
Abstract
Background
Deregulated c-Myc expression is a hallmark of several human cancers where it promotes proliferation and an aggressive tumour phenotype. Myc overexpression is associated with reduced activity of Rel/NF-κB, transcription factors that control the immune response, cell survival, and transformation, and that are frequently altered in cancer. The Rel/NF-κB family member NFKB2 is altered by chromosomal translocations or deletions in lymphoid malignancies and deletion of the C-terminal ankyrin domain of NF-κB2 augments lymphocyte proliferation.
Methods
Precancerous Eμ-Myc-transgenic B cells, Eμ-Myc lymphomas and human Burkitt lymphoma samples were assessed for Nfkb2 expression. The contribution of Nfkb2 to Myc-driven apoptosis, proliferation, and lymphomagenesis was tested genetically in vivo.
Results
Here we report that the Myc oncoprotein suppresses Nfkb2 expression in vitro in primary mouse fibroblasts and B cells, and in vivo in the Eμ-Myc transgenic mouse model of human Burkitt lymphoma (BL). NFKB2 suppression by Myc was also confirmed in primary human BL. Promoter-reporter assays indicate that Myc-mediated suppression of Nfkb2 occurs at the level of transcription. The contribution of Nfkb2 to Myc-driven lymphomagenesis was tested in vivo, where Nfkb2 loss was shown to accelerate lymphoma development in Eμ-Myc transgenic mice, by impairing Myc's apoptotic response.
Conclusions
Nfkb2 is suppressed by c-Myc and harnesses Myc-driven lymphomagenesis. These data thus link Myc-driven lymphomagenesis to the non-canonical NF-κB pathway.
Similar content being viewed by others

Background
Members of the Rel/NF-κB family of transcription factors, including RelA (p65), RelB, c-Rel, NF-κB1 [p105/p50] and NF-κB2 [p100/p52], form homodimers and heterodimers that control aspects of T and B cell development, proliferation and survival. The activity of NF-κB dimers is held in check by dedicated inhibitors coined IκBα or IκBβ, which bind to and sequester NF-κB in the cytoplasm. Signals that activate NF-κB promote phosphorylation and ubiquitin-mediated destruction of the IκBs, which are substrates of the upstream IκB kinases Ikkα or Ikkβ (reviewed in [1–3]).
Regulation of NF-κB2 is unique amongst NF-κB family members. First, p100 NF-κB2 protein harbors a C-terminal ankyrin repeat domain that has intrinsic IκB activity, whereas its Rel homology domain directs its nuclear translocation, dimerization, and DNA binding functions. Second, p100 is proteolytically processed to p52, an event that requires Nik (NF-κB-inducing kinase) and Ikkα [4, 5] activity. Finally, targeted deletion of Nfkb2 in mice leads to defects in B cell development and in lymphoid organogenesis [6].
NF-κB promotes cell survival and proliferation, and alterations in this pathway, via chromosomal translocation or amplification, mutations, and deletions are common in cancer [7–9]. Important roles for NFKB2 in lymphomagenesis have been implicated from studies in both mouse and man. First, NFKB2 is a common target of chromosomal rearrangements in human B lymphomas, and these typically truncate the protein, generating constitutively active nuclear forms [10, 11] that have increased transcriptional activity compared to p52 [12, 13]. Furthermore, loss of the C-terminal ankyrin domain of NF-κB2 in mice results in enlarged lymph nodes and augments lymphocyte proliferation [14].
The c-MYC gene (hereafter Myc) is the cellular homolog of v-Myc, the transforming gene of the MC29 avian leukemia virus [15]. Myc oncoproteins (c-Myc, N-Myc and L-Myc) are activated in ~70% of human malignancies [16] and they function as basic helix-loop-helix-leucine zipper transcription factors that coordinate cell growth, division and metabolism [17–19]. In normal tissue, the regulation of Myc transcription and turnover is tightly controlled by mitogenic and growth inhibitory cues [20], and in tumour cells these controls are frequently lost, either by chromosomal translocations or amplifications, or indirectly by mutations in regulatory pathways.
The Rel/NF-κB pathway is suppressed in Myc-driven human Burkitt lymphoma (BL) [21] and in Myc overexpressing precancerous B cells [22]. Here we report that Myc suppresses Nfkb2 expression in B cells and show that NF-κB2 contributes to Myc's apoptotic response that harnesses lymphomagenesis.
Methods
Mice and tumour analyses
Nfkb2 null mice (C57BL/6) [6] (obtained from Christopher Hunter, University of Pennsylvania, Philadelphia, PA, USA) were bred with Eμ-Myc transgenic mice (C57Bl/6) [23]. F1 Eμ-Myc;Nfkb2 +/- offspring were bred to Nfkb2 +/-mice to obtain the desired Eμ-Myc;Nfkb2 +/+(n = 80) and Eμ-Myc;Nfkb2 -/-(n = 44) cohort. Note that numbers of Eμ-Myc;Nfkb2 -/-mice generated were lower than those of Eμ-Myc;Nfkb2 +/+mice due to integration of the Myc transgene on chromosome 19 where the Nfkb2 gene resides (assessed by FISH analysis, data not shown). Animals were observed for signs of morbidity and tumour development. Lymphomas were harvested, snap-frozen in liquid nitrogen, and then processed for RNA and protein analyses.
With Institutional Review Board approval, and following informed consent, lymphomas from 17 BL patients were banked. RNA and protein were extracted from these tumours (kindly provided by Drs. John Sandlund and Mihaela Onciu, St. Jude Children's Research Hospital, SJCRH, Memphis, TN). As a control, pooled peripheral blood mononuclear cells (PBMC) from healthy donors were enriched using CD19-MicroBeads (Miltenyi Biotech, Bergisch-Gladbach, Germany) and RNA and protein were prepared.
Cell culture
Primary bone marrow (BM)-derived pre-B cells were cultured as described [24]. B cells and mouse embryonic fibroblasts (MEFs, cultured from E13.5-E14.5 embryos) were infected with MSCV-Myc-ER™-IRES-GFP or MSCV-Myc-IRES-GFP retroviruses as described [25]. To activate Myc, Myc-ER™-expressing cells were treated with 2-μM 4-hydroxy-tamoxifen (4-HT) and lysed for protein and RNA analyses. P493-6 human B cells were kindly provided by G. Bornkamm (Helmholtz Zentrum München, Munich, Germany) and were cultured with tetracycline (Tet) or estrogen (both from Sigma-Aldrich, Taufkirchen, Germany) as described [26]. To suppress Myc transcription, cells were treated with Tet (Sigma, Taufkirchen, Germany) for 60 hr; to reactivate Myc cells were then resuspended in medium lacking Tet. Transient transfections of HeLa cells were performed using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen, Carlsbad, CA).
FACS analysis and magnetic-activated cell sorting (MACS) of B cells
Rates of proliferation of bone marrow (BM) and splenic B220+sIgM+ and B220+sIgM- cells were determined using a Flow Kit as described by the manufacturer (BD Biosciences Pharmingen, San Diego, CA). Briefly, animals were injected intraperitoneally with 100 μl of 10 mg/ml BrdU in sterile PBS. Animals were humanely sacrificed 12 hr following injection and BM and spleen were harvested. Red cells were lysed using ammonium chloride/potassium bicarbonate solution. Cells were then resuspended, incubated with antibody against B220 and sIgM (BD Biosciences Pharmingen, San Diego, CA), washed and collected by centrifugation. One million cells were further processed and stained with anti-BrdU-FITC antibody, and 5 × 105 cells were stained with Propidium iodide and Annexin-V FITC antibody (Annexin-V-Fluor Kit, Roche Applied Sciences, Indianapolis, IN). Following incubation cells were washed, resuspended in PBS, and then analysed by FACS. The remainder of the BM and spleen cells were enriched for B cells by magnetic cell sorting with B220 MicroBeads according to the manufacturer's instructions (Miltenyi Biotech, Bergisch-Gladbach, Germany) and were lysed for immunoblot or real-time PCR analysis.
RNA preparation and analyses
RNA was prepared using the RNeasy kit (Qiagen, Valencia, CA). For Affymetrix analyses, cRNA was synthesized using the One-Cycle Target Labeling and Control Reagent package (Affymetrix Inc., Santa Clara, CA) and the reaction was probed to the 430A mouse Affymetrix chip. The GCRMA normalization algorithm was applied to all Affymetrix Chips using GeneSpring GX (v7.3). GeneSpring Hierarchical Clustering (Similarity measure: Pearson Correlation, Clustering Algorithm: Average Linkage) was applied to those probe sets with signal higher than median in at least one sample. For real-time PCR, cDNA was prepared from 1 μg RNA using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). Real-time PCR was performed using an iCycler machine (Bio-Rad) and the iTaq SYBR green kit (Bio-Rad). Data analysis was performed by comparing Ct values with a control sample set as 1. Sequences of primers are available upon request.
Immunoblotting
Protein extracts (20 or 50 μg per lane) were electrophoretically separated on SDS-PAGE gels, transferred to membranes (Protran, Schleicher & Schuell, Dassel, Germany) and blotted with antibodies specific for NF-κB2, c-Myc, IKKα, Miz-1 (Santa Cruz Inc., Santa Cruz, CA) and β-Actin (Sigma Chemicals, St. Louis, MO). Equal loading of protein was demonstrated by Ponceau Red staining of the nitrocellulose membranes following transfer.
Reporter assay
HeLa cells were co-transfected with an Nfkb2 promoter-reporter construct (firefly luciferase, gift of R. M. Schmid, Munich, Germany) and p65 (R. M. Schmid), c-Myc or GFP expression plasmids. Relative luciferase activity was determined as described by the manufacturer (Promega, Madison, WI), by calculating the ratio of firefly to co-transfected renilla luciferase activity.
Results
The Rel/NF-κB pathway is suppressed in Myc-induced lymphomas
In the Eμ-Myc model of human B cell lymphoma disease progression is characterized by a pre-neoplastic state in which high proliferative rates of Eμ-Myc B cells are offset by high levels of apoptosis, which is then disabled during progression to the malignant state [24, 27]. The Rel/NF-κB pathway is suppressed in Myc-transgenic precancerous B cells [22] and in human B lymphoma [21], yet the underlying targets in this response are not resolved. To address this issue we initially assessed the expression of NF-κB components in B220+ splenic B cells of 4 week-old precancerous Eμ-Myc transgenic mice (n = 5) and their wild type littermate controls (n = 5), and in several (n = 13) Eμ-Myc lymphomas. As expected, there were clear increases in the levels of the established Myc targets Cad [28] and Rcl [29], in precancerous Eμ-Myc B cells, and even more so in Eμ-Myc lymphomas, compared to their levels in wild type splenic B cells (Figure 1, lower panel). In contrast, there were reduced levels of nearly all mRNAs encoding Rel/Nfkb factors, their inhibitors (Nfkbia and Nfkbib), and their upstream regulators in pre-malignant Eμ-Myc B cells compared to their levels expressed in wild type B cells (Figure 1). Strikingly, this response was significantly augmented in frank Eμ-Myc lymphomas (Figure 1). Thus, Myc-mediated suppression of the Rel/NF-κB pathway is augmented following the switch to the neoplastic state.

Myc targets the Rel/NF-κB network in precancerous B cells and lymphomas. Hierarchical clustering of Rel/Nfkb family members, upstream kinases that regulate the NF-κB pathway, and selected control genes was performed using RNA prepared from B220+ splenic B cells from five weanling-age wild type mice (wt), from five Eμ-Myc transgenic mice (Eμ-Myc), and from thirteen Eμ-Myc lymphomas. Probe set signals were normalized to the mean across wt samples, and values of each individual sample are represented by a colour, with green corresponding to expression below, red corresponding to expression above, and black corresponding to expression equal to the wt mean expression.
Nfkb2RNA and protein levels are suppressed by c-Myc
NFKB2 is the most frequently mutated Rel/Nfkb member in human lymphoid malignancies [10, 13]. We thus assessed the effects of Myc on Nfkb2 expression in B cells by real time PCR and western blot analyses of age-matched littermate B220+ wild type and precancerous Eμ-Myc B cells. Consistent with the profiling data, Nfkb2 mRNA and NF-κB p100 and p52 protein levels were markedly reduced in Eμ-Myc bone marrow (BM) and splenic B220+ B cells compared to wild type B cells (Figure 2a, b). Nfkb2 suppression was not due major differences in B cell subsets in the BM or spleens of wild type versus pre-malignant Eμ-Myc mice (data not shown). Further, there was a marked suppression of Nfkb2 transcripts in all Eμ-Myc lymphomas (Figure 2c), and in all human Burkitt lymphomas compared to pooled CD19+ control B cells from healthy donors (Figure 2e). As expected, Myc transcript and protein levels remained highly elevated in manifest lymphoma [22, 30]. Finally, both NF-κB p100 and p52 levels were reduced in the majority of Eμ-Myc lymphomas (Figure 2d). Thus, Nfkb2 expression is suppressed in Myc-driven lymphomas in mice and man.

Nfkb2 suppression in precancerous Myc-transgenic B cells and Myc-driven lymphomas. a) real-time PCR analysis of c-Myc, Nfkb2 and p21 mRNA levels in bone marrow (BM) and splenic (spleen) B220+ wild type (wt, grey bars) and precancerous Eμ-Myc (Eμ, black bars) B cells. Levels of mRNA are standardized to the expression of Ubiquitin (Ub), which is not regulated by Myc. * indicates p < 0.05. b) immunoblot analyses of the indicated proteins in control and precancerous Eμ-Myc B220+ B cells from bone marrow (BM) and spleen. c) real-time PCR analysis of Nfkb2 expression in ten Eμ-Myc lymphomas or splenic wild type (wt) B220+ B cells. d) immunoblot analysis of NF-κB2 p100 and p52 expression in wild type (wt) and Eμ-Myc precancerous B220+ B cells and Eμ-Myc lymphomas. Nfkb2 -/- and Nfkb1 -/- B220+ B cells served as controls. e) real-time PCR analysis of NFKB2 expression in seventeen human Burkitt lymphoma samples compared to CD19+ control B cells. Levels of mRNAs were normalized to the expression of Ubiquitin (Ub).
Nfkb2transcription is repressed by Myc
The regulation of Nfkb2 gene expression is complex and involves positive and negative regulatory mechanisms, including autoregulation [31, 32]. In part its expression is controlled by the activity of Ikkα [33]. However, we did not observe significant changes of Ikkα protein in Eμ-Myc lymphomas (Figure 2d). Thus, other mechanisms must mediate the suppression of Nfkb2 by Myc in B cells.
To confirm that Nfkb2 expression in B cells was indeed responsive to Myc, primary BM-derived B cells from wild type mice were cultured and infected with the MSCV-Myc-ER™-IRES-GFP retrovirus that expresses the tamoxifen-regulated, estrogen receptor chimeric Myc transgene, Myc-ER™ [34], along with the gene encoding green fluorescence protein (GFP) through the agency of an internal ribosome entry site (IRES). GFP + cells were isolated by FACS and expanded in culture in IL-7 medium. These cells were then treated with 4-hydroxytamoxifen (4-HT) to activate Myc-ER™. Notably, Myc activation led to reductions in the levels of Nfkb2 transcripts (Figure 3a). To exclude possible indirect effects of Myc-induced apoptosis on Nfkb2 expression, we also evaluated human P493-6 B cells, which harbor a tetracycline (Tet) promoter-regulated, conditional human c-Myc transgene [26] that is robustly induced following removal of Tet. Notably the induction of c-Myc in this system led to marked reduction in Nfkb2 mRNA levels (Figure 3b). Finally, using the Myc-ER™ system, we also assessed Nfkb2 mRNA in primary mouse embryonic fibroblasts (MEFs). Again, Nfkb2 mRNA levels were reduced following activation of Myc-ER™ (Additional File 1). Therefore, the activation or overexpression of Myc suppresses Nfkb2 mRNA and protein expression.

Myc represses Nfkb2 transcription. a) SYBR-green real-time PCR analysis of Nfkb2 mRNA levels in primary, ex vivo cultured B cells infected with MSCV-Myc-ER™-IRES-GFP (Myc-ER) virus or MSCV-IRES-GFP (GFP) control virus. GFP-positive cells were sorted by flow cytometry, cultured, and treated with 4-hydroxytamoxifen (4-HT) for the indicated times. Levels of mRNAs are standardized to the expression of Ubiquitin (Ub). * indicates p < 0.05. b) SYBR-green real-time PCR analysis of NFKB2 expression in human P493-6 cells treated with tetracycline (T, Myc-off state) ± estrogen (E, EBNA2-on state) for the indicated times. c) HeLa cells were co-transfected with an Nfkb2 promoter-reporter construct (firefly luciferase) and the indicated expression plasmids. The relative luciferase activity was determined by calculatiing the ratio of firefly to co-transfected renilla luciferase activity. * indicates p < 0.05, ** indicates p < 0.01. d) expression of the indicated transcripts measured by real-time PCR following infection of primary MEFs with Miz-1 or control (Hygro) retroviruses. Levels of the indicated mRNA were standardized to the expression of Ubiquitin (Ub).
To test whether Nfkb2 transcription was repressed by Myc, we assessed the effects of Myc on Nfkb2 promoter activity using luciferase reporter-based plasmids [32] in HeLa cells. Notably, Myc significantly repressed Nfkb2-promoter activity, and co-expression of Myc also inhibited the robust induction of the Nfkb2 promoter by p65/RelA (Figure 3c). Myc often represses transcription by binding to and inhibiting the functions of the transcriptional activator Miz-1 [35]. Since the Nfkb2 promoter region contains an Initiator element (INR)-like sequence and INRs mediate Miz-1-induced transcription [36], we evaluated whether Miz-1 overexpression induced Nfkb2 transcripts. As expected, the direct Miz-1 target genes p15 INK4band p21 [37, 38] were induced by Miz-1 in primary MEFs, and Nfkb2 transcript levels were also increased (Figure 3d). Thus, the Myc-mediated suppression of Nfkb2 transcription may involve disruption of Miz-1 functions.
Nfkb2impairs Myc-induced lymphomagenesis
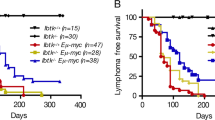
The remarkable changes in the expression of components of the Rel/NF-κB signalling pathway, and particularly the suppression of Nfkb2 by c-Myc, suggested that NF-kB2 might play critical roles in Myc-driven tumorigenesis. If Myc-mediated reductions in Nfkb2 expression in Eμ-Myc B cells were important for Myc-mediated lymphomagenesis, we reasoned that total loss of Nfkb2 should affect lymphoma development. To test this hypothesis, Eμ-Myc transgenic mice were mated to Nfkb2 -/- mice [6] and F1 offspring were bred to Nfkb2 +/- mice to obtain the desired Eμ-Myc;Nfkb2 +/+ and Eμ-Myc;Nfkb2 -/- cohorts. These littermates were then observed for lymphoma onset. Eμ-Myc transgenic mice usually succumb to aggressive lymphoma within 4-8 months of birth [23]. As expected, non-transgenic Nfkb2 -/- littermates showed no signs of tumour development throughout their lifespan (data not shown). Importantly, Eμ-Myc;Nfkb2 -/- transgenic displayed a moderately accelerated course of lymphoma development and, accordingly, had a shorter lifespan, with a median survival of 171 days compared to 205 days median survival in their Eμ-Myc;Nfkb2 +/+ littermates (Figure 4a, p = 0.0307). The lymphomas that arose in Eμ-Myc;Nfkb2 -/- transgenics were phenotypically identical to those that arose in Eμ-Myc;Nfkb2 +/+ mice, and full necropsy and histopathological examination demonstrated similar dissemination of disease in Eμ-Myc;Nfkb2 +/+ versus Eμ-Myc;Nfkb2 -/- mice (data not shown). Thus, Nfkb2 loss accelerates Myc-driven lymphomagenesis without overtly affecting the disease phenotype.

Nfkb2 loss accelerates Myc-induced lymphomagenesis by impairing apoptosis. a) Effects of Nfkb2 deficiency on Myc-induced lymphomagenesis. The survival of Eμ-Myc transgenic littermates of the indicated Nfkb2 genotypes is shown. The differences in the rates of tumor incidence between the Nfkb2 +/+and the Nfkb2 -/-littermates is statistically significant (p = 0.0307). b) Eμ-Myc transgenic littermates of the indicated Nfkb2 genotypes were injected with BrdU, and cells from bone marrow and spleen were harvested 12 hr later. BrdU-incorporation was analyzed using an antibody-dependent fluorescence assay. The bars show the mean percentage of cells in S phase ± SEM (three independent experiments). c) The apoptotic index of the indicated genotypes was analyzed using an antibody dependent fluorescence assay. Annexin V+ B220+ cells of sIgM- or sIgM+ phenotype are shown. The bars represent the mean ± SEM from three independent experiments. * indicates p < 0.05.
Loss of the tumour suppressors p53 or Arf, which mediate Myc's apoptotic response [25], dramatically accelerates Myc-driven tumorigenesis [24, 39]. By contrast, loss of regulators of the Myc-to-p27Kip1 pathway that regulates Myc's proliferative response, such as E2f1 and Cks1, markedly delays lymphoma development and prevents dissemination of disease [27, 30, 40]. We therefore assessed the effects of Nfkb2 loss on Myc's proliferative and apoptotic responses. To evaluate effects on Myc's proliferative response BrdU was injected intraperitoneally into 4-week old Eμ-Myc;Nfkb2 +/+ and Eμ-Myc;Nfkb2 -/- littermates and after 12 hr B220+sIgM+ and B220+sIgM- cells were assessed for their S phase indices. Loss of Nfkb2 had essentially no effects on Eμ-Myc B cell proliferation in either BM or spleen (Figure 4b). To address the effects of Nfkb2 loss on Myc-induced apoptosis, the apoptotic indices of pre-cancerous B220+ B cells from Eμ-Myc;Nfkb2 +/+ and Eμ-Myc;Nfkb2 -/- mice were determined by staining with anti-Annexin V-FITC and propidium iodide. There was a significant reduction in the apoptotic indices of sIgM- splenic Eμ-Myc;Nfkb2 -/- B cells compared to matched sIgM- splenic B cells from Eμ-Myc;Nfkb2 +/+ mice (Figure 4c). The sIgM- population of B cells are those most prone to Myc-induced apoptosis in the Eμ-Myc model [41]. Thus, the accelerated lymphoma development manifest in Eμ-Myc;Nfkb2 -/- mice is associated with an impaired apoptotic response.
Discussion
Members of the Rel/NF-κB family can, depending on cell context, act as either tumour suppressors or oncogenes [42]. A hallmark of human Burkitt lymphoma, and of pre-malignant B cells and lymphomas of Eμ-Myc mice, is suppression of the Rel/NF-κB signaling pathway [21, 22, 43]. These findings, and those reported herein, suggest that Myc-induced suppression of this important immune and inflammatory regulatory network contributes to lymphomagenesis.
Myc executes most of its functions as a transcription factor that regulates a large cast of target genes (see: Myc Cancer Gene http://www.myc-cancer-gene.org; reviewed in [44, 45]). Genes suppressed by Myc include those encoding proteins involved in apoptosis [19] and growth arrest [46]. Various mechanisms of transcriptional repression by Myc are operational. Amongst these, Myc-mediated inhibition of Miz-1 transcriptional activity by Myc:Max heterodimers is clearly a central pathway by which Myc overrides cell growth control [44–46]. Growth arrest genes, including p21 [47], p15 [37] and Gadd45 [48], are elevated in c-Myc -/- cells and are suppressed in cells that overexpress Myc. In B lymphocytes Myc is required for normal B cell proliferation and CD40-mediated proliferation [49]. CD40 signalling has been shown to activate the non-canonical NF-κB pathway [50]. It was therefore tempting to speculate that the suppression of Nfkb2 by Myc controls the expansion of B cells under normal and/or stressful conditions. Unexpectedly, in the Eμ-Myc model precancerous B cell proliferation was not significantly reduced upon Nfkb2 loss.
The recently described autoimmune disease in mice having constitutive NF-κB2 p52 in lymphocytes is associated with increased B cell proliferation and a defective response to apoptotic stimuli [51]. Further, in transgenic mice that express the lymphoma-associated NF-κB2 mutant p80HT, B cell proliferation is not affected but lymphomas do occur, and B cells from these mice are resistant to apoptosis [52]. In the Eμ-Myc model loss of Nfkb2 does not affect Myc's proliferative response, but it does disable the apoptotic response in sIgM- Eμ-Myc B cells, which are the subset that is most prone to Myc-induced apoptosis in this mouse model [41]. Thus, at least in the context of Myc overexpression, which is clearly a hallmark of most rapidly dividing human malignancies including lymphoma and leukemia, NF-κB2 contributes to the apoptosis response and, accordingly, Nfkb2 loss accelerates Myc-driven lymphomagenesis. In addition to Myc suppression of the non-canonical pathway discovered herein, others have recently attributed tumour suppressive functions to classical NF-κB activity in the context of Myc-induced lymphoma [53]. Therefore, both the canonical and the non-canonical NF-κB pathway function as tumour suppressors in Myc-transformed murine lymphoma and most likely human Burkitt lymphoma.
Conclusions
Our work identifies Nfkb2 as a Myc repression target, and Myc appears to regulate both basal and stimulated Nfkb2 transcription. Suppression of the non-canonical NF-κB pathway provides a selective advantage to Myc-transformed lymphomas and thus contributes to lymphomagenesis. These findings have implications for the development of therapies against Myc-dependent tumours.
References
Gilmore TD: Introduction to NF-κB: players, pathways, perspectives. Oncogene. 2006, 25 (51): 6680-6684. 10.1038/sj.onc.1209954.
Hayden MS, Ghosh S: Signaling to NF-κB. Genes Dev. 2004, 18 (18): 2195-2224. 10.1101/gad.1228704.
Ghosh S, Karin M: Missing pieces in the NF-κB puzzle. Cell. 2002, 109 (Suppl): S81-96. 10.1016/S0092-8674(02)00703-1.
Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, et al: Activation by IKKα of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001, 293 (5534): 1495-1499. 10.1126/science.1062677.
Xiao G, Harhaj EW, Sun SC: NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Mol Cell. 2001, 7 (2): 401-409. 10.1016/S1097-2765(01)00187-3.
Caamano JH, Rizzo CA, Durham SK, Barton DS, Raventos-Suarez C, Snapper CM, Bravo R: Nuclear factor (NF)- κB2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J Exp Med. 1998, 187 (2): 185-196. 10.1084/jem.187.2.185.
Orlowski RZ, Baldwin AS: NF-κB as a therapeutic target in cancer. Trends Mol Med. 2002, 8 (8): 385-389. 10.1016/S1471-4914(02)02375-4.
Karin M, Cao Y, Greten FR, Li ZW: NF-κB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002, 2 (4): 301-310. 10.1038/nrc780.
Courtois G, Gilmore TD: Mutations in the NF-κB signaling pathway: implications for human disease. Oncogene. 2006, 25 (51): 6831-6843. 10.1038/sj.onc.1209939.
Rayet B, Gelinas C: Aberrant rel/nfkb genes and activity in human cancer. Oncogene. 1999, 18 (49): 6938-6947. 10.1038/sj.onc.1203221.
Perkins ND: Oncogenes, tumor suppressors and p52 NF-κB. Oncogene. 2003, 22 (48): 7553-7556. 10.1038/sj.onc.1207139.
Chang CC, Zhang J, Lombardi L, Neri A, Dalla-Favera R: Rearranged NFKB-2 genes in lymphoid neoplasms code for constitutively active nuclear transactivators. Mol Cell Biol. 1995, 15 (9): 5180-5187.
Kim KE, Gu C, Thakur S, Vieira E, Lin JC, Rabson AB: Transcriptional regulatory effects of lymphoma-associated NFKB2/lyt10 protooncogenes. Oncogene. 2000, 19 (10): 1334-1345. 10.1038/sj.onc.1203432.
Ishikawa H, Carrasco D, Claudio E, Ryseck RP, Bravo R: Gastric hyperplasia and increased proliferative responses of lymphocytes in mice lacking the COOH-terminal ankyrin domain of NF-κB2. J Exp Med. 1997, 186 (7): 999-1014. 10.1084/jem.186.7.999.
Sheiness D, Fanshier L, Bishop JM: Identification of nucleotide sequences which may encode the oncogenic capacity of avian retrovirus MC29. J Virol. 1978, 28 (2): 600-610.
Boxer LM, Dang CV: Translocations involving c-myc and c-myc function. Oncogene. 2001, 20 (40): 5595-5610. 10.1038/sj.onc.1204595.
Kelly K, Cochran BH, Stiles CD, Leder P: Cell-specific regulation of the c-myc gene by lymphocyte mitogens and platelet-derived growth factor. Cell. 1983, 35 (3 Pt 2): 603-610. 10.1016/0092-8674(83)90092-2.
Henriksson M, Luscher B: Proteins of the Myc network: essential regulators of cell growth and differentiation. Adv Cancer Res. 1996, 68: 109-182. full_text.
Nilsson JA, Cleveland JL: Myc pathways provoking cell suicide and cancer. Oncogene. 2003, 22 (56): 9007-9021. 10.1038/sj.onc.1207261.
Grandori C, Cowley SM, James LP, Eisenman RN: The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu Rev Cell Dev Biol. 2000, 16: 653-699. 10.1146/annurev.cellbio.16.1.653.
Dave SS, Fu K, Wright GW, Lam LT, Kluin P, Boerma EJ, Greiner TC, Weisenburger DD, Rosenwald A, Ott G, et al: Molecular diagnosis of Burkitt's lymphoma. N Engl J Med. 2006, 354 (23): 2431-2442. 10.1056/NEJMoa055759.
Keller U, Nilsson JA, Maclean KH, Old JB, Cleveland JL: Nfkb 1 is dispensable for Myc-induced lymphomagenesis. Oncogene. 2005, 24 (41): 6231-6240. 10.1038/sj.onc.1208779.
Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL: The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985, 318 (6046): 533-538. 10.1038/318533a0.
Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL: Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes & development. 1999, 13 (20): 2658-2669. 10.1101/gad.13.20.2658.
Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF: Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes & development. 1998, 12 (15): 2424-2433. 10.1101/gad.12.15.2424.
Pajic A, Spitkovsky D, Christoph B, Kempkes B, Schuhmacher M, Staege MS, Brielmeier M, Ellwart J, Kohlhuber F, Bornkamm GW, et al: Cell cycle activation by c-myc in a burkitt lymphoma model cell line. Int J Cancer. 2000, 87 (6): 787-793. 10.1002/1097-0215(20000915)87:6<787::AID-IJC4>3.0.CO;2-6.
Baudino TA, Maclean KH, Brennan J, Parganas E, Yang C, Aslanian A, Lees JA, Sherr CJ, Roussel MF, Cleveland JL: Myc-mediated proliferation and lymphomagenesis, but not apoptosis, are compromised by E2f1 loss. Mol Cell. 2003, 11 (4): 905-914. 10.1016/S1097-2765(03)00102-3.
Frank SR, Schroeder M, Fernandez P, Taubert S, Amati B: Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes & development. 2001, 15 (16): 2069-2082. 10.1101/gad.906601.
Lewis BC, Shim H, Li Q, Wu CS, Lee LA, Maity A, Dang CV: Identification of putative c-Myc-responsive genes: characterization of rcl, a novel growth-related gene. Molecular and cellular biology. 1997, 17 (9): 4967-4978.
Keller UB, Old JB, Dorsey FC, Nilsson JA, Nilsson L, Maclean KH, Chung L, Yang C, Spruck C, Boyd K, et al: Myc targets Cks1 to provoke the suppression of p27(Kip1), proliferation and lymphomagenesis. Embo J. 2007, 26 (10): 2562-2574. 10.1038/sj.emboj.7601691.
Liptay S, Schmid RM, Nabel EG, Nabel GJ: Transcriptional regulation of NF-κB2: evidence for κB-mediated positive and negative autoregulation. Molecular and cellular biology. 1994, 14 (12): 7695-7703.
Oswald F, Liptay S, Adler G, Schmid RM: NF-κB2 is a putative target gene of activated Notch-1 via RBP-Jκ. Mol Cell Biol. 1998, 18 (4): 2077-2088.
Qing G, Xiao G: Essential role of IκB kinase-α in the constitutive processing of NF-κB2 p100. The Journal of biological chemistry. 2005, 280 (11): 9765-9768. 10.1074/jbc.C400502200.
Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI: A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic acids research. 1995, 23 (10): 1686-1690. 10.1093/nar/23.10.1686.
Peukert K, Staller P, Schneider A, Carmichael G, Hanel F, Eilers M: An alternative pathway for gene regulation by Myc. Embo J. 1997, 16 (18): 5672-5686. 10.1093/emboj/16.18.5672.
Wanzel M, Herold S, Eilers M: Transcriptional repression by Myc. Trends Cell Biol. 2003, 13 (3): 146-150. 10.1016/S0962-8924(03)00003-5.
Staller P, Peukert K, Kiermaier A, Seoane J, Lukas J, Karsunky H, Moroy T, Bartek J, Massague J, Hanel F, et al: Repression of p15INK4b expression by Myc through association with Miz-1. Nat Cell Biol. 2001, 3 (4): 392-399. 10.1038/35070076.
Wu S, Cetinkaya C, Munoz-Alonso MJ, von der Lehr N, Bahram F, Beuger V, Eilers M, Leon J, Larsson LG: Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene. 2003, 22 (3): 351-360. 10.1038/sj.onc.1206145.
Schmitt CA, McCurrach ME, de Stanchina E, Wallace-Brodeur RR, Lowe SW: INK4a/ARF mutations accelerate lymphomagenesis and promote chemoresistance by disabling p53. Genes Dev. 1999, 13 (20): 2670-2677. 10.1101/gad.13.20.2670.
Old JB, Kratzat S, Hoellein A, Graf S, Nilsson JA, Nilsson L, Nakayama KI, Peschel C, Cleveland JL, Keller UB: Skp2 directs Myc-mediated suppression of p27Kip1 yet has modest effects on Myc-driven lymphomagenesis. Mol Cancer Res. 2010, 8 (3): 353-362. 10.1158/1541-7786.MCR-09-0232.
Nilsson JA, Keller UB, Baudino TA, Yang C, Norton S, Old JA, Nilsson LM, Neale G, Kramer DL, Porter CW, et al: Targeting ornithine decarboxylase in Myc-induced lymphomagenesis prevents tumor formation. Cancer Cell. 2005, 7 (5): 433-444. 10.1016/j.ccr.2005.03.036.
Perkins ND: NF-κB: tumor promoter or suppressor?. Trends Cell Biol. 2004, 14 (2): 64-69. 10.1016/j.tcb.2003.12.004.
Hummel M, Bentink S, Berger H, Klapper W, Wessendorf S, Barth TF, Bernd HW, Cogliatti SB, Dierlamm J, Feller AC, et al: A biologic definition of Burkitt's lymphoma from transcriptional and genomic profiling. N Engl J Med. 2006, 354 (23): 2419-2430. 10.1056/NEJMoa055351.
Adhikary S, Eilers M: Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol. 2005, 6 (8): 635-645. 10.1038/nrm1703.
Lee LA, Dang CV: Myc target transcriptomes. Curr Top Microbiol Immunol. 2006, 302: 145-167. full_text.
Gartel AL, Shchors K: Mechanisms of c-myc-mediated transcriptional repression of growth arrest genes. Exp Cell Res. 2003, 283 (1): 17-21. 10.1016/S0014-4827(02)00020-4.
Coller HA, Grandori C, Tamayo P, Colbert T, Lander ES, Eisenman RN, Golub TR: Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc Natl Acad Sci USA. 2000, 97 (7): 3260-3265. 10.1073/pnas.97.7.3260.
Bush A, Mateyak M, Dugan K, Obaya A, Adachi S, Sedivy J, Cole M: c-myc null cells misregulate cad and gadd45 but not other proposed c-Myc targets. Genes Dev. 1998, 12 (24): 3797-3802. 10.1101/gad.12.24.3797.
de Alboran IM, O'Hagan RC, Gartner F, Malynn B, Davidson L, Rickert R, Rajewsky K, DePinho RA, Alt FW: Analysis of c-MYC function in normal cells via conditional gene-targeted mutation. Immunity. 2001, 14 (1): 45-55. 10.1016/S1074-7613(01)00088-7.
Homig-Holzel C, Hojer C, Rastelli J, Casola S, Strobl LJ, Muller W, Quintanilla-Martinez L, Gewies A, Ruland J, Rajewsky K, et al: Constitutive CD40 signaling in B cells selectively activates the noncanonical NF-κB pathway and promotes lymphomagenesis. J Exp Med. 2008, 205 (6): 1317-1329. 10.1084/jem.20080238.
Wang Z, Zhang B, Yang L, Ding J, Ding HF: Constitutive production of NF-κB2 p52 is not tumorigenic but predisposes mice to inflammatory autoimmune disease by repressing Bim expression. J Biol Chem. 2008, 283 (16): 10698-10706. 10.1074/jbc.M800806200.
Zhang B, Wang Z, Li T, Tsitsikov EN, Ding HF: NF-κB2 mutation targets TRAF1 to induce lymphomagenesis. Blood. 2007, 110 (2): 743-751. 10.1182/blood-2006-11-058446.
Klapproth K, Sander S, Marinkovic D, Baumann B, Wirth T: The IKK2/NF-κB pathway suppresses MYC-induced lymphomagenesis. Blood. 2009, 114 (12): 2448-2458. 10.1182/blood-2008-09-181008.
Yang W, Shen J, Wu M, Arsura M, FitzGerald M, Suldan Z, Kim DW, Hofmann CS, Pianetti S, Romieu-Mourez R, et al: Repression of transcription of the p27(Kip1) cyclin-dependent kinase inhibitor gene by c-Myc. Oncogene. 2001, 20 (14): 1688-1702. 10.1038/sj.onc.1204245.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/10/348/prepub
Acknowledgements
This work was supported by Deutsche Forschungsgemeinschaft grant KE222/5-1 and EHA grant 2007/06 (UK). JLC acknowledges support of NIH grant CA076379, the Cancer Center (CORE) Support Grant CA21765 of St. Jude Children's Research Hospital (SJCRH), the American Lebanese Syrian Associated Charities (ALSAC) of SJCRH, and monies from the State of Florida to Scripps Florida. We thank Sara Norton and Katja Urlbauer for expert technical assistance, Drs. Mihaela Onciu and John Sandlund (SJCRH) for providing samples from Burkitt lymphoma patients and the Animal Resource Center, the Hartwell Center and the FACS Core Facility of SJCRH for support. We thank Bristol-Myers Squibb (Princeton, NJ), for the permission to use the Nfkb2-null mice, which were obtained from Dr. Christopher Hunter, (University of Pennsylvania, Philadelphia, PA, USA). We also thank Dr. Martin Eilers, (University of Würzburg, Würzburg, Germany), for providing the Miz-1 expression plasmid and Dr. Roland Schmid (TU München, Munich, Germany), for the Nfkb2 promoter reporter plasmid and the RelA/p65 expression plasmid.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
UK, CP and JLC designed the experiments. UK, JH and JAN carried out experiments as well as the statistical data analysis. MAH and MF performed gene expression analyses. UK and JLC wrote the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12885_2010_2147_MOESM1_ESM.PPT
Additional file 1: Myc suppresses Nfkb2 expression in primary, early passage MEFs. SYBR-green real-time PCR analysis of Nfkb2 RNA levels in primary MEFs infected with MSCV-Myc-ER™-IRES-GFP (Myc-ER) retrovirus. GFP-positive cells were sorted by flow cytometry and treated with 2 μM 4-HT for the indicated times. The known Myc targets E2f1 [27] and p27 [54] were included as controls. The levels of the mRNAs were standardized to the expression of Ubiquitin (Ub).* indicates p < 0.05. (PPT 158 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Keller, U., Huber, J., Nilsson, J.A. et al. Myc suppression of Nfkb2 accelerates lymphomagenesis. BMC Cancer 10, 348 (2010). https://doi.org/10.1186/1471-2407-10-348
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-10-348