883-892.
Dupont Jensen J, Laenkholm AV, Knoop A, Ewertz M, Bandaru R, Liu W, Hackl W, Barrett JC, Gardner H: PIK3CA mutations may be discordant between primary and corresponding metastatic disease in breast cancer. Clin Cancer Res. 2011, 17: 667-677.
Fisher R, Pusztai L, Swanton C: Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer. 2013, 108: 479-485.
Jeselsohn R, Yelensky R, Buchwalter G, Frampton GM, Meric-Bernstam F, Gonzalez-Angulo AM, Ferrer-Lozano J, Perez-Fidalgo A, Cristofanilli M, Gomez H, Arteaga CL, Giltnane J, Balko JM, Cronin MT, Jarosz M, Sun J, Hawryluk M, Lipson D, Otto G, Ross J, Dvir A, Soussan-Gutman L, Wolf I, Rubinek T, Gilmore L, Schnitt SJ, Come SE, Pusztai L, Stephens PJ, Brown M, Miller VA: Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor positive breast cancer. Clin Cancer Res. 2014, 20: 1757-1767.
Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A, Delaney A, Gelmon K, Guliany R, Senz J, Steidl C, Holt RA, Jones S, Sun M, Leung G, Moore R, Severson T, Taylor GA, Teschendorff AE, Tse K, Turashvili G, Varhol R, Warren RL, Watson P, Zhao Y, Caldas C, Huntsman D, Hirst M, Marra MA, Aparicio S: Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009, 461: 809-813.
Janku F: Tumor heterogeneity in the clinic: is it a real problem?. Ther Adv Med Oncol. 2014, 6: 43-51.
Pantel K, Alix-Panabières C: Real-time liquid biopsy in cancer patients: fact or fiction?. Cancer Res. 2013, 73: 6384-6388.
Heitzer E, Auer M, Gasch C, Pichler M, Ulz P, Hoffmann EM, Lax S, Waldispuehl-Geigl J, Mauermann O, Lackner C, Höfler G, Eisner F, Sill H, Samonigg H, Pantel K, Riethdorf S, Bauernhofer T, Geigl JB, Speicher MR: Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res. 2013, 73: 2965-2975.
Banys M, Krawczyk N, Fehm T: The role and clinical relevance of disseminated tumor cells in breast cancer. Cancers (Basel). 2014, 6: 143-152.
Møller EK, Kumar P, Voet T, Peterson A, Van Loo P, Mathiesen RR, Fjelldal R, Grundstad J, Borgen E, Baumbusch LO, Naume B, Børresen-Dale AL, White KP, Nord S, Kristensen VN: Next-generation sequencing of disseminated tumor cells. Front Oncol. 2013, 3: 320-
Mathiesen RR, Fjelldal R, Liestøl K, Due EU, Geigl JB, Riethdorf S, Borgen E, Rye IH, Schneider IJ, Obenauf AC, Mauermann O, Nilsen G, Christian Lingjaerde O, Børresen-Dale AL, Pantel K, Speicher MR, Naume B, Baumbusch LO: High-resolution analyses of copy number changes in disseminated tumor cells of patients with breast cancer. Int J Cancer. 2012, 131: E405-E415.
Solakoglu O, Maierhofer C, Lahr G, Breit E, Scheunemann P, Heumos I, Pichlmeier U, Schlimok G, Oberneder R, Kollermann MW, Kollermann J, Speicher MR, Pantel K: Heterogeneous proliferative potential of occult metastatic cells in bone marrow of patients with solid epithelial tumors. Proc Natl Acad Sci U S A. 2002, 99: 2246-2251.
Bednarz-Knoll N, Alix-Panabières C, Pantel K: Clinical relevance and biology of circulating tumor cells. Breast Cancer Res. 2011, 13: 228-
Talasaz AH, Powell AA, Huber DE, Berbee JG, Roh KH, Yu W, Xiao W, Davis MM, Pease RF, Mindrinos MN, Jeffrey SS, Davis RW: Isolating highly enriched populations of circulating epithelial cells and other rare cells from blood using a magnetic sweeper device. Proc Natl Acad Sci U S A. 2009, 106: 3970-3975.
Ameri K, Luong R, Zhang H, Powell AA, Montgomery KD, Espinosa I, Bouley DM, Harris AL, Jeffrey SS: Circulating tumour cells demonstrate an altered response to hypoxia and an aggressive phenotype. Br J Cancer. 2010, 102: 561-569.
Powell AA, Talasaz AH, Zhang H, Coram MA, Reddy A, Deng G, Telli ML, Advani RH, Carlson RW, Mollick JA, Sheth S, Kurian AW, Ford JM, Stockdale FE, Quake SR, Pease RF, Mindrinos MN, Bhanot G, Dairkee SH, Davis RW, Jeffrey SS: Single cell profiling of circulating tumor cells: transcriptional heterogeneity and diversity from breast cancer cell lines. PloS One. 2012, 7: e33788-
Network CGA: Comprehensive molecular portraits of human breast tumours. Nature. 2012, 490: 61-70.
Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, Carey M, Hu Z, Guan Y, Sahin A, Symmans WF, Pusztai L, Nolden LK, Horlings H, Berns K, Hung MC, van de Vijver MJ, Valero V, Gray JW, Bernards R, Mills GB, Hennessy BT: An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008, 68: 6084-6091.
Kalinsky K, Jacks LM, Heguy A, Patil S, Drobnjak M, Bhanot UK, Hedvat CV, Traina TA, Solit D, Gerald W, Moynahan ME:2: PIK3CA mutation associates with improved outcome in breast cancer. Clin Cancer Res. 2009, 15: 5049-5059.
Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE: High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004, 304: 554-
Deng G, Herrler M, Burgess D, Manna E, Krag D, Burke JF: Enrichment with anticytokeratin alone or combined with anti-EpCAM antibodies significantly increases the sensitivity for circulating tumor cell detection in metastatic breast cancer patients. Breast Cancer Res. 2008, 10: R69-
Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LW, Hayes DF: Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med. 2004, 351: 781-791.
Braun S, Pantel K, Müller P, Janni W, Hepp F, Kentenich CR, Gastroph S, Wischnik A, Dimpfl T, Kindermann G, Riethmüller G, Schlimok G: Cytokeratin-positive cells in the bone marrow and survival of patients with stage I, II, or III breast cancer. N Engl J Med. 2000, 342: 525-533.
Liang X, Lau QC, Salto-Tellez M, Putti TC, Loh M, Sukumar S: Mutational hotspot in exon 20 of PIK3CA in breast cancer among Singapore Chinese. Cancer Biol Ther. 2006, 5: 544-548.
Higgins MJ, Jelovac D, Barnathan E, Blair B, Slater S, Powers P, Zorzi J, Jeter SC, Oliver GR, Fetting J, Emens L, Riley C, Stearns V, Diehl F, Angenendt P, Huang P, Cope L, Argani P, Murphy KM, Bachman KE, Greshock J, Wolff AC, Park BH: Detection of tumor PIK3CA status in metastatic breast cancer using peripheral blood. Clin Cancer Res. 2012, 18: 3462-3469.
Hollestelle A, Elstrodt F, Nagel JH, Kallemeijn WW, Schutte M: Phosphatidylinositol-3-OH kinase or RAS pathway mutations in human breast cancer cell lines. Mol Cancer Res. 2007, 5: 195-201.
Kataoka Y, Mukohara T, Shimada H, Saijo N, Hirai M, Minami H: Association between gain-of-function mutations in PIK3CA and resistance to HER2-targeted agents in HER2-amplified breast cancer cell lines. Ann Oncol. 2010, 21: 255-262.
Wu G, Xing M, Mambo E, Huang X, Liu J, Guo Z, Chatterjee A, Goldenberg D, Gollin SM, Sukumar S, Trink B, Sidransky D: Somatic mutation and gain of copy number of PIK3CA in human breast cancer. Breast Cancer Res. 2005, 7: R609-
Muller CI, Miller CW, Hofmann WK, Gross ME, Walsh CS, Kawamata N, Luong QT, Koeffler HP: Rare mutations of the PIK3CA gene in malignancies of the hematopoietic system as well as endometrium, ovary, prostate and osteosarcomas, and discovery of a PIK3CA pseudogene. Leukemia Res. 2007, 31: 27-32.
Bidard FC, Fehm T, Ignatiadis M, Smerage JB, Alix-Panabières C, Janni W, Messina C, Paoletti C, Müller V, Hayes DF, Piccart M, Pierga JY: Clinical application of circulating tumor cells in breast cancer: overview of the current interventional trials. Cancer Metastasis Rev. 2013, 32: 179-188.
Smerage JB, Barlow WE, Hortobagyi GN, Winer EP, Leyland-Jones B, Srkalovic G, Tejwani S, Schott AF, O'Rourke MA, Lew DL, Doyle GV, Gralow JR, Livingston RB, Hayes DF: Circulating tumor cells and response to chemotherapy in metastatic breast cancer: SWOG S0500. J Clin Oncol. 2014, Epub ahead of print
Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM, Concannon KF, Donaldson MC, Sequist LV, Brachtel E, Sgroi D, Baselga J, Ramaswamy S, Toner M, Haber DA, Maheswaran S: Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013, 339: 580-584.
Chen CL, Mahalingam D, Osmulski P, Jadhav RR, Wang CM, Leach RJ, Chang TC, Weitman SD, Kumar AP, Sun L, Gaczynska ME, Thompson IM, Huang TH: Single-cell analysis of circulating tumor cells identifies cumulative expression patterns of EMT-related genes in metastatic prostate cancer. Prostate. 2013, 73: 813-826.
Yokobori T, Iinuma H, Shimamura T, Imoto S, Sugimachi K, Ishii H, Iwatsuki M, Ota D, Ohkuma M, Iwaya T, Nishida N, Kogo R, Sudo T, Tanaka F, Shibata K, Toh H, Sato T, Barnard GF, Fukagawa T, Yamamoto S, Nakanishi H, Sasaki S, Miyano S, Watanabe T, Kuwano H, Mimori K, Pantel K, Mori M: Plastin3 is a novel marker for circulating tumor cells undergoing the epithelial-mesenchymal transition and is associated with colorectal cancer prognosis. Cancer Res. 2013, 73: 2059-2069.
Zhang L, Ridgway LD, Wetzel MD, Ngo J, Yin W, Kumar D, Goodman JC, Groves MD, Marchetti D: The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci Transl Med. 2013, 5: 180ra48-Erratum in: Sci Transl Med 2013, 5:189er5
Di Nicolantonio F, Arena S, Tabernero J, Grosso S, Molinari F, Macarulla T, Russo M, Cancelliere C, Zecchin D, Mazzucchelli L, Sasazuki T, Shirasawa S, Geuna M, Frattini M, Baselga J, Gallicchio M, Biffo S, Bardelli A: Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J Clin Invest. 2010, 120: 2858-2866.
Lohr JG, Adalsteinsson VA, Cibulskis K, Choudhury AD, Rosenberg M, Cruz-Gordillo P, Francis JM, Zhang CZ, Shalek AK, Satija R, Trombetta JJ, Lu D, Tallapragada N, Tahirova N, Kim S, Blumenstiel B, Sougnez C, Lowe A, Wong B, Auclair D, Van Allen EM, Nakabayashi M, Lis RT, Lee GS, Li T, Chabot MS, Ly A, Taplin ME, Clancy TE, Loda M, et al: Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat Biotechnol. 2014, 32: 479-484.
Alix-Panabières C, Pantel K: Technologies for detection of circulating tumor cells: facts and vision. Lab Chip. 2014, 14: 57-62.
Krebs MG, Metcalf RL, Carter L, Brady G, Blackhall FH, Dive C: Molecular analysis of circulating tumour cells-biology and biomarkers. Nat Rev Clin Oncol. 2014, 11: 129-144.
Lowes LE, Allan AL: Recent advances in the molecular characterization of circulating tumor cells. Cancers (Basel). 2014, 6: 595-624.
Van Loo P, Voet T: Single cell analysis of cancer genomes. Curr Opin Genet Dev. 2014, 24C: 82-91.
Schneck H, Blassl C, Meier-Stiegen F, Neves RP, Janni W, Fehm T, Neubauer H: Analysing the mutational status of PIK3CA in circulating tumor cells from metastatic breast cancer patients. Mol Oncol. 2013, 7: 976-986.
Harb W, Fan A, Tran T, Danila DC, Keys D, Schwartz M, Ionescu-Zanetti C: Mutational analysis of circulating tumor cells using a novel microfluidic collection device and qPCR assay. Transl Oncol. 2013, 6: 528-538.
Gutiérrez C, Rodriguez J, Patiño-García A, García-Foncillas J, Salgado J: KRAS mutational status analysis of peripheral blood isolated circulating tumor cells in metastatic colorectal patients. Oncol Lett. 2013, 6: 1343-1345.
Fabbri F, Carloni S, Zoli W, Ulivi P, Gallerani G, Fici P, Chiadini E, Passardi A, Frassineti GL, Ragazzini A, Amadori D: Detection and recovery of circulating colon cancer cells using a dielectrophoresis-based device: KRAS mutation status in pure CTCs. Cancer Lett. 2013, 335: 225-231.
Mostert B, Jiang Y, Sieuwerts AM, Wang H, Bolt-de Vries J, Biermann K, Kraan J, Lalmahomed Z, van Galen A, de Weerd V, van der Spoel P, Ramírez-Moreno R, Verhoef C, Ijzermans JN, Wang Y, Gratama JW, Foekens JA, Sleijfer S, Martens JW: KRAS and BRAF mutation status in circulating colorectal tumor cells and their correlation with primary and metastatic tumor tissue. Int J Cancer. 2013, 133: 130-141.
Tórtola S, Steinert R, Hantschick M, Peinado MA, Gastinger I, Stosiek P, Lippert H, Schlegel W, Reymond MA: Discordance between K-ras mutations in bone marrow micrometastases and the primary tumor in colorectal cancer. J Clin Oncol. 2001, 19: 2837-2843.
Raimondi C, Nicolazzo C, Gradilone A, Giannini G, De Falco E, Chimenti I, Varriale E, Hauch S, Plappert L, Cortesi E, Gazzaniga P: Circulating tumor cells: exploring intratumor heterogeneity of colorectal cancer. Cancer Biol Ther. 2014, 15: 496-503.
Klein CA, Seidl S, Petat-Dutter K, Offner S, Geigl JB, Schmidt-Kittler O, Wendler N, Passlick B, Huber RM, Schlimok G, Baeuerle PA, Riethmüller G: Combined transcriptome and genome analysis of single micrometastatic cells. Nat Biotechnol. 2002, 20: 387-392.
Czyż ZT, Hoffmann M, Schlimok G, Polzer B, Klein CA: Reliable single cell array CGH for clinical samples. PLoS One. 2014, 9: e85907-
Magbanua MJ, Sosa EV, Roy R, Eisenbud LE, Scott JH, Olshen A, Pinkel D, Rugo HS, Park JW: Genomic profiling of isolated circulating tumor cells from metastatic breast cancer patients. Cancer Res. 2013, 73: 30-40.
Gasch C, Bauernhofer T, Pichler M, Langer-Freitag S, Reeh M, Seifert AM, Mauermann O, Izbicki JR, Pantel K, Riethdorf S: Heterogeneity of epidermal growth factor receptor status and mutations of KRAS/PIK3CA in circulating tumor cells of patients with colorectal cancer. Clin Chem. 2013, 59: 252-260.
Ni X, Zhuo M, Su Z, Duan J, Gao Y, Wang Z, Zong C, Bai H, Chapman AR, Zhao J, Xu L, An T, Ma Q, Wang Y, Wu M, Sun Y, Wang S, Li Z, Yang X, Yong J, Su XD, Lu Y, Bai F, Xie XS, Wang J: Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proc Natl Acad Sci U S A. 2013, 110: 21083-21088.
Sakaizawa K, Goto Y, Kiniwa Y, Uchiyama A, Harada K, Shimada S, Saida T, Ferrone S, Takata M, Uhara H, Okuyama R: Mutation analysis of BRAF and KIT in circulating melanoma cells at the single cell level. Br J Cancer. 2012, 106: 939-946.
Ulmer A, Schmidt-Kittler O, Fischer J, Ellwanger U, Rassner G, Riethmüller G, Fierlbeck G, Klein CA: Immunomagnetic enrichment, genomic characterization, and prognostic impact of circulating melanoma cells. Clin Cancer Res. 2004, 10: 531-537.
Ross AA, Cooper BW, Lazarus HM, Mackay W, Moss TJ, Ciobanu N, Tallman MS, Kennedy MJ, Davidson NE, Sweet D, Winter C, Akard L, Jansen J, Copelan E, Meagher RC, Herzig RH, Klumpp TR, Kahn DG, Warner NE: Detection and viability of tumor cells in peripheral blood stem cell collections from breast cancer patients using immunocytochemical and clonogenic assay techniques. Blood. 1993, 82: 2605-2610.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/14/456/prepub
Acknowledgments
The authors thank Ms. Loralee Lobato for her help with clinical coordination and maintenance of the patient database. The authors thank Marc A. Coram, Katharina E. Effenberger, Michael Herrler, and Klaus Pantel for helpful scientific discussions, and Kyra Heirich for assistance during the manuscript revision. This study was supported in part by National Institutes of Health grants R01GM085601 (SSJ), P01HG000205 (RWD) and U54GM62119 (RWD), the John and Marva Warnock Cancer Research Fund, the Andrew and Debra Rachleff Fund, the Hubei 100 Professional Program (GD), the Wuhan 3551 Talents Program (GD), and the Longzhong Talents Plan (GD).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
Drs. Stefanie Jeffrey, Ashley Powell, Michael Mindrinos, and Ronald Davis are co-inventors of the MagSweeper device used for tumor cell isolation in this study. Dr Jeffrey has donated her royalties to a non-profit institution. The authors declare that they have no competing interests.
Authors’ contributions
GD, SK, MM, RD, and SJ conceived of the study and participated in its design and coordination. MT contributed patient samples. GD, AP and HZ isolated CTCs, and GD isolated single tumor cells from all other tissues. GD performed the cell culture experiments and immunostains. GD, SK, and MM performed and analyzed the sequencing assays. GD, SK, MM, and SJ drafted the manuscript. All authors have read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited.
About this article

Cite this article
Deng, G., Krishnakumar, S., Powell, A.A. et al. Single cell mutational analysis of PIK3CA in circulating tumor cells and metastases in breast cancer reveals heterogeneity, discordance, and mutation persistence in cultured disseminated tumor cells from bone marrow. BMC Cancer 14, 456 (2014). https://doi.org/10.1186/1471-2407-14-456
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-14-456