
Abstract
Background
The HER2 gene has been established as a valid biological marker for the treatment of breast cancer patients with trastuzumab and probably other agents, such as paclitaxel and anthracyclines. The TOP2A gene has been associated with response to anthracyclines. Limited information exists on the relationship of HER2/TOP2A gene status in the presence of centromere 17 (CEP17) gain with outcome of patients treated with anthracycline-containing adjuvant chemotherapy.
Methods
Formalin-fixed paraffin-embedded tumor tissue samples from 1031 patients with high-risk operable breast cancer, enrolled in two consecutive phase III trials, were assessed in a central laboratory by fluorescence in situ hybridization for HER2/TOP2A gene amplification and CEP17 gain (CEP17 probe). Amplification of HER2 and TOP2A were defined as a gene/CEP17 ratio of >2.2 and ≥2.0, respectively, or gene copy number higher than 6. Additionally, HER2, TopoIIa, ER/PgR and Ki67 protein expression was assessed by immunohistochemistry (IHC) and patients were classified according to their IHC phenotype. Treatment consisted of epirubicin-based adjuvant chemotherapy followed by hormonal therapy and radiation, as indicated.
Results
HER2 amplification was found in 23.7% of the patients and TOP2A amplification in 10.1%. In total, 41.8% of HER2-amplified tumors demonstrated TOP2A co-amplification. The median (range) of HER2, TOP2A and CEP17 gain was 2.55 (0.70-45.15), 2.20 (0.70-26.15) and 2.00 (0.70-26.55), respectively. Forty percent of the tumors had CEP17 gain (51% of those with HER2 amplification). Adjusting for treatment groups in the Cox model, HER2 amplification, TOP2A amplification, CEP17 gain and HER2/TOP2A co-amplification were not associated with time to relapse or time to death.
Conclusion
HER2 amplification, TOP2A amplification, CEP17 gain and HER2/TOP2A co-amplification were not associated with outcome in high-risk breast cancer patients treated with anthracycline-based adjuvant chemotherapy.
Trial registration
Australian New Zealand Clinical Trials Registry (ANZCTR) ACTRN12611000506998 and ACTRN12609001036202
Similar content being viewed by others

Background
Breast cancer is the most frequent non-skin malignancy and the second leading cause of cancer death in American and European women [1, 2]. Adjuvant chemotherapy is administered to most patients with high-risk operable breast cancer, since it prolongs disease-free survival (DFS) and overall survival (OS) [3]. Anthracyclines and taxanes are considered to be two of the most efficient drugs in this setting [4, 5]. Despite intensive clinical research devoted to the role of adjuvant chemotherapy, the majority of patients do not benefit from its use and a small but considerable percentage of them suffer from long-term life-threatening toxicities, such as acute leukemia, myelodysplasia or irreversible congestive heart failure [6, 7]. To select candidate patients for such aggressive treatments, robust prognostic markers in human breast cancer are needed. Investigators intensively evaluate well-established oncogenes or chromosome aberrations, using large tumor repositories, in an effort to widen their knowledge on the molecular mechanisms, gene interrelationships or gene function underlying breast cancer.
It has long been established that breast cancer is often characterized by gains or losses of specific chromosomes, leading to activation of oncogenes or inactivation of tumor suppressor genes [8]. Chromosome 17 is the second most gene-dense chromosome in the human genome, housing important genes for breast cancer pathophysiology, such as BRCA1, HER2, RAD51C, RARA, TOP2A and TP53[9]. Changes of chromosome 17 copy number (aneusomy) are extremely frequent in breast cancer [10]. These chromosome aberrations (reviewed in ref. [11]) are tightly linked to important cell functions, such as proliferation, apoptosis, angiogenesis and motility. Increased numbers of centromere 17 copies are seen in 10% to 50% of breast tumors [12–14], depending on the criteria used, and this is more common in tumors with HER2 gene amplification. However, it has to be stated that an increase in chromosome 17 signals seen with fluorescence in situ hybridization (FISH) does not always correspond to true polysomy of the whole chromosome, but may rather represent a focal pericentromeric gain or partial polysomy [15]. Other abnormalities of chromosome 17 include losses and gains of genetic material in both the p and q arms, focal copy number gains and losses and other structural rearrangements [15, 16]. Indeed, recent studies using different techniques, such as comparative genomic hybridization (CGH) [17, 18], multiplex ligation-dependent probe amplification (MLPA) [19], single nucleotide polymorphism arrays (SNP arrays) [15], or FISH using alternative chromosome 17 reference genes (RARA, TP53, SMS) [20] suggested that true chromosome 17 polysomy is a rare event in breast cancer. In fact, in most of the cases, polysomy, as detected by FISH or chromogen in situ hybridization (CISH), actually reflects a gain or amplification in the pericentromeric region of the chromosome [21]. For these reasons the term “CEP17 gain” instead of “chromosome 17 polysomy”, is used here, referring to its detection by the centromere 17 enumeration probe (CEP17 probe).
CEP17 gain has been incriminated for the inconsistencies seen in cases with HER2 gene amplification defined by absolute gene copy numbers, versus gene amplification defined by the ratio of HER2 gene copy number to CEP17. Misclassification of HER2 gene status based on dual-color FISH assays, due to CEP17 gain, may have important therapeutic implications since a number of patients considered being HER2-negative by the second definition could be denied trastuzumab.
Importantly, recently published data from retrospectively assessed (although prospectively collected) tumors, by triple color FISH, from 1762 patients who participated in the National Epirubicin Adjuvant Trial (NEAT/BR9601) suggested that CEP17 duplication was associated with increased relapse-free and overall survival in patients treated with an anthracycline compared to CMF [22].
The HER2 oncogene is located on the long arm of chromosome 17 (17q12) [23]. HER2 amplification and/or protein overexpression has been identified in 15% to 25% of invasive breast tumors [24] and is associated with worse prognosis [25]. HER2 gene amplification has been shown to predict benefit from the use of several chemotherapeutic agents, including anthracyclines and paclitaxel [26, 27]. Notably, a meta-analysis provided compelling evidence that the use of anthracyclines benefits exclusively those patients with HER2 amplification [28]. However, other investigators could not confirm these data [29, 30], suggesting that other genes, also located on chromosome 17, may regulate anthracycline responsiveness [26].
One such gene is the topoisomerase II alpha gene (TOP2A), which is located ~700 kb telomerically to HER2 and encodes the alpha isozyme of the human topoisomerase II. In general, topoisomerases are responsible for transcription, replication and chromosome condensation and segregation during cell division [31, 32]. TOP2A in particular is considered a molecular target for anthracyclines and other chemotherapeutic agents [33, 34]. The TOP2A gene is amplified in 30%-40% of the tumors with HER2 gene amplification, while deletions are frequently observed [35]. TOP2A gene amplification [36] and, perhaps, topoisomerase II alpha (TopoIIa) protein overexpression [37] may benefit high-risk breast cancer patients treated with anthracyclines.
Information regarding the interaction of HER2/TOP2A gene status in the presence of CEP17 gain with the outcome of breast cancer patients is limited. This urged us to investigate the prognostic role of HER2 and TopoIIa protein expression, as well as HER2 and TOP2A gene status along with CEP17 gain in a large cohort of breast cancer patients. This is a prospective-retrospective study as described by Simon [38], performed in the context of two randomized, consecutively conducted, phase III trials (HE10/97 and HE10/00) with epirubicin-based adjuvant chemotherapy with or without paclitaxel [39–42].
Methods
Clinical studies
The HE10/97 trial [39] was a randomized phase III trial (ACTRN12611000506998) in patients with high-risk node-negative or intermediate/high-risk node-positive operable breast cancer, comparing four cycles of epirubicin (E) followed by four cycles of intensified CMF (E-CMF) with three cycles of E, followed by three cycles of paclitaxel (T, Taxol®, Bristol Myers-Squibb, Princeton, NJ) followed by three cycles of intensified CMF (E-T-CMF). All cycles were given every two weeks with G-CSF support. Dose intensity of all drugs in both treatment arms was identical, but cumulative doses and duration of chemotherapy period differed. Totally, 595 eligible patients entered the study in a period of 3.5 years (1997–2000).
The HE10/00 trial [40, 41] was a randomized phase III trial (ACTRN12609001036202), in which patients were treated with E-T-CMF (exactly as in the HE10/97 trial) or with four cycles of epirubicin/paclitaxel (ET) combination (given on the same day) every three weeks followed by three cycles of intensified CMF every two weeks (ET-CMF). By study design, the cumulative doses and the chemotherapy duration were identical in the two arms but dose intensity of epirubicin and paclitaxel was double in the E-T-CMF arm. A total of 1086 eligible patients with node-positive operable breast cancer were accrued in a period of 5 years (2000–2005).
HER2-positive patients received trastuzumab upon relapse, as previously described [43]. Treatment schedules for the two studies, baseline characteristics and clinical outcomes of both trials have already been described in detail [39–42]. Primary tumor diameter, axillary nodal status and tumor grade were obtained from the pathology report. Clinical protocols were approved by local regulatory authorities, while the present translational research protocol was approved by the Bioethics Committee of the Aristotle University of Thessaloniki School of Medicine. All patients signed a study-specific written informed consent before randomization, which in addition to giving consent for the trial allowed the use of biological material for future research purposes.
Tissue microarray (TMA) construction
Formalin-fixed paraffin-embedded (FFPE) tumor tissue samples from 1031 patients (61.3% of 1681 randomized patients) were collected from both trials, retrospectively in the first (HE10/97) and prospectively in the second (HE10/00). The REMARK diagram [44] for the study is shown in Figure 1. Hematoxylin-eosin stained sections from the tissue blocks were reviewed by two experienced breast cancer pathologists (M.B. and D.T.) and the most representative tumor areas were marked for the construction of the TMA blocks with the use of a manual arrayer (Model I, Beecher Instruments, San Prairie, WI), as previously described [45, 46]. Each case was represented by 2 tissue cores, 1.5 mm in diameter, obtained from the most representative areas of primary invasive tumors or in some cases (9.6%) from synchronous axillary lymph node metastases and re-embedded in 51 microarray blocks. Each TMA block contained 38 to 66 tissue cores from the original tumor tissue blocks, while cores from various neoplastic, non-neoplastic and reactive tissues were also included, serving as controls for slide-based assays. Cases not represented, damaged or inadequate on the TMA sections were re-cut from the original blocks and these sections were used for protein and gene analysis. Histological grade was evaluated according to the Scarff, Bloom and Richardson system.

REMARK diagram.
Immunohistochemistry (IHC)
Immunohistochemical labeling was performed according to standard protocols on serial 2.5 μm thick sections from the original blocks or the TMA blocks. All cases were also stained for vimentin (clone V9, Dako, Glostrup, Denmark) and cytokeratin 8/18 (clone 5D3, Novocastra™, Leica Biosystems, Newcastle, U.K), which were used as control stains for tissue immunoreactivity and fixation, as well as identification of tumor cells. Tissue samples negative for the above antibodies were excluded from the study. To assure optimal reactivity, immunostaining was applied 7 to 10 days after sectioning at the Laboratory of Molecular Oncology of the Hellenic Foundation for Cancer Research, Aristotle University of Thessaloniki School of Medicine. The staining procedures for HER2 (A0485 polyclonal antibody, Dako), estrogen receptor (ER, clone 6 F11, Novocastra™, Leica Biosystems), progesterone receptor (PgR, clone 1A6, Novocastra™, Leica Biosystems) and Ki67 (clone MIB-1, Dako) were performed using a Bond Max™ autostainer (Leica Microsystems, Wetzlar, Germany), as previously described [47]. TopoIIa protein expression was evaluated using the KiS1 monoclonal antibody (Dako), as previously described [48] with slight modifications (antibody dilution: 1:200; detection system: Envision™, Dako).
Interpretation of the IHC results
The evaluation of all IHC sections was done by two experienced breast cancer pathologists (M.B. and A.B.), blinded as to the patients’ clinical characteristics and survival data, according to existing established criteria, as previously described [43]. Briefly, HER2 protein expression was scored in a scale from 0 to 3+, the latter corresponding to uniform, intense membrane staining in >30% invasive tumor cells [49]; ER and PgR were evaluated using the Histoscore method (max score: 400) and were considered positive if staining was present in ≥1% of tumor cell nuclei [50]; for Ki67, the expression was defined as low (<14%) or high (≥14%) based on the percentage of stained/unstained nuclei from the tumor areas [51]; and, for TopoIIa immunostaining, a tumor was considered to be positive if moderate to intense nuclear staining was detected in >5% of tumor cells [52]. The mean percentage of stained cells from the two cores was calculated, while in cases with different intensities, the higher intensity score obtained from the two cores was used. If one of the tissue cores was lost or damaged the overall score was determined from the remaining one. When whole tissue sections were used, the entire tumor area was evaluated.
Fluorescence in situ hybridization (FISH)
TMA sections or whole sections (5 μm thick) were cut for FISH analysis, using the ZytoLight® SPEC HER2/TOP2A/CEP17 triple-color probe kit (ZytoVision, Bremerhaven, Germany). The FISH was performed according to the manufacturer’s protocol with minor modifications. Four carcinoma cell lines (MDA-MB-231, MDA-MB-175, MDA-MB-453 and SK-BR-3) from the Oracle HER2 Control Slide (Leica Biosystems), with a known HER2 gene status, were also used as a control for the FISH assays and analyzed for HER2 and TOP2A genomic status.
For all probes, sequential (5 planes at 1.0 μm) digital images were captured using the Plan Apo VC 100x/1.40 oil objective (Nikon, Japan) using specific filters for each probe. The resulting images were reconstructed using specifically developed software for cytogenetics (XCyto-Gen, ALPHELYS, Plaisir, France). Processed sections were considered eligible for FISH evaluation according to the ASCO/CAP criteria [49]. For the evaluation of HER2/TOP2A/CEP17 status, non-overlapping nuclei from the invasive part of the tumor were randomly selected, according to morphological criteria using DAPI staining, and scored (M.B and E.T). The virtual slides of HER2, ER or PgR stains, created as previously described [47], were used for selecting the invasive part of the tumor in each TMA. Twenty tumor nuclei were counted according to Press et al. [53]. The HER2 gene was considered to be amplified when the HER2/CEP17 ratio was >2.2 [49], or the mean HER2 copy number was >6 [54] and deleted when the ratio was <0.8.
The TOP2A gene was considered to be amplified when the TOP2A/CEP17 ratio was ≥2.0 and deleted when the ratio was <0.8 [36]. Cases with ≥3 CEP17 hybridization signals detected in >30% of counted nuclei were classified as CEP17 gain [55]. Re-classification of CEP17 gain was performed for the current analysis in comparison to the previous report [56].
In cases with ratios at or near the cut-off (1.8-2.2 for amplifications and 0.7-0.9 for deletions), additional 20 or 40 nuclei were counted and the ratio was recalculated. In cases with a borderline ratio at 60 nuclei, additional FISH assays were performed in whole sections [57]. In addition, tumors were classified according to the number of gene copies as normal (≤4 copies), low gain (4–6 copies) or high gain (>6 copies) tumors. The first category included tumors with possible gene losses, diploid, or with replicated DNA; the second, tumors with possible polysomy for the gene of interest; and, the third, tumors with unequivocal gene amplification. All primary image data of the TMA and whole tumor sections have been digitally scanned and made publicly available at: http://www.hecog-images.gr/HER2/TOP2A/CEN17/FISH_HE10/97_HE10/00.
Statistical analysis
Categorical data are presented as numbers and corresponding percentages, while continuous data are presented as median and range values. The Fisher’s exact or Pearson chi-square tests were used for group comparison of categorical data, while for continuous data the Mann–Whitney test was used. DFS was defined as the time interval from study entry to first locoregional recurrence, first distant metastasis, contralateral breast cancer, secondary neoplasm, death from the disease, or death from any cause, whichever occurred first [58]. OS was measured from study entry until death from any cause. Surviving patients were censored at the date of last contact. Kaplan-Meier curves and log-rank tests were used for comparing time to event distributions.
Cox proportional hazard regression analyses, adjusted for treatment, were performed for the examined markers, as well as for the combination of HER2/TOP2A gene status to assess prognostic significance on DFS and OS. In multivariate analysis, a backward selection procedure with p > 0.10 as a removal criterion based on the likelihood ratio test was performed to identify significant clinicopathological variables among the following: age (≥50 vs. <50), treatment group (E-CMF vs. ET-CMF vs. E-T-CMF), menopausal status (postmenopausal vs. premenopausal), histological grade (III-undifferentiated vs. I-II), Ki67 protein expression (high vs. low), tumor size (>5 cm vs. 2 to 5 cm vs. <2 cm), number of positive axillary nodes (≥4 vs. 0–3), ER/PgR status (positive vs. negative), adjuvant hormonotherapy (yes vs. missing vs. no) and type of operation (breast-conserving surgery vs. modified radical mastectomy). Treatment group and the examined markers were included in the final model, in order to examine whether they added independent prognostic information to the model containing the significant clinicopathological parameters.
The results of this study are presented according to reporting recommendations for tumor marker prognostic studies [44]. No adjustments for multiple comparisons were done. Statistical analyses were performed using the following statistical software: SPSS for Windows (version 15.0, IBM Corporation, Armonk, NY) and SAS for Windows (version 9.3, SAS Institute Inc., Cary, NC).
Results
A total of 1031 patients with available FFPE tumor tissue blocks were included in the analysis at a median follow-up of 106 months (updated in March 2012). Results at median follow-up of 92 months were presented at the 2011 San Antonio Breast Cancer Symposium [56].
Selected patient and tumor characteristics are presented in Table 1. The majority of the patients were postmenopausal (53.1%), had ≥4 positive nodes (60.4%) and tumors of ductal histology (77.6%), while approximately half of the patients had tumors of high grade (50.2%). The basic clinicopathological characteristics were similar between patients with and without available tissue blocks in each study, except for the number of positive nodes, radiotherapy treatment and histological grade (above II) (Additional file 1: Table S1). Patients with available tissue blocks had a higher incidence of ≥4 positive nodes (p = 0.022 and p = 0.027 for HE10/97 and HE10/00, respectively). This fact was probably reflected in the corresponding more frequent use of radiotherapy treatment (p = 0.024 and p = 0.021), while higher histological grade was more frequent in patients with available blocks only in the HE10/00 trial (p = 0.026).
Incidence and associations between examined biological markers
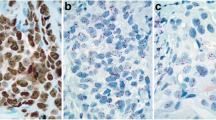
Representative FISH images for HER2, TOP2A and CEP17 are shown in Figure 2. The distribution of centrally assessed tumor markers by FISH and IHC are presented in Table 2. Cases with HER2 deletions (n = 27, 2.6%) were grouped together with HER2 normal tumors, for analysis purposes. Amplification, classified according to gene/CEP17 ratios, was found for HER2 in 23.7% and for TOP2A in 10.1% of the tumors. The incidence of amplified tumors was lower when amplification was considered according to the cut-off of >6 copies for each gene (Table 2). Ten cases were equivocal for HER2 (with HER2/CEP17 ratios between 1.8-2.2 and ≤6 gene copies) and they were also included in the HER2 normal tumors, for analysis purposes. CEP17 gain was seen in approximately 40% of tumors (Table 2). Histograms of the distribution of HER2, TOP2A and CEP17 copy numbers are presented in Figure 3.

Representative FISH images in invasive breast carcinoma (IBC) cases, using the HER2 / TOP2A /CEP17 triple-color probes. In the first four panels (A-D) an IBC case is shown with normal status of the HER2 gene (A), TOP2A gene (B) and CEP17 (C). An IBC case (E-H) showing simultaneous amplification of the HER2 and TOP2A genes (E-F), as well as CEP17 gain (G). The third IBC case presented in panels (I-L) showed amplification of the HER2 gene (I), normal status of the TOP2A gene (J) and CEP17 gain (K). In the last case, co-amplification of the HER2 (M) and TOP2A genes (N) was found in tumor cells, accompanied by high-level CEP17 gain (O). The last panel for each case (panels D, H, L and P) depicts a merged image of the three-colored probes. Magnification x1000. CEP17, centromere 17 enumeration probe.

Distribution of HER2 , TOP2A and CEP17 copies (A, B and C). Red line represents 6 gene copies (for A and B) and 3 copies for C.
Examining the association of markers with clinicopathological parameters, CEP17 gene gain was found to be associated only with postmenopausal status (48.1% in no gain vs. 60.6% in gain, p < 0.001) (Additional file 1: Table S2). HER2 gene amplification was associated with higher histological grade (45% in non-amplified vs. 66% in amplified, p < 0.001), ductal carcinoma (75% in non-amplified vs. 87% in amplified, p < 0.001), negative receptor status (16% in non-amplified vs. 44% in amplified, p < 0.001) and high Ki67 (64% in non-amplified vs. 79% in amplified, p < 0.001), while TOP2A amplification was associated with higher histological grade (58% in deleted vs. 49% in non-amplified vs. 62% in amplified, p = 0.023) and negative receptor status (37% in deleted vs. 20% in non-amplified vs. 40% in amplified, p < 0.001).
Overall, 24% of the patients had a HER2-positive status, based on either HER2 gene/CEP17 ratio of >2.2 or gene copy number of >6 or an IHC score of 3+. Interestingly, 27 tumors with HER2 IHC scores of 0 (7 cases) or 1+ (20 cases), were found to be amplified either by gene gain >6 (n = 3) or FISH ratio >2.2 (n = 24) (Additional file 1: Table S3). In addition, among cases with HER2 IHC scores of 0 or 1+, there were 17 tumors (2.4%) with HER2 deletion. It is worth noting that among 204 cases with HER2/CEP17 ratio >2.2 (i.e., amplified by ratio criteria), 184 (90%) also had >6 HER2 gene copies (i.e., amplified by gene copy criteria).
Tumors with CEP17 gain were also HER2 amplified in about one third of the cases (N = 120), while they were TOP2A amplified in 15% of the cases (N = 59) (Table 3). Among 244 HER2 amplified tumors, 51% had CEP17 gain. Similar percentages were observed for CEP17 gain in TOP2A amplified (58%) and deleted tumors (65%). Overall, tumors with low HER2 or TOP2A copy numbers had CEP17 gain in 37% and 36%, respectively (Table 3). In addition, among 827 tumors with HER2/CEP17 ratio ≤2.2, 327 (40%) had CEP17 gain. Among 10 equivocal cases with HER2/CEP17 ratios between 1.8-2.2 there was only one case with CEP17 gain.
The distribution of TOP2A and CEP17 by breast cancer tumor subtypes is presented in Table 4. Among 126 triple-negative breast cancer (TNBC) tumors, no amplifications of TOP2A were found. CEP17 gain was more frequent in Luminal-HER2 and HER2-enriched tumors.
Associations of TOP2A gene status and TopoIIa protein expression are shown in detail (Additional file 1: Table S4). TOP2A deletions did not result in lower TopoIIa expression. Among 953 cases with paired TOP2A gene status and protein expression data, there were 28 tumors with TOP2A gene deletion and simultaneous protein expression. No association was found between TopoIIa protein expression and TOP2A gene amplification (p = 0.22).
Significant associations were observed between CEP17 gene status and HER2 protein expression, as well as TopoIIa protein expression (Additional file 1: Table S5). More specifically, CEP17 gain was more frequent in HER2 2+ and 3+ tumors and in tumors expressing TopoIIa.
In total, 42% of HER2 amplified tumors demonstrated TOP2A co-amplification (Additional file 1: Table S6). Among the HER2 non-amplified cases, 28 deletions (3.6%) and only two amplifications of the TOP2A gene were identified.
Associations of examined markers with prognosis
DFS and OS did not differ significantly between treatment groups. At a median follow-up of 106 months (range 0.1-167), the 5-year DFS rates were 75%, 69% and 75%, while the OS rates were 88%, 81% and 86%, for the E-T-CMF, E-CMF and ET-CMF groups, respectively (Additional file 1: Table S7).
HER2 amplification, TOP2A amplification, TopoIIa protein expression, CEP17 gain and HER2/TOP2A co-amplification were not associated with either relapse or death (Figures 4, 5, 6 and 7). Similarly, when examining combined TOP2A gene pathology (deletion and amplification) vs. normal TOP2A, no effect on patient outcome was observed. This did not change when adjusting for treatment group in the Cox regression model. HER2 and TOP2A gene copy numbers (amplified vs. low gain vs. low-normal-replicated) were also not associated with DFS or OS. Stratifying by CEP17 status, differences in outcome by HER2 gene status (amplified vs. non-amplified tumors) and by TOP2A gene status (amplified vs. deleted vs. non-amplified tumors) were examined. No such differences were observed for either DFS or OS. The predictive role of all examined markers for paclitaxel treatment were also evaluated, performing Cox model analysis with interaction terms of each gene with treatment (paclitaxel vs. no paclitaxel). None of the markers tested was predictive for paclitaxel treatment.

Kaplan-Meier curves for DFS and OS according to HER2 gene status (log-rank test p-values).

Kaplan-Meier curves for DFS and OS according to TOP2A gene status (log-rank test p-values).

Kaplan-Meier curves for DFS and OS according to CEP17 gain (log-rank test p-values).

Kaplan-Meier curves for DFS and OS according to HER2/TOP2A co-amplification (log-rank test p-values).
Multivariate analyses for the examined biological markers, in the presence of significant clinical parameters and treatment group, are presented by forest plots (Figure 8). Clinicopathological factors associated with increased risk for both relapse and death were tumor size of more than 5 cm (p = 0.009 for DFS and p = 0.001 for OS) and four or more positive nodes (p < 0.001 for both DFS and OS). Hormonal therapy was associated with improved DFS and OS (p = 0.028 and p = 0.002, respectively), while breast-conserving surgery was associated with improved DFS only (p = 0.011) and high histological grade with poor OS only (p = 0.039). No association was found with DFS or OS for any of the examined chromosome 17 markers. Finally, none of the examined markers were associated with either DFS or OS in the context of univariate or multivariate analyses, when excluding the lymph node samples.

Multivariate analyses presented by forest plots in terms of DFS (A) and OS (B).
Discussion
In the present study we investigated the prognostic role of CEP17 gain in relation to HER2 and TOP2A gene status and protein expression in 1031 patients with operable breast cancer. All these patients were treated with epirubicin-based adjuvant chemotherapy in the context of two consecutively conducted phase III trials [39–41]. In a previous study published by our group for the HE10/00 and HE10/97 cohorts [42], patients with either luminal B, luminal-HER2 or HER2-enriched tumors performed worse than those with luminal A tumors, while patients with triple-negative tumors had the worst outcome. In addition, it was observed that the HER2-enriched subtype was predictive of response to paclitaxel-containing treatments. These prognostic and predictive HER2-related effects were breast cancer subtype specific and were not maintained in the present study. An earlier observation reported for this cohort, of HER2 amplification being predictive for OS benefit from adjuvant treatment with paclitaxel [56], was not confirmed in the current analysis with updated follow-up. In both analyses however, the ability to detect any predictive impact of HER2/TOP2A amplification or CEP17 gain in the presence of taxanes was limited (only 1 of the 4 trial arms did not include taxanes). The present results concerning HER2 are in line with reports on the prognostic value of this marker [59, 60]. A recent meta-analysis suggests that patients with both HER2 amplified and non-amplified tumors may benefit from anthracyclines [61, 62]. This could not be investigated in the current study, since all patients had been treated with anthracyclines.
Among HER2 amplified tumors, 42% exhibited TOP2A co-amplification, which is within the reported range of 35%-50% for this genomic alteration [26, 29, 63, 64]. TOP2A deletions were more common in HER2 amplified tumors, comprising approximately 10% of the HER2 amplified cases. TOP2A gene pathology (amplification, deletion and combinations of both) has been reported as a favorable prognostic and predictive marker in adjuvant-treated breast cancer patients [36, 65]. However, in the present study we did not observe any association between patient outcome and TOP2A amplification, deletion, or both, in accordance with the recent meta-analysis mentioned above [62].
The clinical importance of CEP17 gain, as detected by FISH, in human breast cancer remains a controversial issue. From the biological perspective, CEP17 gain and chromosome 17 polysomy do not represent the same situation, since the first one corresponds to the fluorescent signals of a 5.6 kb region, while the second reflects aberrant numbers of the whole chromosome, which should be demonstrated with spectral karyotyping (SKY) or other cytogenetic approaches. The CEP17 FISH probe detects the alpha-satellite repeat region at the centromere of chromosome 17, at 17p11.1-17q11.1. The specificity of CEP17 remains undetermined, while this probe and a centromeric probe detecting additional neighboring regions on 17p11.2-12 yield different results concerning chromosome 17 status and, therefore, different HER2 gene amplification status, when the latter is assessed as HER2/CEP17 ratio of >2.2 [66]. This may reflect the presence of the probed satellite repeats outside the centromeric region, which happen during evolution [67] and probably during cancer clone evolution, as well. Another problem for assessing low copy gains is that during DNA synthesis and in the G2/M phases, the targeted regions will appear double (three to four copies instead of two, taking into account the nuclear truncation effect during paraffin block sectioning). The cut-off used in the present study for the classification of CEP17 gains has been shown to correct for the maximally four centromeric signals that would be expected in this situation [55]. With this cut-off, we detected CEP17 gain in approximately 40% of all carcinomas examined, which is in the range of published results when using the FISH method in all-type breast carcinoma series (10-50%) [12–14, 54, 68, 69].
Measurement of CEP17 probe signals reflects the condition of the corresponding centromeric area and can by no means reflect gains of the entire chromosome 17 or “chromosome 17 polysomy”, as often reported in the literature. In the same treatment settings, depending on how CEP17 signals are classified and interpreted, and also depending on the drugs administered, the effect of CEP17 status on patient outcome may vary. Thus, in the adjuvant setting, by using the same FISH probe, duplication of the CEP17 region of chromosome 17 seems to be predictive of benefit from anthracyclines [22, 70] or of borderline association with clinical response to the same drugs [71]. Furthermore, CEP17 gain in the absence of HER2/TOP2A amplification has been reported by one recent study to be an unfavorable prognostic marker [72]; however, no prognostic value was identified for CEP17 gain in other studies [55, 64, 69], which is in line with our present findings.
CEP17 appears to be related with disease prognosis when this marker is combined with HER2 status. Whether “polysomy” 17 drives HER2 amplification or the opposite is true, as was recently suggested [54, 73], remains unanswered; it is, however, noteworthy that “polysomy” 17 is rarely observed in circulating tumor cells from patients with metastatic breast cancer and when present, it corresponds to HER2-negative primary tumors [74]. In the absence of HER2 amplification, CEP17 “polysomy” has been reported to confer a more favorable prognosis [75] or to be associated with aggravating prognostic markers [76]. In addition to these contradictory results, herein we did not observe any interaction between CEP17 and HER2 status. Methodological differences in the assessment of these parameters and cohort-fitted results should account for the diversity of data regarding the role of CEP17/HER2 status on adjuvant-treated breast cancer patients.
Although more than half of the tumors with CEP17 gain were not HER2 or TOP2A amplified, we did observe a higher than double incidence of CEP17 gain in tumors with aberrant HER2 and TOP2A genes (amplification, deletion, and, especially, high copy gains) in comparison to tumors with a normal status of these genes. These data are in line with “polysomy” 17 correlating with multiple copies of HER2 but not with HER2 amplification [77], while they further justify the higher incidence of CEP17 gain in the luminal-HER2 and HER2-enriched subtypes, as described in this study.
Equivocal HER2 IHC findings were observed in cases with chromosome 17 “polysomy” and correspondingly increased HER2 gene copy numbers [54, 78]. In the present study, the incidence of CEP17 gains was strongly related to HER2 IHC grades, but it did not contribute to the further assessment of HER2 IHC 2+ cases. With respect to FISH equivocal cases, herein we used very stringent criteria involving both gene/CEP17 ratios and gene copy numbers. Increased CEP17 ratios might result in false negative HER2/CEP17 ratios of ≤2.2; on the other hand, their coincidence with increased HER2 copies is not equivalent to HER2-positive disease [79]. The few (n = 10, <0.01% of the entire tumor series) FISH equivocal tumors for HER2 gene status were HER2 IHC 0 to 2+. CEP17 gain was observed in only one such case. Hence, at least in the present series, CEP17 gain did not aid further in the classification of equivocal HER2 gene status cases. In addition, we observed that most of the tumors with low-gain of HER2 and/or TOP2A copies indeed had CEP17 gains as well. However, in order to evaluate the impact of these concomitant alterations on patient outcome, larger patient series with this sub-category of tumors would be needed.
Conclusions
The present combined chromosome 17 marker analysis by FISH represents one of the largest of its type in early high-risk breast cancer. With the cut-offs used for the characterization of CEP17 gain, as well as HER2 and TOP2A gene amplification, these chromosome 17 markers, individually or in conjunction, did not appear to be related with patient outcome.
Abbreviations
- CGH:
-
Comparative genomic hybridization
- ANZCTR:
-
Australian New Zealand Clinical Trials Registry
- BRCA1:
-
Breast cancer type 1 susceptibility protein
- CEP17:
-
Centromere 17 enumeration probe
- CISH:
-
Chromogen in situ hybridization
- CMF:
-
Cyclophosphamide, Methotrexate, Fluorouracil
- DFS:
-
Disease-free survival
- DNA:
-
Deoxyribonucleic acid
- E:
-
Epirubicin
- ER:
-
Estrogen receptor
- FFPE:
-
Formalin-fixed, paraffin-embedded
- FISH:
-
Fluorescence in situ hybridization
- G-CSF:
-
Granulocyte-colony stimulating factor
- HeCOG:
-
Hellenic Cooperative Oncology Group
- HER2:
-
Human epidermal growth factor receptor 2
- HR:
-
Hazard ratio
- HT:
-
Hormonal therapy
- IHC:
-
Immunohistochemistry
- Ki67:
-
Antigen Ki67
- MLPA:
-
Multiplex ligation-dependent probe amplification
- OS:
-
Overall survival
- PgR:
-
Progesterone receptor
- RAD51C:
-
RAD51 homolog C
- RARA:
-
Retinoic acid receptor, alpha
- REMARK:
-
Reporting Recommendations for Tumor Marker Prognostic Studies
- RNA:
-
Ribonucleic acid
- SMS:
-
Spermine synthase
- SNP:
-
Single nucleotide polymorphism
- T:
-
Taxol (Paclitaxel)
- TMA:
-
Tissue microarray
- TNBC:
-
Triple-negative breast cancer
- TOP2A:
-
Topoisomerase II alpha (gene expression)
- TopoIIa:
-
Topoisomerase II alpha (protein expression)
- TP53:
-
Tumor protein 53.
References
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ: Cancer statistics. CA Cancer J Clin. 2009, 59 (4): 225-249. 10.3322/caac.20006.
La Vecchia C, Bosetti C, Lucchini F, Bertuccio P, Negri E, Boyle P, Levi F: Cancer mortality in Europe, 2000–2004, and an overview of trends since 1975. Ann Oncol. 2010, 21 (6): 1323-1360. 10.1093/annonc/mdp530.
Early Breast Cancer Trialists' Collaborative Group (EBCTCG): Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005, 365 (9472): 1687-1717.
Levine MN, Whelan T: Adjuvant chemotherapy for breast cancer–30 years later. N Engl J Med. 2006, 355 (18): 1920-1922. 10.1056/NEJMe068204.
Smith I, Chua S: Medical treatment of early breast cancer. III: chemotherapy. Bmj. 2006, 332 (7534): 161-162.
Doyle JJ, Neugut AI, Jacobson JS, Grann VR, Hershman DL: Chemotherapy and cardiotoxicity in older breast cancer patients: a population-based study. J Clin Oncol. 2005, 23 (34): 8597-8605. 10.1200/JCO.2005.02.5841.
Hershman D, Neugut AI, Jacobson JS, Wang J, Tsai WY, McBride R, Bennett CL, Grann VR: Acute myeloid leukemia or myelodysplastic syndrome following use of granulocyte colony-stimulating factors during breast cancer adjuvant chemotherapy. J Natl Cancer Inst. 2007, 99 (3): 196-205. 10.1093/jnci/djk028.
Thompson F, Emerson J, Dalton W, Yang JM, McGee D, Villar H, Knox S, Massey K, Weinstein R, Bhattacharyya A: Clonal chromosome abnormalities in human breast carcinomas. I. Twenty-eight cases with primary disease. Genes Chromosomes Cancer. 1993, 7 (4): 185-193. 10.1002/gcc.2870070402.
Zody MC, Garber M, Adams DJ, Sharpe T, Harrow J, Lupski JR, Nicholson C, Searle SM, Wilming L, Young SK: DNA sequence of human chromosome 17 and analysis of rearrangement in the human lineage. Nature. 2006, 440 (7087): 1045-1049. 10.1038/nature04689.
Botti C, Pescatore B, Mottolese M, Sciarretta F, Greco C, Di Filippo F, Gandolfo GM, Cavaliere F, Bovani R, Varanese A: Incidence of chromosomes 1 and 17 aneusomy in breast cancer and adjacent tissue: an interphase cytogenetic study. J Am Coll Surg. 2000, 190 (5): 530-539. 10.1016/S1072-7515(00)00252-0.
Reinholz MM, Bruzek AK, Visscher DW, Lingle WL, Schroeder MJ, Perez EA, Jenkins RB: Breast cancer and aneusomy 17: implications for carcinogenesis and therapeutic response. Lancet Oncol. 2009, 10 (3): 267-277. 10.1016/S1470-2045(09)70063-4.
Salido M, Tusquets I, Corominas JM, Suarez M, Espinet B, Corzo C, Bellet M, Fabregat X, Serrano S, Sole F: Polysomy of chromosome 17 in breast cancer tumors showing an overexpression of ERBB2: a study of 175 cases using fluorescence in situ hybridization and immunohistochemistry. Breast Cancer Res. 2005, 7 (2): 26-
Dal Lago L, Durbecq V, Desmedt C, Salgado R, Verjat T, Lespagnard L, Ma Y, Veys I, Di Leo A, Sotiriou C: Correction for chromosome-17 is critical for the determination of true Her-2/neu gene amplification status in breast cancer. Mol Cancer Ther. 2006, 5 (10): 2572-2579. 10.1158/1535-7163.MCT-06-0129.
Hyun CL, Lee HE, Kim KS, Kim SW, Kim JH, Choe G, Park SY: The effect of chromosome 17 polysomy on HER-2/neu status in breast cancer. J Clin Pathol. 2008, 61 (3): 317-321.
Vranic S, Teruya B, Repertinger S, Ulmer P, Hagenkord J, Gatalica Z: Assessment of HER2 gene status in breast carcinomas with polysomy of chromosome 17. Cancer. 2011, 117 (1): 48-53. 10.1002/cncr.25580.
Yeh IT, Martin MA, Robetorye RS, Bolla AR, McCaskill C, Shah RK, Gorre ME, Mohammed MS, Gunn SR: Clinical validation of an array CGH test for HER2 status in breast cancer reveals that polysomy 17 is a rare event. Mod Pathol. 2009, 22 (9): 1169-1175. 10.1038/modpathol.2009.78.
Viale G: Be precise! The need to consider the mechanisms for CEP17 copy number changes in breast cancer. J Pathol. 2009, 219 (1): 1-2. 10.1002/path.2593.
Marchio C, Lambros MB, Gugliotta P, Di Cantogno LV, Botta C, Pasini B, Tan DS, Mackay A, Fenwick K, Tamber N: Does chromosome 17 centromere copy number predict polysomy in breast cancer? A fluorescence in situ hybridization and microarray-based CGH analysis. J Pathol. 2009, 219 (1): 16-24. 10.1002/path.2574.
Moelans CB, de Weger RA, van Diest PJ: Absence of chromosome 17 polysomy in breast cancer: analysis by CEP17 chromogenic in situ hybridization and multiplex ligation-dependent probe amplification. Breast Cancer Res Treat. 2010, 120 (1): 1-7. 10.1007/s10549-009-0539-2.
Tse CH, Hwang HC, Goldstein LC, Kandalaft PL, Wiley JC, Kussick SJ, Gown AM: Determining true HER2 gene status in breast cancers with polysomy by using alternative chromosome 17 reference genes: implications for anti-HER2 targeted therapy. J Clin Oncol. 2011, 29 (31): 4168-4174. 10.1200/JCO.2011.36.0107.
Moelans CB, Reis-Filho JS, van Diest PJ: Implications of rarity of chromosome 17 polysomy in breast cancer. Lancet Oncol. 2011, 12 (12): 1087-1089. 10.1016/S1470-2045(11)70234-0.
Bartlett JM, Munro AF, Dunn JA, McConkey C, Jordan S, Twelves CJ, Cameron DA, Thomas J, Campbell FM, Rea DW: Predictive markers of anthracycline benefit: a prospectively planned analysis of the UK National Epirubicin Adjuvant Trial (NEAT/BR9601). Lancet Oncol. 2010, 11 (3): 266-274. 10.1016/S1470-2045(10)70006-1.
Pauletti G, Godolphin W, Press MF, Slamon DJ: Detection and quantitation of HER-2/neu gene amplification in human breast cancer archival material using fluorescence in situ hybridization. Oncogene. 1996, 13 (1): 63-72.
Tanner M, Jarvinen P, Isola J: Amplification of HER-2/neu and topoisomerase IIalpha in primary and metastatic breast cancer. Cancer Res. 2001, 61 (14): 5345-5348.
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL: Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987, 235 (4785): 177-182. 10.1126/science.3798106.
Jarvinen TA, Liu ET: Simultaneous amplification of HER-2 (ERBB2) and topoisomerase IIalpha (TOP2A) genes–molecular basis for combination chemotherapy in cancer. Curr Cancer Drug Targets. 2006, 6 (7): 579-602. 10.2174/156800906778742497.
Hayes DF, Thor AD, Dressler LG, Weaver D, Edgerton S, Cowan D, Broadwater G, Goldstein LJ, Martino S, Ingle JN: HER2 and response to paclitaxel in node-positive breast cancer. N Engl J Med. 2007, 357 (15): 1496-1506. 10.1056/NEJMoa071167.
Gennari A, Sormani MP, Pronzato P, Puntoni M, Colozza M, Pfeffer U, Bruzzi P: HER2 status and efficacy of adjuvant anthracyclines in early breast cancer: a pooled analysis of randomized trials. J Natl Cancer Inst. 2008, 100 (1): 14-20. 10.1093/jnci/djm252.
Di Leo A, Larsimont D, Gancberg D, Jarvinen T, Beauduin M, Vindevoghel A, Michel J, Focan CH, Ries F, Gobert PH: HER-2 and topo-isomerase IIalpha as predictive markers in a population of node-positive breast cancer patients randomly treated with adjuvant CMF or epirubicin plus cyclophosphamide. Ann Oncol. 2001, 12 (8): 1081-1089. 10.1023/A:1011669223035.
Konecny GE, Thomssen C, Luck HJ, Untch M, Wang HJ, Kuhn W, Eidtmann H, du Bois A, Olbricht S, Steinfeld D: Her-2/neu gene amplification and response to paclitaxel in patients with metastatic breast cancer. J Natl Cancer Inst. 2004, 96 (15): 1141-1151. 10.1093/jnci/djh198.
Kellner U, Sehested M, Jensen PB, Gieseler F, Rudolph P: Culprit and victim – DNA topoisomerase II. Lancet Oncol. 2002, 3 (4): 235-243. 10.1016/S1470-2045(02)00715-5.
Beser AR, Tuzlali S, Guzey D, Dolek Guler S, Hacihanefioglu S, Dalay N: HER-2, TOP2A and chromosome 17 alterations in breast cancer. Pathol Oncol Res. 2007, 13 (3): 180-185. 10.1007/BF02893497.
Champoux JJ: DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem. 2001, 70: 369-413. 10.1146/annurev.biochem.70.1.369.
Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG: A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science. 2006, 312 (5781): 1798-1802. 10.1126/science.1127196.
Jarvinen TA, Tanner M, Barlund M, Borg A, Isola J: Characterization of topoisomerase II alpha gene amplification and deletion in breast cancer. Genes Chromosomes Cancer. 1999, 26 (2): 142-150. 10.1002/(SICI)1098-2264(199910)26:2<142::AID-GCC6>3.0.CO;2-B.
Knoop AS, Knudsen H, Balslev E, Rasmussen BB, Overgaard J, Nielsen KV, Schonau A, Gunnarsdottir K, Olsen KE, Mouridsen H: Retrospective analysis of topoisomerase IIa amplifications and deletions as predictive markers in primary breast cancer patients randomly assigned to cyclophosphamide, methotrexate, and fluorouracil or cyclophosphamide, epirubicin, and fluorouracil: Danish Breast Cancer Cooperative Group. J Clin Oncol. 2005, 23 (30): 7483-7490. 10.1200/JCO.2005.11.007.
Schindlbeck C, Mayr D, Olivier C, Rack B, Engelstaedter V, Jueckstock J, Jenderek C, Andergassen U, Jeschke U, Friese K: Topoisomerase IIalpha expression rather than gene amplification predicts responsiveness of adjuvant anthracycline-based chemotherapy in women with primary breast cancer. Cancer Res Clin Oncol. 2010, 136 (7): 1029-1037. 10.1007/s00432-009-0748-4.
Simon RM, Paik S, Hayes DF: Use of archived specimens in evaluation of prognostic and predictive biomarkers. J Natl Cancer Inst. 2009, 101 (21): 1446-1452. 10.1093/jnci/djp335.
Fountzilas G, Skarlos D, Dafni U, Gogas H, Briasoulis E, Pectasides D, Papadimitriou C, Markopoulos C, Polychronis A, Kalofonos HP: Postoperative dose-dense sequential chemotherapy with epirubicin, followed by CMF with or without paclitaxel, in patients with high-risk operable breast cancer: a randomized phase III study conducted by the Hellenic Cooperative Oncology Group. Ann Oncol. 2005, 16 (11): 1762-1771. 10.1093/annonc/mdi366.
Fountzilas G, Dafni U, Gogas H, Linardou H, Kalofonos HP, Briasoulis E, Pectasides D, Samantas E, Bafaloukos D, Stathopoulos GP: Postoperative dose-dense sequential chemotherapy with epirubicin, paclitaxel and CMF in patients with high-risk breast cancer: safety analysis of the Hellenic Cooperative Oncology Group randomized phase III trial HE 10/00. Ann Oncol. 2008, 19 (5): 853-860. 10.1093/annonc/mdm539.
Gogas H, Dafni U, Karina M, Papadimitriou C, Batistatou A, Bobos M, Kalofonos HP, Eleftheraki AG, Timotheadou E, Bafaloukos D: Postoperative dose-dense sequential versus concomitant administration of epirubicin and paclitaxel in patients with node-positive breast cancer: 5-year results of the Hellenic Cooperative Oncology Group HE 10/00 phase III Trial. Breast Cancer Res Treatment. 2012, 132 (2): 609-619. 10.1007/s10549-011-1913-4.
Fountzilas G, Dafni U, Bobos M, Batistatou A, Kotoula V, Trihia H, Malamou-Mitsi V, Miliaras S, Chrisafi S, Papadopoulos S: Differential response of immunohistochemically defined breast cancer subtypes to anthracycline-based adjuvant chemotherapy with or without paclitaxel. PLoS One. 2012, 7 (6): e37946-10.1371/journal.pone.0037946.
Razis E, Bobos M, Kotoula V, Eleftheraki AG, Kalofonos HP, Pavlakis K, Papakostas P, Aravantinos G, Rigakos G, Efstratiou I: Evaluation of the association of PIK3CA mutations and PTEN loss with efficacy of trastuzumab therapy in metastatic breast cancer. Breast Cancer Research Treat. 2011, 128 (2): 447-456. 10.1007/s10549-011-1572-5.
McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM: REporting recommendations for tumor MARKer prognostic studies (REMARK). Breast Cancer Res Treat. 2006, 100 (2): 229-235. 10.1007/s10549-006-9242-8.
Kononen J, Bubendorf L, Kallioniemi A, Barlund M, Schraml P, Leighton S, Torhorst J, Mihatsch MJ, Sauter G, Kallioniemi OP: Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med. 1998, 4 (7): 844-847. 10.1038/nm0798-844.
Skacel M, Skilton B, Pettay JD, Tubbs RR: Tissue microarrays: a powerful tool for high-throughput analysis of clinical specimens: a review of the method with validation data. Appl Immunohistochem Mol Morphol. 2002, 10 (1): 1-6. 10.1097/00022744-200203000-00001.
Fountzilas G, Kourea HP, Bobos M, Televantou D, Kotoula V, Papadimitriou C, Papazisis KT, Timotheadou E, Efstratiou I, Koutras A: Paclitaxel and bevacizumab as first line combined treatment in patients with metastatic breast cancer: the Hellenic Cooperative Oncology Group experience with biological marker evaluation. Anticancer Res. 2011, 31 (9): 3007-3018.
Christodoulou C, Kostopoulos I, Kalofonos HP, Lianos E, Bobos M, Briasoulis E, Gogas H, Razis E, Skarlos DV, Fountzilas G: Trastuzumab combined with pegylated liposomal doxorubicin in patients with metastatic breast cancer. phase II Study of the Hellenic Cooperative Oncology Group (HeCOG) with biomarker evaluation. Oncology. 2009, 76 (4): 275-285. 10.1159/000207504.
Wolff AC, Hammond ME, Schwartz JN, Hagerty KL, Allred DC, Cote RJ, Dowsett M, Fitzgibbons PL, Hanna WM, Langer A: American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol. 2007, 25 (1): 118-145.
Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, Fitzgibbons PL, Francis G, Goldstein NS, Hayes M: American society of clinical oncology/college of American pathologists guideline recommendations for immunohistochemical testing of Estrogen and progesterone receptors in breast cancer. J Clin Oncol. 2010, 28 (16): 2784-2795. 10.1200/JCO.2009.25.6529.
Cheang MC, Chia SK, Voduc D, Gao D, Leung S, Snider J, Watson M, Davies S, Bernard PS, Parker JS: Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst. 2009, 101 (10): 736-750. 10.1093/jnci/djp082.
Bhargava R, Lal P, Chen B: HER-2/neu and topoisomerase IIa gene amplification and protein expression in invasive breast carcinomas: chromogenic in situ hybridization and immunohistochemical analyses. Am J Clin Pathol. 2005, 123 (6): 889-895. 10.1309/PCFK8YTQPYWD534F.
Press MF, Sauter G, Buyse M, Bernstein L, Guzman R, Santiago A, Villalobos IE, Eiermann W, Pienkowski T, Martin M: Alteration of topoisomerase II-alpha gene in human breast cancer: association with responsiveness to anthracycline-based chemotherapy. J Clin Oncol. 2011, 29 (7): 859-867. 10.1200/JCO.2009.27.5644.
Vanden Bempt I, Van Loo P, Drijkoningen M, Neven P, Smeets A, Christiaens MR, Paridaens R, De Wolf-Peeters C: Polysomy 17 in breast cancer: clinicopathologic significance and impact on HER-2 testing. J Clin Oncol. 2008, 26 (30): 4869-4874. 10.1200/JCO.2007.13.4296.
Perez EA, Reinholz MM, Hillman DW, Tenner KS, Schroeder MJ, Davidson NE, Martino S, Sledge GW, Harris LN, Gralow JR: HER2 and chromosome 17 effect on patient outcome in the N9831 adjuvant trastuzumab trial. J Clin Oncol. 2010, 28 (28): 4307-4315. 10.1200/JCO.2009.26.2154.
Dafni U, Bobos M, Tsolaki E, Batistatou A, Koletsa F, Televantou D, Gogas H, Linardou H, Pectasides D, Kalogeras KT, Galani E, Koutras A, Papadimitriou CA, Fountzilas G: Effect of HER2/Topoisomerase II alpha (TOP2A) Gene Status or Protein Expression and Chromosome 17 (CEP17) Polysomy on the Outcome of Breast Cancer Patients Treated with Anthracycline-Containing Dose-Dense Sequential Adjuvant Chemotherapy with or without Paclitaxel. A pooled analysis of two Hellenic Cooperative Oncology Group (HeCOG) phase III trials. Cancer Res. 2011, 71 (24 Suppl): Abstract nr PD05–02-
Sauter G, Lee J, Bartlett JM, Slamon DJ, Press MF: Guidelines for human epidermal growth factor receptor 2 testing: biologic and methodologic considerations. J Clin Oncol. 2009, 27 (8): 1323-1333. 10.1200/JCO.2007.14.8197.
Hudis CA, Barlow WE, Costantino JP, Gray RJ, Pritchard KI, Chapman JA, Sparano JA, Hunsberger S, Enos RA, Gelber RD: Proposal for standardized definitions for efficacy end points in adjuvant breast cancer trials: the STEEP system. J Clin Oncol. 2007, 25 (15): 2127-2132. 10.1200/JCO.2006.10.3523.
Clark GM, McGuire WL: Follow-up study of HER-2/neu amplification in primary breast cancer. Cancer Res. 1991, 51 (3): 944-948.
Allred DC, Clark GM, Tandon AK, Molina R, Tormey DC, Osborne CK, Gilchrist KW, Mansour EG, Abeloff M, Eudey L: HER-2/neu in node-negative breast cancer: prognostic significance of overexpression influenced by the presence of in situ carcinoma. J Clin Oncol. 1992, 10 (4): 599-605.
Bonnefoi HR: Anthracyclines, HER2, and TOP2A: the verdict. Lancet Oncol. 2011, 12 (12): 1084-1085. 10.1016/S1470-2045(11)70241-8.
Di Leo A, Desmedt C, Bartlett JM, Piette F, Ejlertsen B, Pritchard KI, Larsimont D, Poole C, Isola J, Earl H: HER2 and TOP2A as predictive markers for anthracycline-containing chemotherapy regimens as adjuvant treatment of breast cancer: a meta-analysis of individual patient data. Lancet Oncol. 2011, 12 (12): 1134-1142. 10.1016/S1470-2045(11)70231-5.
Hicks DG, Tubbs RR: Assessment of the HER2 status in breast cancer by fluorescence in situ hybridization: a technical review with interpretive guidelines. Hum Pathol. 2005, 36 (3): 250-261. 10.1016/j.humpath.2004.11.010.
Nielsen KV, Ejlertsen B, Moller S, Jensen MB, Balslev E, Muller S, Knoop A, Mouridsen HT: Lack of independent prognostic and predictive value of centromere 17 copy number changes in breast cancer patients with known HER2 and TOP2A status. Mol Oncol. 2012, 6 (1): 88-97. 10.1016/j.molonc.2011.11.006.
O’Malley FP, Chia S, Tu D, Shepherd LE, Levine MN, Bramwell VH, Andrulis IL, Pritchard KI: Topoisomerase II alpha and responsiveness of breast cancer to adjuvant chemotherapy. J Natl Cancer Inst. 2009, 101 (9): 644-650. 10.1093/jnci/djp067.
Varga Z, Tubbs RR, Wang Z, Sun Y, Noske A, Kradolfer D, Bosshard G, Jochum W, Moch H, Ohlschlegel C: Co-amplification of the HER2 gene and chromosome 17 centromere: a potential diagnostic pitfall in HER2 testing in breast cancer. Breast Cancer Res Treat. 2012, 132 (3): 925-935. 10.1007/s10549-011-1642-8.
She X, Horvath JE, Jiang Z, Liu G, Furey TS, Christ L, Clark R, Graves T, Gulden CL, Alkan C: The structure and evolution of centromeric transition regions within the human genome. Nature. 2004, 430 (7002): 857-864. 10.1038/nature02806.
Baehner FL, Achacoso N, Maddala T, Shak S, Quesenberry CP, Goldstein LC, Gown AM, Habel LA: Human epidermal growth factor receptor 2 assessment in a case–control study: comparison of fluorescence in situ hybridization and quantitative reverse transcription polymerase chain reaction performed by central laboratories. J Clin Oncol. 2010, 28 (28): 4300-4306. 10.1200/JCO.2009.24.8211.
Zaczek A, Markiewicz A, Supernat A, Bednarz-Knoll N, Brandt B, Seroczynska B, Skokowski J, Szade J, Czapiewski P, Biernat W: Prognostic Value of TOP2A Gene Amplification and Chromosome 17 Polysomy in Early Breast Cancer. Pathol Oncol Res. 2012, 18 (4): 885-894. 10.1007/s12253-012-9518-8.
Munro AF, Cameron DA, Bartlett JM: Targeting anthracyclines in early breast cancer: new candidate predictive biomarkers emerge. Oncogene. 2010, 29 (38): 5231-5240. 10.1038/onc.2010.286.
Pritchard KI, Munro A, O’Malley FP, Tu D, Li X, Levine MN, Shepherd L, Chia S, Bartlett JM: Chromosome 17 centromere (CEP17) duplication as a predictor of anthracycline response: evidence from the NCIC Clinical Trials Group (NCIC CTG) MA.5 Trial. Breast Cancer Res Treat. 2012, 131 (2): 541-551. 10.1007/s10549-011-1840-4.
Kim A, Shin HC, Bae YK, Kim MK, Kang SH, Lee SJ, Lee EH: Multiplication of Chromosome 17 Centromere Is Associated with Prognosis in Patients with Invasive Breast Cancers Exhibiting Normal HER2 and TOP2A Status. J Breast Cancer. 2012, 15 (1): 24-33. 10.4048/jbc.2012.15.1.24.
Vanden Bempt I, Drijkoningen M, De Wolf-Peeters C: The complexity of genotypic alterations underlying HER2-positive breast cancer: an explanation for its clinical heterogeneity. Curr Opin Oncol. 2007, 19 (6): 552-557. 10.1097/CCO.0b013e3282f0ad8e.
Hayashi N, Nakamura S, Yagata H, Shimoda Y, Ota H, Hortobagyi GN, Cristofanilli M, Ueno NT: Chromosome 17 polysomy in circulating tumor cells in patients with metastatic breast cancer: a case series. Int J Clin Oncol. 2011, 16 (5): 596-600. 10.1007/s10147-010-0173-3.
Torrisi R, Rotmensz N, Bagnardi V, Viale G, Curto BD, Dell’orto P, Veronesi P, Luini A, D’Alessandro C, Cardillo A: HER2 status in early breast cancer: relevance of cell staining patterns, gene amplification and polysomy 17. Eur J Cancer. 2007, 43 (16): 2339-2344. 10.1016/j.ejca.2007.07.033.
Krishnamurti U, Hammers JL, Atem FD, Storto PD, Silverman JF: Poor prognostic significance of unamplified chromosome 17 polysomy in invasive breast carcinoma. Mod Pathol. 2009, 22 (8): 1044-1048. 10.1038/modpathol.2009.61.
Watters AD, Going JJ, Cooke TG, Bartlett JM: Chromosome 17 aneusomy is associated with poor prognostic factors in invasive breast carcinoma. Breast Cancer Res Treat. 2003, 77 (2): 109-114. 10.1023/A:1021399923825.
Bose S, Mohammed M, Shintaku P, Rao PN: Her-2/neu gene amplification in low to moderately expressing breast cancers: possible role of chromosome 17/Her-2/neu polysomy. Breast J. 2001, 7 (5): 337-344. 10.1046/j.1524-4741.2001.21018.x.
Ross JS: Human epidermal growth factor receptor 2 testing in 2010: does chromosome 17 centromere copy number make any difference?. J Clin Oncol. 2010, 28 (28): 4293-4295. 10.1200/JCO.2010.29.6673.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/13/163/prepub
Acknowledgements
Presented in part at the 34th Annual San Antonio Breast Cancer Symposium, December 6–10, 2011.
The authors are indebted to all patients and their families for their trust and participation in the HE10/97 and HE10/00 trials and for the provision of biological material for research purposes.
The authors also wish to thank all HeCOG personnel (data managers, research assistants and monitors) for their dedication, M. Moschoni for data coordination, T. Spinari for collection of FFPE tissue blocks and S. Dallidou for secretarial assistance.
Supported by an internal Hellenic Cooperative Oncology Group (HeCOG) translational research grant (HE TRANS_BR).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
On behalf of the Hellenic Foundation for Cancer Research, Athens, Greece, the senior author (GF) has pending patent applications with Siemens Healthcare Diagnostics, Tarrytown, NY. The rest of the authors declare that they have no competing interests.
Authors’ contributions
GF conceived of the study, participated in its design and coordination, contributed to the analysis and interpretation of data and drafted the manuscript. UD conceived of the study, participated in its design, contributed to the analysis and interpretation of data and drafted the manuscript. MB carried out the IHC and FISH analysis, contributed to the analysis and interpretation of data and drafted the manuscript. VK conceived of the study, participated in its design, carried out the molecular studies, contributed to the analysis and interpretation of data and drafted the manuscript. AB carried out the immunoassays and contributed to the analysis and interpretation of data. IX participated in the acquisition of data and contributed to the collection of the tumor tissue samples analyzed in the study. CP participated in the acquisition of data and contributed to the collection of the tumor tissue samples analyzed in the study. TK carried out the molecular studies and contributed to the analysis and interpretation of data. ET carried out the FISH assays and contributed to the analysis and interpretation of data. DT carried out the immunoassays and contributed to the analysis and interpretation of data. ET participated in the acquisition of data and contributed to the collection of the tumor tissue samples analyzed in the study. AK participated in the acquisition of data and contributed to the collection of the tumor tissue samples analyzed in the study. GK participated in the acquisition of data. ES participated in the acquisition of data and contributed to the collection of the tumor tissue samples analyzed in the study. NP participated in the acquisition of data and contributed to the collection of the tumor tissue samples analyzed in the study. CK participated in the acquisition of data and contributed to the collection of the tumor tissue samples analyzed in the study. IS participated in the acquisition of data. NP participated in the acquisition of data and contributed to the collection of the tumor tissue samples analyzed in the study. HG participated in the acquisition of data and contributed to the collection of the tumor tissue samples analyzed in the study. HL participated in the acquisition of data and contributed to the collection of the tumor tissue samples analyzed in the study. KTK conceived of the study, participated in its design, contributed to the analysis and interpretation of data and drafted the manuscript. DP conceived of the study, participated in its design and contributed to the analysis and interpretation of data. MAD conceived of the study, participated in its design, contributed to the analysis and interpretation of data and drafted the manuscript. All authors read and approved the final manuscript.
George Fountzilas, Urania Dafni contributed equally to this work.
Electronic supplementary material
12885_2012_3786_MOESM1_ESM.doc
Additional file 1: Table S1: Comparison of basic clinicopathological characteristics between patients with and without available tissue material (paraffin blocks) per study. Table S2. Association of CEP17, HER2 and TOP2A markers with basic clinicopathological parameters. Table S3. Distribution of HER2 gene status according to HER2 protein expression. Table S4. Distribution of TOP2A gene status according to TopoIIa protein expression. Table S5. Distribution of CEP17 status according to HER2 and TopoIIa protein expression. Table S6. Association of HER2 and TOP2A gene status assessed by FISH. Table S7. Survival data for the total study population and according to randomization group. (DOC 100 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Fountzilas, G., Dafni, U., Bobos, M. et al. Evaluation of the prognostic role of centromere 17 gain and HER2/topoisomerase II alpha gene status and protein expression in patients with breast cancer treated with anthracycline-containing adjuvant chemotherapy: pooled analysis of two Hellenic Cooperative Oncology Group (HeCOG) phase III trials. BMC Cancer 13, 163 (2013). https://doi.org/10.1186/1471-2407-13-163
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-13-163