
Abstract
Background
The discordance between steady-state levels of mRNAs and protein has been attributed to posttranscriptional control mechanisms affecting mRNA stability and translation. Traditional methods of genome wide microarray analysis, profiling steady-state levels of mRNA, may miss important mRNA targets owing to significant posttranscriptional gene regulation by RNA binding proteins (RBPs).
Methods
The ribonomic approach, utilizing RNA immunoprecipitation hybridized to microarray (RIP-Chip), provides global identification of putative endogenous mRNA targets of different RBPs. HuR is an RBP that binds to the AU-rich elements (ARE) of labile mRNAs, such as proto-oncogenes, facilitating their translation into protein. HuR has been shown to play a role in cancer progression and elevated levels of cytoplasmic HuR directly correlate with increased invasiveness and poor prognosis for many cancers, including those of the breast. HuR has been described to control genes in several of the acquired capabilities of cancer and has been hypothesized to be a tumor-maintenance gene, allowing for cancers to proliferate once they are established.
Results
We used HuR RIP-Chip as a comprehensive and systematic method to survey breast cancer target genes in both MCF-7 (estrogen receptor positive, ER+) and MDA-MB-231 (estrogen receptor negative, ER-) breast cancer cell lines. We identified unique subsets of HuR-associated mRNAs found individually or in both cell types. Two novel HuR targets, CD9 and CALM2 mRNAs, were identified and validated by quantitative RT-PCR and biotin pull-down analysis.
Conclusion
This is the first report of a side-by-side genome-wide comparison of HuR-associated targets in wild type ER+ and ER- breast cancer. We found distinct, differentially expressed subsets of cancer related genes in ER+ and ER- breast cancer cell lines, and noted that the differential regulation of two cancer-related genes by HuR was contingent upon the cellular environment.
Similar content being viewed by others


Background
Over the past decade array technologies have provided several new means for profiling global changes in gene expression. The power of DNA microarrays is perhaps best illustrated in the way it has been used to differentiate treatment responses in patient populations. Individualized and targeted therapy for several tumors, based upon underlying differences at the molecular level among gene expression profiles, is beginning to replace the traditional morphological-based treatment paradigm [1–3]. Genome wide microarray analyses, however, are inherently flawed since they globally profile the steady-state levels of mRNA, referred to as the transcriptome. Cellular protein expression levels, however, do not directly correlate with steady-state levels of mRNAs. It is well accepted in the RNA field that there is a poor correlation between steady-state RNA levels and protein. This discordance has been attributed to posttranscriptional control mechanisms affecting mRNA stability and translation. Steady-state mRNA levels of genes, controlled partially or totally at this level, may be misleading. Gygi and colleagues have shown that correlation between mRNA and protein levels could not be predicted from only mRNA steady-state levels [4]. They observed that some genes had the same mRNA levels but protein levels varied more than 20 fold. Conversely, some proteins were of equal expression but their respective mRNA level varied by more than 30-fold. They concluded that "transcript levels provide little predictive value with respect to the extent of protein expression" [4]. Additionally, Idekar and colleagues have described similar results for the galactose gene [5].
Although our understanding of transcriptional gene regulation is advanced, posttranscriptional gene regulation remains largely unexplored. It is becoming clear, however, that this is an important mode of gene regulation, especially for proinflammatory genes. These genes appear to be posttranscriptionally regulated by RNA binding proteins (RBPs) which interact with AU-rich elements (AREs) in the 3' untranslated region (UTR) of mRNAs. Approximately 3,000 human genes contain AREs, representing 8% of the human genome [6]. Many of these genes which possess AREs are in areas of transient biological responses, including cell growth and differentiation, immune responses, signal transduction, transcriptional and translational control, hematopoiesis, apoptosis, nutrient transport, and metabolism [6, 7].
New methodologies have provided global identification of in vivo mRNA targets of different RBPs. One of these, termed the ribonomic approach, involves the immunoprecipitation of ribonucleoprotein complexes (RNPs) with antibodies against different RBPs, extraction of mRNA, and hybridization to microarrays [8–10]. This approach, also referred to as RIP-Chip, enables investigators to identify groups of posttranscriptionally regulated mRNAs coordinately controlled by RBPs during various biological processes. A new paradigm, the posttranscriptional operon hypothesis, has been developed which states that RBPs coordinately regulate the expression of biologically related molecules [11, 12]. This paradigm is being confirmed by the work of many different laboratories as our understanding of posttranscriptional regulation broadens and putative operons are described [8, 13–17]. HuR is an RBP that binds to AREs of many proto-oncogenes and labile mRNAs. It has emerged as a key regulatory factor which stabilizes and translationally enhances its targets mRNAs, and affects their transport from the nucleus to the cytoplasm [18–20]. HuR belongs to the ELAV (embryonic lethal abnormal vision) family found in mammalian cells containing four members: HuR, HuB, HuC, and HuD. HuR is the only ubiquitously expressed member. The other family members are found primarily in the central nervous system and gonadal tissue [18]. Many HuR targets are cytokines, chemokines, and other early-response genes [21, 22].
Of the hallmarks of cancer originally described by Hanahan and Weinberg, HuR has been demonstrated to control expression of genes in multiple areas of malignant transformation [23]. Consequently, HuR has been suggested to function as a tumor maintenance gene, permissive for malignant transformation, tumor growth, and perhaps metastasis [24]. HuR has been described in the literature as controlling the expression of many cancer-relevant genes, including those that encode these proteins: Prothymosin-α, Bcl-2, Mcl-1, SirT1, TGF-β, MMP-9, MTC-1, uPA, VEGF-α, HIF1-α and cyclins A1 (CCN A1), B1 and D1 [25–35]. Increased levels of HuR have been associated with a more aggressive breast cancer and a worse prognosis [36–38]. Of significance, HuR has been described as posttranscriptionally regulating the expression of many breast cancer relevant genes including those that encode Glut-1, ERα, COX-2, IL-8, Cyclin E1, and most recently BRCA-1 [36, 39–44]. HuR RIP-Chip analysis has recently identified Thrombospondin 1 as a key HuR target in the MCT-1 transformed estrogen receptor positive (ER+) cell line MCF-7 [45]. Its interactions, however, are complex and, at times, HuR may interact with miRNAs such as Let-7 to translationally suppress the expression of C-MYC mRNA [46].
Since HuR has been described as regulating the expression of many cancer relevant genes, we asked whether it may coordinately regulate breast cancer genes in ER+ and ER- breast cancer. We performed a HuR RIP-Chip analysis on MDA-MB-231 (ER-) and MCF-7 (ER+) cell lines to identify cancer-relevant genes, not known to be regulated by HuR, and potential novel breast cancer targets. Our studies indicated that HuR was associated with unique subsets of mRNAs in each cell line as well as a subset of HuR associated mRNA targets common to both. We chose two cancer-associated genes, CD9 and CALMODULIN 2 (CALM2), highly expressed in both cell lines, and functionally validated the role of HuR in regulating their expression. Unexpectedly, HuR differentially regulated the same target, CD9, in both cell lines in an opposite manner. Moreover, we found presumptive differential regulation of CALM2 by HuR, as HuR interacted only with CALM2 mRNA, but not with family members CALM1 and CALM3 mRNAs. We discovered that HuR interacts with many breast cancer-relevant genes not previously known to be controlled by HuR, and target genes which have not been shown to be cancer related. This latter category may indeed represent novel cancer genes discovered by HuR RIP-Chip analysis.
Methods
Cells in culture
The MDA-MB-231 (MB-231) and MCF-7 cell lines were obtained from American Type Culture Collection (Manassas, VA). The cell lines were maintained at 37°C in a humidified atmosphere of 95% air and 5% CO2. MB-231 cells were grown in RPMI (GIBCO®, Invitrogen™, Carlsbad, CA) containing 10% fetal calf serum (Hyclone, Thermo Fisher Scientific, Waltham, MA), 0.5 mM L-glutamine (GIBCO®), 25 mg/ml glucose (Sigma-Aldrich), HEPES (GIBCO®) and sodium pyruvate (GIBCO®). MCF-7 cells were grown in DMEM (GIBCO®) supplemented with 10% fetal calf serum.
HuR Immunoprecipitations (RIP-Chip)
HuR RIP-Chip was performed as previously described [8, 47, 48]. Briefly, lysates were prepared from exponentially growing MB-231 and MCF-7 cells. Equal amounts of protein lysates were used (100-300 μg). HuR monoclonal antibody 3A2 (made in our laboratory from the 3A2 hybridoma, generously provided by Dr. Joan Steitz, Yale University, New Haven, CT), or isotype control IgG1 (BD Biosciences, San Jose, CA), were pre-coated onto Protein A Sepharose beads (PAS) and extensively washed. Lysates from each cell initially were pre-absorbed with 30 μg of IgG1 and then removed by addition of PAS beads. Individual pull-downs were performed at 4°C for only 1-2 hr to minimize potential re-assortment of mRNAs.
RNA amplification
The entire amount of recovered RNA per immunoprecipitation was amplified using the WT-Ovation™ Pico RNA Amplification System protocol (NuGen, San Carlos, CA). Forty ng of total RNA was used as starting material to generate at least 6 μg of cDNA. Amplified cDNA was purified using Zymo Research Clean and Concentrator™-25 (Zymo Research, Orange, CA). Three μg of amplified and purified cDNA was incubated at 50°C for 30 minutes with 5 μl of UNG buffer and 5 μl UNG enzyme and 60 minutes with 5 μl labeling buffer and 5 μl ARP (biotin) solution as described in NuGen's labeling protocol for the Illumina BeadArray platform. All samples (total RNA, amplified cDNA, and biotin labeled amplified cDNA) were quantitated using a Nanodrop™ (Thermo Fisher Scientific, Waltham, MA) spectrophotometer. RNA quality and integrity were assessed on selected samples with the Experion™ automated electrophoresis system (Bio-Rad, Hercules, CA).
Microarray
Biotin-labeled, amplified cDNA (1.5 μg) was hybridized to a Sentrix® Human-6 v.2 Whole Genome Expression BeadChips (Sentrix Human WG-6; Illumina, San Diego, CA). Each chip tested 6 samples and contained 47,293 gene targets, representing 18,025 distinct RefSeq genes that are not pseudogenes. A total of 3 chips were used for this experiment. The chips were hybridized at 48°C for 20 hr in the hybridization buffer provided by the manufacturer. After hybridization, the chips were washed and stained with streptavidin-C3. The chips were scanned on the BeadArray Reader, as described by Illumina at http://www.illumina.com. The Illumina BeadStudio software was used to assess fluorescent hybridization signals.
Quantitative RT-PCR
Selected genes were validated by quantitative RT-PCR. Briefly, cDNA was generated from the same samples as previously described for the microarray experiments using 10 ng total RNA and the SuperScript™ III Platinum® Two-Step qRT-PCR Kit with SYBR® Green (Invitrogen Carlsbad, CA). RT-PCR was performed on the StepOne™ Real-Time PCR System (Applied Biosystems, Foster City, CA). Each sample was run in triplicate for these genes and the cDNA was divided equally per reaction in a 20 μl volume. The PCR conditions were: 50°C for 2 minutes and 95°C for 2 minutes, followed by 40 cycles of 95°C for 15 seconds alternating with 60°C for 30 seconds. Melting curve analysis was performed on every reaction to confirm a single amplicon. For each cell line, differences in gene expression were determined using the equation 2-ΔΔCt, where the C t value for either the HuR or IgG IP was subtracted from the C t value of the GAPDH control to yield the ΔC t value. For each cell line, the ΔC t value for the HuR and IgG IP were computed in triplicate and averaged to give one ΔΔC t value per sample. Primers used:
Human RT GAPDH Forward 5' AGCCTCAAGATCATCAGCAATGCC 3'
Reverse 5' TGTGGTCATGAGTCCTTCCACGAT 3'
Human RT HuR Forward 5' ATGAAGACCACATGGCCGAAGACT 3'
Reverse 5' AGTTCACAAAGCCATAGCCCAAGC 3'
Human RT CD9 Forward 5' TCAGACCAAGAGCATCTTCGAGCA 3'
Reverse 5' ACCAAGAGGAAGCCGAAGAACAGT 3'
Human RT CALM2 Forward 5' CTGACCAACTGACTGAAGAGCAGA 3'
Reverse 5' TTCTGTGGGATTCTGCCCAAGAG 3'
Cloning strategy of HA HuR
Hemagglutinin (HA) tagged human HuR was cloned into the NheI and XhoI sites of the pZeoSV2 (-) vector (Invitrogen). The plasmids were sequenced in both directions to confirm identity. Cells were transfected with either pZeo HA HuR or pZeo empty vector using Lipofectamine 2000 (Invitrogen). After five days transfected media was removed and replaced with fresh medium containing 200 μg/ml of Zeocin antibiotic (Invitrogen). Cells were selected for a ten day period. After ten days, the selected cells were maintained in 50 μg/ml of Zeocin to maintain pZeo HA HuR and empty vector expression. No viable cells remained in the untransfected well. Cells were then cloned by limiting dilution.
Lentiviral RNAi HuR knock-down
In order to knockdown HuR, PSICOOLIGOMAKER v1.5 http://web.mit.edu/ccr/labs/jacks/was used to identify optimal shRNAs sequences to HuR. We tested multiple sequences and chose GGATCCTCTGGCAGATGT, identified and designated shRNA H760. Annealed sense and antisense DNA (Integrated DNA Technologies, Inc, IDT, Coralville, IA), along with stem loops to create hairpin, were cloned into the HpaI and XhoI restriction sites in the Lentilox pll3.7 vector (ATCC). After sequence verification, lentivirus was packaged in 293FT cells using ViraPower™ Lentiviral Expression Systems (Invitrogen) following manufacturer's protocol. Both MB-231 and MCF-7 cells were seeded at a density of 100,000 cells in 100 mm tissue culture plates with 10 ml of media. The following day lentivirus, expressing either GFP and no shRNA (empty lentilox control) or GFP and HuR shRNA H760, was added at a multiplicity of infection (MOI) of 10 along with polybrene (8 μg/ml) (Sigma-Aldrich Corp, St. Louis, MO). After five days, cells were harvested by trypsinization and sorted for GFP expression using BD FACSDiva (BD Bioscience). Cells were cloned by limiting dilution and GFP expression was assessed using FACScan (BD Bioscience) and CellQuest software (BD Bioscience). GFP expression was >98% and indicated homogenous cell population.
SDS-PAGE and Western Blot Analysis
Western analysis was performed as described previously with slight modifications [47]. Briefly, cells were harvested and lysed in triple-detergent RIPA buffer with protease inhibitor cocktail (Roche, Pleasanton, CA). For nuclear and cytoplasmic fractionation, the NE-PER kit was used (Pierce, Rockford, IL). Protein quantity was determined by Bradford Assay. Forty μg of protein was electrophoresed on a 12% SDS-polyacrylamide gel and transferred to a nitrocellulose membrane. The membrane was blocked with 5% nonfat milk at room temperature for 1 hr and incubated with anti-β-tubulin (1 μg/ml, Sigma-Aldrich) at 4°C overnight. After washing, the membrane was incubated with monoclonal anti-HuR clone 3A2 antibody (1 μg/ml) at room temperature for 1 hr, or anti-CD9 antibody (1:100) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) at 4°C overnight. The secondary antibody used was sheep anti-mouse Ig horse radish peroxidase (1:4000) (GE Healthcare, Piscataway, NJ), incubated at room temperature for 1 hr. Specific proteins were detected using chemiluminescence (GE Healthcare). HuR knock-down was determined to be >90% using Bio-Rad's Quantity One software (Bio-Rad) normalizing to β-tubulin, and HuR over-expression was quantitated in a similar manner.
Biotin Pull-down
Biotinylated transcripts were synthesized using cDNA that was prepared from MB-231 cells. Templates were prepared using forward primers that contained the T7 RNA polymerase promoter sequence (CCAAGCTTCTAATACGACTCACTATAGGGAGA [T7]). Primers used for the preparation of biotinylated transcripts spanning the CD9 CR, and 3'UTR (NM_001769) and CALM2 CR and 3'UTR (NM_001743.3) were as follows:
CD9 CR 118-560: [T7] TCAAAGGAGGCACCAAGTGCAT and AACGCATAGTGGATGGCTTTCA
CD93'UTR798-1231: [T7] AGTCAGCTTACATCCCTGAGCA and GACATTGTCATAATTTTTTATTATGTATC
CALM2 CR 72-515: [T7] GCTGACCAACTGACTGAAGA and CTTTGCTGTCATCATTTGTACAAA
CALM2 3'UTR 518-1128: [T7] AGACCTTGTACAGAATGTGTTAA and GGGTAAATTGTAATTTTTTTATTGGAA
GAPDH 3'UTR: [T7] CCTCAACGACCACTTTGTCA and GGTTGAGCACAGGG TACTTTATT
The PCR-amplified fragments were purified and used as templates for in vitro synthesis of the corresponding biotinylated RNAs by MAXIscript kit (Ambion®, Applied Biosystems). Biotin pull-down assays were performed by incubating 40 μg of MB-231 cell lysates with equimolar of biotinylated transcripts for 1 hr at room temperature. The complexes were isolated using paramagnetic streptavidin-conjugated Dynabeads (Dynal®, Invitrogen), and bound proteins in the pull-down material were analyzed by Western blotting using an antibody recognizing HuR (Santa Cruz). After secondary-antibody incubations, the signals were visualized by chemiluminescence (Amersham Biosciences, GE Healthcare).
Statistical Analysis of Microarray Data
Analysis of microarray gene expression data was primarily performed using the Linear Models for Microarray Data (limma) package [49] and the lumi package [50], available through the Bioconductor project [51] for use with R statistical software [52]. After data pre-processing was completed (Appendix), the statistical analysis was performed using moderated t-statistics applied to the log-transformed (base 2) normalized intensity for each gene using an Empirical Bayes approach [53]. Three contrasts of interest were computed and tested. The first was the difference between HuR pull-down and IgG background for the MB-231 cell line. Genes which exhibited significantly greater expression in the pull-down were considered to be in the HuR pellet for the MB-231 cell line. The second contrast was similar to the first, but for the MCF-7 cell line. The third and most important contrast was the difference between the first and second contrast, and can be viewed as a test of statistical interaction between HuR and cell line. For a given gene, this term can be interpreted as reflection of the synergistic relationship between HuR and estrogen in breast cancer. Adjustment for multiple testing was made using the false discovery rate (FDR) method of Benjamini and Hochberg [54] with an FDR of 10% as our cutoff for declaring significance. To facilitate interpretation, log fold changes were transformed back to fold change on the data scale (fluorescent intensity).
Gene ontology (GO) analyses were carried out on the list of significant genes based on the third contrast described above. The purpose of the analyses was to test the association between Gene Ontology Consortium categories [55] and differentially expressed HuR pellet genes between MB-231 and MCF-7. Using our defined gene universe (Appendix), GOstats [56] was used to carry out conditional hypergeometric tests. These tests exploit the hierarchical nature of the relationships among the GO terms for conditioning [57]. We carried out GO analyses for over-representation of biological process (BP), molecular function (MF), and cellular component (CC) ontologies, and computed the nominal hypergeometric probability for each GO category. These results were used to assess whether the number of selected genes associated with a given term was larger than expected under the null hypothesis, and a p-value cutoff of 0.01 was used. GO categories containing less than 10 genes from our gene universe were not considered to be reliable indicators, and are not reported.
Results
HuR immunoprecipitation from ER+ and ER- breast cancer cell lines
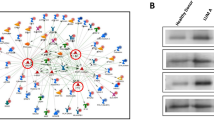
We first determined HuR protein expression levels in breast cancer cell lines. HuR is expressed in both the ER- and the ER+ cell lines, MB-231 and MCF-7, respectively (Figure 1A). RNA immunoprecipitation, using HuR monoclonal antibody 3A2, recovered HuR (Figure 1A) and revealed, by quantitative RT-PCR, a significant enrichment of up to fifteen fold for a known HuR target, β-ACTIN mRNA, as compared to isotype control (IgG1) and normalized to a non-target, GAPDH mRNA (Figure 1B). These data showed that HuR RIP specifically immunoprecipitate HuR protein and associated mRNAs, though absolute quantitative conclusions cannot be drawn since different amounts of lysates were used and efficiency of immunoprecipitation from different cell lines may differ.

Immunoprecipitation and RIP in MB-231 and MCF-7 breast cancer cells. Immunoprecipitations were performed from MB-231 or MCF-7 cell lysates using anti-HuR monoclonal antibody (3A2) and IgG1 isotype control. A. IP Western of HuR revealed expected size band as detected by 3A2. Panel on right reveals amounts of HuR in lysates used from both cell lines. B. Verification by quantitative RT-PCR showed fifteen and eleven fold enrichments of B-ACTIN, a known HuR target, in the 3A2 IPs from MB231 and MCF-7, respectively. All ΔΔCT values were normalized to GAPDH. Experiments were done in duplicate (n = 2).
RIP-Chip from ER+ and ER- breast cancer cell lines identifies unique sets of associated mRNAs
RIP-Chip was performed on cytoplasmic lysates from both breast cancer cell lines with HuR antibody and isotype control in order to determine HuR associated mRNAs. Each immunoprecipitation was done at least three independent times with matching controls. Signals from isotype control were subtracted out. Recovered mRNA was amplified and hybridized to Illumina Sentrix Human arrays consisting of 47,000 genes. Figure 2 represents a composite array generated by combining hybridizations to twelve different arrays (log2 scale). Three groups of HuR-associated target genes were identified: MB-231 targets in the left upper quadrant; both MB-231 and MCF-7 targets in the right upper quadrant; MCF-7 targets in the right lower quadrant. As expected, most of the mRNAs did not associate with HuR and were located in the lower left quadrant. There were 395 and 64 annotated genes, at least 2 fold or more enriched, associated with either MB-231 or MCF-7 cells, respectively, and 182 genes associated with both cell lines. A complete list can be found in Additional File 1, Figure S2. The raw data files are available in the NCBI database at the following link: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=pdsnrqmiawukqlm&acc=GSE17820, NCBI Accession number GSE17820). These genes generally fell into three groups. Group 1 consisted of cancer-associated genes which were known HuR targets, such as PTMA mRNA. Group 2 consisted of genes which played a role in cancer but were not known to be HuR targets. Group 3 consisted of genes with an unknown function in cancer, but which may be regulated by HuR. These data revealed that HuR was associated with distinct subsets of mRNAs in ER+ and ER- breast cancer cells.

HuR RIP-CHIP identifies distinct genetic profiles in ER+ and ER- breast cancer cells. HuR immunoprecipitations were performed from MB-231 or MCF-7 cell lysates using HuR antibody and IgG1 isotype control hybridized to Illumina Sentrix arrays (47,000 genes). Control signals were subtracted. Results represent cumulative data from 12 different arrays. Experiments were done in triplicate (n = 3) for each cell line with matching controls. Scales are log2.
Gene Ontology (GO) analyses of differentially expressed significant genes between ER+ and ER- cells were categorized into Biological Process (BP), Cellular Component (CC), and Molecular Function (MF). GO analyses allow for the identification of gene families that may play significant roles related to these categories in expression profiles. Most of the differentially expressed genes (155) were found to be more abundant than expected in 14 BP categories (Figure 3A). Three MF categories consisted of 100 genes with most of these (83) related to protein binding and transcription activator activity. The CC categories contained the least (34) and were primarily associated with the Golgi apparatus. For the complete GO analyses see Additional File 2, Table S1. In Table S1 we list the top HuR associated mRNAs in the different categories which were approximately 5 fold enriched or greater. As can be seen in Figure 3B, a partial listing of some of these genes (in bold) are candidate members to multiple areas of cancer control, as suggested by Hanahan and Weinberg. We note that though β-ACTIN mRNA was amongst the most abundant of HuR-associated mRNAs in MCF-7 cells, β-ACTIN mRNA levels were only 3.93-fold higher in HuR IP compared to IgG IP in MB-231 cells. Therefore, since this was less than the 5-fold cut-off we employed for Table S1, it is not listed. Thus, these results may have identified novel HuR-controlled genes which may play roles in breast carcinogenesis in a cancer subtype-specific fashion.

GO Classification of genes found by RIP CHIP of potential HuR targets and their relationship to the Acquired Capabilities of Cancer Model. A. Differentially expressed genes which are more represented in the Biological Processes (BP) GO category than expected. B. Original representation showing subsets of transcripts found to be targets of association with HuR (normal type). New transcripts found in this study with RIP-Chip (bold type). Enhanced expression upon binding to HuR influences several of the acquired capabilities of cancer cells described by Hanahan and Weinberg [23, 24].
Validation of HuR targets CD9 and CALM2 by real-time PCR and biotin pull-down analyses
In order to validate HuR binding to genes identified in Figure 2, we chose two known cancer associated genes, CD9 and CALM2, highly expressed in both cell lines. Two independent approaches confirmed the physical interaction between HuR, CD9 and CALM2 mRNAs. Precipitated mRNA from the RIP-Chip experiments were analyzed by quantitative RT-PCR. Both CD9 and CALM2 mRNAs were enriched in the HuR RIP by as much as 160 fold (Figures 4A and 4B), but not the isotype control IP. We further confirmed HuR binding to CD9 and CALM2 mRNAs by biotin pull-downs. The relevant portion of the mRNA was transcribed with biotin tags and incubated with lysates from the two cell lines to probe for interactions with protein. The mixture was then separated by pull-down using streptavidin-coated beads and HuR levels were analyzed by Western blot analysis. As seen in Figure 5, HuR specifically interacts with CD9 and CALM2 mRNAs in the 3'UTR regions, but not within the coding region (CR) or with a control biotinylated RNA corresponding to the 3'UTR of the housekeeping control GAPDH mRNA, which is not a target of HuR.

Validation of target CALM2 and CD9 mRNAs by quantitative RT-PCR. Quantitative RT-PCR using cell lysates, HuR antibody, and IgG1 from RIP-CHIP analysis confirmed results identifying CALM2 mRNA (A) and CD9 mRNA (B) as HuR targets. Change in gene expression is represented as fold increase in HuR immunoprecipitation as compared to IgG1. GAPDH mRNA was used as an endogenous control. Error bars represent SEM. p value is < 0.005. Experiments were done in triplicate (n = 3).

Biotin Pull-down of CD9 and CALM2 . A. Scheme of Coding region (CR) and 3'UTR fragments for biotin pull-down assay. The sequences were obtained from Entrez data base. CR and 3'UTR fragments selected for amplification by PCR are as noted. B. 1% agarose gel electrophoresis showing PCR amplified products of the coding regions and 3'UTR's for CD9 (442 bp and 432 bp, respectively) and CALM2 (443 bp and 610 bp, respectively). C. Biotin pull-down assay using lysates prepared from MB-231 cells. The binding of HuR to biotinylated 3'UTR transcripts from both CD9 and CALM2 mRNAs was specific. HuR did not bind a biotinylated control (GAPDH 3'UTR); and did not bind to biotinylated transcripts spanning the CR of CD9 or CALM2. Experiments were done in duplicate (n = 2).
HuR differentially regulates CD9 and CALM2 in MB-231 and MCF-7 cell lines
To gain insight into the biological effects of these associations, we studied the consequences of stably increasing or decreasing HuR abundance. Individual MB-231 clones which over- and under-express HuR were established by limiting dilution (Figure 6A and 6B). MB-231 cells over-expressed HuR by about 140% (Figure 6A). HuR knock-down using lentiviral shRNA resulted in ~95% reduction in HuR expression (Figure 6B). Surprisingly, over-expression of HuR in MB-231 cells caused decreases in both CD9 protein and mRNA levels (Figures 6C and 6D). HuR knock-down, however, resulted in increases in both CD9 mRNA and protein levels (Figures 6C and 6E). This is the opposite of what we predicted, since HuR is generally regarded as a stabilizer of mRNA. In contrast, over-expression of HuR in MB-231 cells did not significantly alter the levels of CALM2 mRNA (Figure 6D). Figure 6F depicts a graphical analysis which reveals that HuR over-expression decreases both CD9 mRNA and protein levels, as compared to controls (dashed line set at 100%). Whereas, HuR shRNA knock-down results in increases in both CD9 mRNA and protein levels above control levels.

HuR differentially regulates CD9 and CALM2 in MB-231. A. Epitope HA tagged HuR is over-expressed by 142% and 138% respectively, in stably transfected clones 4E1 and 5F1, as compared to empty vector (EV) control clone 2C7. B. HuR knock-down using lentiviral short hairpin (sh) RNA H760 results in a 94% reduction in steady state levels of protein in clone A7 (LL = lentilox control). C. HuR over-expression results in a 40% reduction in CD9 protein levels as assayed by Western analysis; however, HuR knock-down using lentiviral shRNA results in an increase from 100% to 228% of CD9 levels. D. Over-expression of HuR decreases CD9 mRNA levels but not CALM2 expression. Analysis of steady state CD9 and CALM2 mRNA levels by quantitative RT-PCR reveals significant decreases in CD9 mRNA levels, whereas CALM2 levels are unaffected. Although CALM2 expression appears greater, the change is not significant. E. Knocking down HuR levels by shRNA in MB-231 cells shows significant increases in CD9 and CALM2 mRNA levels by quantitative RT-PCR. Decreased levels of HuR mRNA validate HuR shRNA knock-down. F. Graph showing the effects of HuR on the expression of CD9 mRNA. HuR over-expression results in decreases in both mRNA and protein levels, though the decreases are greater in RNA. Whereas, HuR knock-down by shRNA results in significant increases at both the mRNA and protein levels, with greater change at transcript levels. The dashed line represents levels in control cells. Error bars represent SEM. p value is < 0.005; N.S. = not statistically significant; * = statistically significant. All experiments were done in triplicate (n = 3).
We performed similar analyses with MCF-7 cells, though the over-expression levels of HA HuR were only approximately 10%, since this was a pooled population (we have been unable to obtain MCF-7 clones which over-express HuR). In contrast, we generated MCF-7 clones with reduced HuR levels (93%) using lentiviral shRNA (Figure 7B). Western blot analysis of MCF-7 cells with over-expression of HuR reveals modest increases in CD9 protein levels (Figure 7C). There are also modest decreases in CD9 protein expression in MCF-7 with reduced HuR levels (Figure 7C). mRNA levels of CD9 and CALM2 are essentially unchanged in MCF-7 cells which over-express HuR (Figure 7D). As expected, HuR knock-down in MCF-7 cells using lentiviral shRNA resulted in significant reductions in both CD9 and CALM2 mRNA levels (Figure 7E). The right panel in Figure 7E indicates efficiency of HuR mRNA knock-down which is consistent with the protein data (Figure 7B). These results are summarized in Figure 7F. There are no significant changes seen in CD9 mRNA and CD9 protein for HuR over-expression. There is a more pronounced knock-down, however, in CD9 mRNA in MCF-7 cells with reduced HuR levels.

Effects of over-expressing or reducing HuR on CD9 and CALM2 expression in MCF-7 cells. A. Western analysis of HuR over-expression in heterogenous population of cells reveals approximately 10% over-expression. B. Lentiviral HuR shRNA efficiently knocks down HuR protein by over 90%. C. HuR over- expression and under-expression results in small changes in CD9 protein levels in MCF-7 cells. D. Levels of both CD9 and CALM2 mRNAs are unchanged in cells which over-express HuR; whereas lentiviral knock-down of HuR in MCF-7 cells results in decreases in steady-state mRNA levels (E). The graph in (F) shows minimal changes in CD9 mRNA and protein levels in HuR over-expressing MCF-7 cells. The CD9 mRNA levels, however, are more affected in HuR knock-down. P value is < 0.005; N.S. = not statistically significant; * = statistically significant.
The results of HuR shRNA knock-down in MCF-7 cells were as expected, but opposite of those seen for MB-231 cells. Steady-state mRNA levels of CD9 and CALM2 mRNAs decreased, consistent with the hypothesis that HuR generally stabilizes its mRNA targets. One possible explanation of these disparate results is different levels of total cellular or cytoplasmic HuR. We performed nuclear and cytoplasmic fractionation (Additional File 3, Figure S3). These results demonstrated modest (approximately 10%) greater cytoplasmic levels of HuR in MB-231 cells as compared to MCF-7. The total cellular HuR levels are very similar for both MB-231 and MCF-7 cells. Taken together, these results indicated that HuR appeared to differentially regulate the same mRNAs in a manner dependent upon the cellular milieu.
Discussion
We utilized RIP-Chip technologies to define differentially regulated HuR genes in ER+ and ER- breast cancer. To our knowledge, this is the first report of a side-by-side genome-wide comparison of HuR-associated targets in wild type ER+ and ER- breast cancer cells. Our findings indicated that HuR interacts with small subsets of genes in breast cancer, out of the possible 8% of human genes possessing AREs which are potential HuR targets. Three broad categories of HuR targets were identified. First, there was a subset of targets only found in ER+ breast cancer. Second, there was a unique subset of HuR targets found only in ER- breast cancer. A third subset consisted of HuR-associated mRNAs common to both forms of breast cancer, many of which were previously described as having roles in cancer.
We selected and validated two HuR targets, CD9 and CALM2 mRNAs, which were found in high abundance in both types of breast cancer. Initially, we employed the previously developed "heat map" signature of HuR binding to gain insight into putative HuR target sequences [30]. HuR binding was verified by HuR immunoprecipitations, and analyzed by quantitative RT-PCR and biotin pull-downs. Both CD9 and CALM2 mRNAs were enriched in HuR RIPs compared to isotype control IP reactions. Biotin pull-downs verified the binding of HuR protein specifically to the 3'UTR regions of both mRNAs, as had been predicted. CD9 is a tetraspanin molecule which plays important roles in cellular development, activation, growth and motility. It has been implicated in a variety of cancers, including but not limited to gastric cancers and B cell acute leukemia [58–60].
The role of CALM2 in cancer is less well understood but may be linked to cancer since it is involved in controlling calcium signaling [61, 62]. There are three CALMODULIN genes (CALM1, CALM2 and CALM3) highly expressed in both MB-231 and MCF-7 cell lines (Additional File 4, Figure S1). Interestingly, although they are encoded by different genes at different chromosomal locations, all three encode the same open reading frame but differ in the 5' and 3' UTRs [[61, 63], and [64]]. Only CALM2 mRNA interacts with HuR by RIP analysis. Moreover, previously published reports have indicated the necessity of knocking down all three CALMODULIN mRNAs by siRNA to achieve knock-down of the protein [61]. We conclude that there may be differential HuR associated regulation of these CALMODULIN genes in breast cancer, even though the mechanism needs to be further delineated.
Surprisingly, the regulation of both CD9 and CALM2 target genes was dependent upon the cellular milieu. To test the functional consequences of HuR binding to these two transcripts, we prepared cells that stably expressed higher or lower HuR, compared to the parent cells, in both ER+ and ER- breast cancer cell lines. HuR appears to differentially regulate the expression of CD9 in opposite directions in the two different forms of breast cancer. Specifically, HuR over-expression in ER- breast cancer (MB-231) paradoxically decreased CD9 mRNA and protein levels, whereas HuR knock-down increased the CD9 mRNA levels. This is the opposite of what is predicted for most HuR targets, since HuR is thought to stabilize its mRNA targets and often increases their translation. There did not seem to be similar effects upon CALM2 expression. As expected, knock-down of HuR by shRNA decreased expression of CD9 and CALM2 in ER+ breast cancer (MCF-7). Though there are differences in cytoplasmic HuR levels in MB-231 cells as compared with MCF-7, these are modest (10%). This is in keeping, however, with observations that MB-231 cells are more undifferentiated and more aggressive.
Moreover, analysis of HuR-associated mRNAs in both ER+ and ER- breast cancer revealed three broad categories of genes. First, there were well known cancer genes, such as PTMA, which are regulated by HuR [27]. Second, there were cancer-related genes, such as CD9 and CALMODULIN, which were not known to be HuR regulated until this report. Third, there were other genes identified by HuR association with unknown cancer function. These could potentially represent novel cancer targets. Additional proof of HuR involvement with other known cancer genes, such as CD44 and GATA-3, may represent novel insights into the mechanisms of regulation of these cancer targets (see Additional Files). These results may therefore advance the field by shedding insights into posttranscriptional regulation of known and perhaps unknown cancer target genes.
Though the exact mechanisms of HuR differential regulation of CD9 and CALM2 are presently unclear, it may involve microRNA (miRNA) regulation. In a recent report, we described the recruitment by HuR of miRNA let-7 to translationally silence C-MYC expression [46]. It is clear from the findings of laboratories headed by Filipowicz, Steitz and other investigators, that RBPs and miRNAs are involved in intricate associations to affect downstream translational suppression or activation of target mRNAs to help meet cellular needs [65, 66]. Sharp and colleagues proposed that different interactions between RBPs and miRNAs may have evolved as a protective mechanism for the cell against environmental stress [67].
A remaining question is why HuR selectively binds to certain genes containing AREs. Our previous work has demonstrated the role that HuR plays in myogenesis by stabilizing the expression of three critical genes involved in myogenesis: MYOD, MYOGENIN, and p21 cip1 [68]. HuR over-expression results in precocious muscle differentiation and HuR siRNA knock-down prevents muscle differentiation [69]. It is highly probable that there are more than three HuR targets inside these cells. A specific phenotype potentially arises when HuR levels are altered which may involve interactions with miRNAs, although this theory needs to be fully investigated.
Our findings share some similarity to earlier reports of HuR RIP-Chip analysis of MCF-7 cells stably transfected with MCT-1 [45]. These analyses, however, were not genome-wide and employed transfected cells. Nevertheless, thrombospondin, a known important anti-angiogenic factor, was identified as a HuR-regulated target. Combined with earlier reports of the role of HuR in regulating, VEGF-α and HIF1α, HuR may be controlling a "posttranscriptional mini-operon" involved in angiogenesis [29, 32, 70]. Further studies are being conducted in our laboratory to investigate the role of HuR in breast cancer angiogenesis using xenograft animal models. It will be particularly important to test the role of HuR upon CD9 and CALM2 expression in breast tumors in vivo.
Posttranscriptional gene regulation is increasingly being appreciated as a driver of malignant transformation. The roles of both RBPs and miRNAs (so-called oncomirs) are being recognized in cancer [71]. Many reports have described alterations in miRNA expression profile and function as contributing to breast cancer malignant transformation and metastasis [72–75]. HuR RIP-Chip analysis may shed further light into malignant breast cancer transformation by identifying HuR associated mRNAs.
We believe that there are potential applications for tamoxifen resistance as well. Keen and colleagues have described a potential mechanistic link between HuR expression and tamoxifen drug resistance [76]. As breast cancer cells acquire tamoxifen resistance, there are increased levels of cytoplasmic HuR expression. Increased cytoplasmic HuR levels have previously been described in situations where HuR actively influences expression of cytoplasmic targets [18, 47, 48]. Drug resistance could be reversed by using siRNA to knock-down HuR expression, whereas exogenous over-expression of HuR could cause cells to become resistant to tamoxifen. We therefore propose that HuR may be coordinately regulating genes which may allow a cell to acquire tamoxifen resistance. It will be interesting to further investigate HuR-associated target genes in ER+ cells in this light.
Conclusion
In summary, using RIP-Chip analysis, we have performed for the first time a genome-wide comparison of HuR-associated targets in wild type ER+ and ER- breast cancer. We have identified novel HuR targets and have gained insight into the role HuR plays in regulating known cancer genes. We found distinct, differentially expressed subsets of HuR cancer related genes in ER+ and ER- breast cancer cell lines. Based on our observations, the enhanced expression of these mRNA subsets by HuR can influence many of the acquired capabilities of cancer cells. Further investigation into HuR's role in regulating these genes may provide novel insights into breast cancer diagnosis and therapy.
Appendix
Microarray Data Preprocessing
Data quality was examined by looking at quality controls metrics produced by Illumina's software (BeadStudio v3.1.3.0, Gene Expression Module 3.2.7). The data were then exported for further analyses in R. Image plots of each array were examined for spatial artifacts, and there was no evidence of systematic effects indicative of technical problems with the arrays. Within limma, quantile normalization was used for between chip normalization. Finally, quality control statistics were computed using a variety of Illumina's internal control probes that are replicated on each array. Any probes which were considered "not detectable" across all samples were excluded from further statistical analyses in order to reduce false positives. The determination of "not detectable" was based upon the BeadStudio computed detection p-value being greater than 1%.
Gene Ontology Gene Universe
In defining the gene universe for the analysis, non-specific filtering was used to increase statistical power without biasing the results. We started with all probes on the Illumina array which had both an Entrez gene identifier [77] and a GO annotation, as provided in the lumiHumanAll.db [78] annotation data package and GO.db [79] annotation maps (built using data obtained from NCBI on 4/2/08). This set was then reduced by excluding probes that exhibited little variability (interquartile range (IQR) of <0.1 on log2 scale) across all samples because such probes are generally not informative. Finally, for probes that mapped to the same Entrez identifier, a single probe was chosen in order to insure a surjective map from probe IDs to GO categories (via Entrez identifiers). This was necessary to avoid redundantly counting GO categories which produces false positives. Probes with the largest IQR were chosen to be associated with an Entrez identifier.
Abbreviations
- (ER-):
-
Estrogen receptor negative
- (ER+):
-
estrogen receptor positive
- (RIP):
-
RNA immunoprecipitation
- (RIP-Chip):
-
RNA immunoprecipitation applied to microarrays
- (3' UTR):
-
3' untranslated region
- ELAV1 (embryonic lethal abnormal vision 1 :
-
).
References
Dietel M, Sers C: Personalized medicine and development of targeted therapies: The upcoming challenge for diagnostic molecular pathology. A review. Virchows Arch. 2006, 448 (6): 744-755. 10.1007/s00428-006-0189-2.
Mischel PS, Cloughesy TF, Nelson SF: DNA-microarray analysis of brain cancer: molecular classification for therapy. Nat Rev Neurosci. 2004, 5 (10): 782-792. 10.1038/nrn1518.
Muss HB: Targeted therapy for metastatic breast cancer. N Engl J Med. 2006, 355 (26): 2783-2785. 10.1056/NEJMe068260.
Gygi SP, Rochon Y, Franza BR, Aebersold R: Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999, 19 (3): 1720-1730.
Ideker T, Thorsson V, Ranish JA, Christmas R, Buhler J, Eng JK, Bumgarner R, Goodlett DR, Aebersold R, Hood L: Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science. 2001, 292 (5518): 929-934. 10.1126/science.292.5518.929.
Khabar KS, Bakheet T, Williams BR: AU-rich transient response transcripts in the human genome: expressed sequence tag clustering and gene discovery approach. Genomics. 2005, 85 (2): 165-175. 10.1016/j.ygeno.2004.10.004.
Khabar KS: The AU-rich transcriptome: more than interferons and cytokines, and its role in disease. J Interferon Cytokine Res. 2005, 25 (1): 1-10. 10.1089/jir.2005.25.1.
Intine RV, Tenenbaum SA, Sakulich AL, Keene JD, Maraia RJ: Differential phosphorylation and subcellular localization of La RNPs associated with precursor tRNAs and translation-related mRNAs. Mol Cell. 2003, 12 (5): 1301-1307. 10.1016/S1097-2765(03)00429-5.
Tenenbaum SA, Carson CC, Lager PJ, Keene JD: Identifying mRNA subsets in messenger ribonucleoprotein complexes by using cDNA arrays. Proc Natl Acad Sci USA. 2000, 97 (26): 14085-14090. 10.1073/pnas.97.26.14085.
Tenenbaum SA, Lager PJ, Carson CC, Keene JD: Ribonomics: identifying mRNA subsets in mRNP complexes using antibodies to RNA-binding proteins and genomic arrays. Methods. 2002, 26 (2): 191-198. 10.1016/S1046-2023(02)00022-1.
Keene JD: Organizing mRNA export. Nat Genet. 2003, 33 (2): 111-112. 10.1038/ng0203-111.
Keene JD, Tenenbaum SA: Eukaryotic mRNPs may represent posttranscriptional operons. Mol Cell. 2002, 9 (6): 1161-1167. 10.1016/S1097-2765(02)00559-2.
Gerber AP, Herschlag D, Brown PO: Extensive association of functionally and cytotopically related mRNAs with Puf family RNA-binding proteins in yeast. PLoS Biol. 2004, 2 (3): E79-10.1371/journal.pbio.0020079.
Grigull J, Mnaimneh S, Pootoolal J, Robinson MD, Hughes TR: Genome-wide analysis of mRNA stability using transcription inhibitors and microarrays reveals posttranscriptional control of ribosome biogenesis factors. Mol Cell Biol. 2004, 24 (12): 5534-5547. 10.1128/MCB.24.12.5534-5547.2004.
Hieronymus H, Silver PA: Genome-wide analysis of RNA-protein interactions illustrates specificity of the mRNA export machinery. Nat Genet. 2003, 33 (2): 155-161. 10.1038/ng1080.
Hieronymus H, Yu MC, Silver PA: Genome-wide mRNA surveillance is coupled to mRNA export. Genes Dev. 2004, 18 (21): 2652-2662. 10.1101/gad.1241204.
Rajasekhar VK, Holland EC: Postgenomic global analysis of translational control induced by oncogenic signaling. Oncogene. 2004, 23 (18): 3248-3264. 10.1038/sj.onc.1207546.
Atasoy U, Watson J, Patel D, Keene JD: ELAV protein HuA (HuR) can redistribute between nucleus and cytoplasm and is upregulated during serum stimulation and T cell activation. J Cell Sci. 1998, 111 (Pt 21): 3145-3156.
Fan XC, Steitz JA: Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. Embo J. 1998, 17 (12): 3448-3460. 10.1093/emboj/17.12.3448.
Ma WJ, Cheng S, Campbell C, Wright A, Furneaux H: Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. J Biol Chem. 1996, 271 (14): 8144-8151. 10.1074/jbc.271.14.7942.
Meisner NC, Hackermuller J, Uhl V, Aszodi A, Jaritz M, Auer M: mRNA openers and closers: modulating AU-rich element-controlled mRNA stability by a molecular switch in mRNA secondary structure. Chembiochem. 2004, 5 (10): 1432-1447. 10.1002/cbic.200400219.
Brennan CM, Steitz JA: HuR and mRNA stability. Cell Mol Life Sci. 2001, 58 (2): 266-277. 10.1007/PL00000854.
Hanahan D, Weinberg RA: The hallmarks of cancer. Cell. 2000, 100 (1): 57-70. 10.1016/S0092-8674(00)81683-9.
Lopez de Silanes I, Lal A, Gorospe M: HuR: post-transcriptional paths to malignancy. RNA Biol. 2005, 2 (1): 11-13.
Abdelmohsen K, Lal A, Kim HH, Gorospe M: Posttranscriptional orchestration of an anti-apoptotic program by HuR. Cell Cycle. 2007, 6 (11): 1288-1292.
Abdelmohsen K, Pullmann R, Lal A, Kim HH, Galban S, Yang X, Blethrow JD, Walker M, Shubert J, Gillespie DA, Furneaux H, Gorospe M: Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol Cell. 2007, 25 (4): 543-557. 10.1016/j.molcel.2007.01.011.
Lal A, Kawai T, Yang X, Mazan-Mamczarz K, Gorospe M: Antiapoptotic function of RNA-binding protein HuR effected through prothymosin alpha. Embo J. 2005, 24 (10): 1852-1862. 10.1038/sj.emboj.7600661.
Lal A, Mazan-Mamczarz K, Kawai T, Yang X, Martindale JL, Gorospe M: Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J. 2004, 23 (15): 3092-3102. 10.1038/sj.emboj.7600305.
Levy AP: Hypoxic regulation of VEGF mRNA stability by RNA-binding proteins. Trends Cardiovasc Med. 1998, 8 (6): 246-250. 10.1016/S1050-1738(98)00020-6.
Lopez de Silanes I, Zhan M, Lal A, Yang X, Gorospe M: Identification of a target RNA motif for RNA-binding protein HuR. Proc Natl Acad Sci USA. 2004, 101 (9): 2987-2992. 10.1073/pnas.0306453101.
Nabors LB, Gillespie GY, Harkins L, King PH: HuR, a RNA stability factor, is expressed in malignant brain tumors and binds to adenine- and uridine-rich elements within the 3' untranslated regions of cytokine and angiogenic factor mRNAs. Cancer Res. 2001, 61 (5): 2154-2161.
Sheflin LG, Zou AP, Spaulding SW: Androgens regulate the binding of endogenous HuR to the AU-rich 3'UTRs of HIF-1alpha and EGF mRNA. Biochem Biophys Res Commun. 2004, 322 (2): 644-651. 10.1016/j.bbrc.2004.07.173.
Tran H, Maurer F, Nagamine Y: Stabilization of urokinase and urokinase receptor mRNAs by HuR is linked to its cytoplasmic accumulation induced by activated mitogen-activated protein kinase-activated protein kinase 2. Mol Cell Biol. 2003, 23 (20): 7177-7188. 10.1128/MCB.23.20.7177-7188.2003.
Wang W, Caldwell MC, Lin S, Furneaux H, Gorospe M: HuR regulates cyclin A and cyclin B1 mRNA stability during cell proliferation. EMBO J. 2000, 19 (10): 2340-2350. 10.1093/emboj/19.10.2340.
Wang W, Yang X, Cristofalo VJ, Holbrook NJ, Gorospe M: Loss of HuR is linked to reduced expression of proliferative genes during replicative senescence. Mol Cell Biol. 2001, 21 (17): 5889-5898. 10.1128/MCB.21.17.5889-5898.2001.
Denkert C, Weichert W, Winzer KJ, Muller BM, Noske A, Niesporek S, Kristiansen G, Guski H, Dietel M, Hauptmann S: Expression of the ELAV-like protein HuR is associated with higher tumor grade and increased cyclooxygenase-2 expression in human breast carcinoma. Clin Cancer Res. 2004, 10 (16): 5580-5586. 10.1158/1078-0432.CCR-04-0070.
Heinonen M, Bono P, Narko K, Chang SH, Lundin J, Joensuu H, Furneaux H, Hla T, Haglund C, Ristimaki A: Cytoplasmic HuR expression is a prognostic factor in invasive ductal breast carcinoma. Cancer Res. 2005, 65 (6): 2157-2161. 10.1158/0008-5472.CAN-04-3765.
Heinonen M, Fagerholm R, Aaltonen K, Kilpivaara O, Aittomaki K, Blomqvist C, Heikkila P, Haglund C, Nevanlinna H, Ristimaki A: Prognostic role of HuR in hereditary breast cancer. Clin Cancer Res. 2007, 13 (23): 6959-6963. 10.1158/1078-0432.CCR-07-1432.
Gantt KR, Cherry J, Richardson M, Karschner V, Atasoy U, Pekala PH: The regulation of glucose transporter (GLUT1) expression by the RNA binding protein HuR. J Cell Biochem. 2006, 99 (2): 565-574. 10.1002/jcb.20950.
Guo X, Hartley RS: HuR contributes to cyclin E1 deregulation in MCF-7 breast cancer cells. Cancer Res. 2006, 66 (16): 7948-7956. 10.1158/0008-5472.CAN-05-4362.
Kang SS, Chun YK, Hur MH, Lee HK, Kim YJ, Hong SR, Lee JH, Lee SG, Park YK: Clinical significance of glucose transporter 1 (GLUT1) expression in human breast carcinoma. Jpn J Cancer Res. 2002, 93 (10): 1123-1128.
Pryzbylkowski P, Obajimi O, Keen JC: Trichostatin A and 5 Aza-2' deoxycytidine decrease estrogen receptor mRNA stability in ER positive MCF7 cells through modulation of HuR. Breast Cancer Res Treat. 2008, 111 (1): 15-25. 10.1007/s10549-007-9751-0.
Saunus JM, French JD, Edwards SL, Beveridge DJ, Hatchell EC, Wagner SA, Stein SR, Davidson A, Simpson KJ, Francis GD, Leedman PJ, Brown MA: Posttranscriptional regulation of the breast cancer susceptibility gene BRCA1 by the RNA binding protein HuR. Cancer Res. 2008, 68 (22): 9469-9478. 10.1158/0008-5472.CAN-08-1159.
Suswam EA, Nabors LB, Huang Y, Yang X, King PH: IL-1beta induces stabilization of IL-8 mRNA in malignant breast cancer cells via the 3' untranslated region: Involvement of divergent RNA-binding factors HuR, KSRP and TIAR. Int J Cancer. 2005, 113 (6): 911-919. 10.1002/ijc.20675.
Mazan-Mamczarz K, Hagner PR, Corl S, Srikantan S, Wood WH, Becker KG, Gorospe M, Keene JD, Levenson AS, Gartenhaus RB: Post-transcriptional gene regulation by HuR promotes a more tumorigenic phenotype. Oncogene. 2008, 16;27 (47): 6151-63. 10.1038/onc.2008.215.
Kim HH, Kuwano Y, Srikantan S, Lee EK, Martindale JL, Gorospe M: HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev. 2009, 1;23 (15): 1743-8. 10.1101/gad.1812509.
Atasoy U, Curry SL, Lopez de Silanes I, Shyu AB, Casolaro V, Gorospe M, Stellato C: Regulation of eotaxin gene expression by TNF-alpha and IL-4 through mRNA stabilization: involvement of the RNA-binding protein HuR. J Immunol. 2003, 171 (8): 4369-4378.
Casolaro V, Fang X, Tancowny B, Fan J, Wu F, Srikantan S, Asaki SY, De Fanis U, Huang SK, Gorospe M, Atasoy UX, Stellato C: Posttranscriptional regulation of IL-13 in T cells: role of the RNA-binding protein HuR. The Journal of allergy and clinical immunology. 2008, 121 (4): 853-859. 10.1016/j.jaci.2007.12.1166.
Smyth G: Limma: linear models for microarray data. Bioinformatics and computational Biology Solutions. Edited by: Gentleman RCV, Dudoit S, Irizarry R, Huber W. 2005, New York: Springer, 397-420. full_text.
Du P, Kibbe WA, Lin SM: lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008, 24 (13): 1547-1548. 10.1093/bioinformatics/btn224.
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J: Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004, 5 (10): R80-10.1186/gb-2004-5-10-r80.
Team RDC: R: A language and environment for statistical computing. ISBN 3-900051-07-0. 2006, R Foundation for Statistical Computing Vienna, Austria, [http://www.r-project.org]
Smyth GK: Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004, 3: Article3-
Benjamini Y, Hochberg Y: Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. 1995, Series B 57: 289-300.
Consortium TGO: Gene Ontology: tool for the unification of biology. Nat Genetics. 2000, 25: 25-29. 10.1038/75556.
Falcon S, Gentleman R: Using GOstats to test gene lists for GO term association. Bioinformatics. 2007, 23 (2): 257-258. 10.1093/bioinformatics/btl567.
Alexa A, Rahnenfuhrer J, Lengauer T: Improved scoring of functional groups from gene expression data by decorrelationg GO graph structure. Bioinformatics. 2006, 22: 1600-1607. 10.1093/bioinformatics/btl140.
Lafleur MA, Xu D, Hemler ME: Tetraspanin proteins regulate membrane type-1 matrix metalloproteinase-dependent pericellular proteolysis. Mol Biol Cell. 2009, 20 (7): 2030-2040. 10.1091/mbc.E08-11-1149.
Nakamoto T, Murayama Y, Oritani K, Boucheix C, Rubinstein E, Nishida M, Katsube F, Watabe K, Kiso S, Tsutsui S, Tamura S, Shinomura Y, Hayashi N: A novel therapeutic strategy with anti-CD9 antibody in gastric cancers. J Gastroenterol. 2009, 44 (9): 889-896. 10.1007/s00535-009-0081-3.
Nishida H, Yamazaki H, Yamada T, Iwata S, Dang NH, Inukai T, Sugita K, Ikeda Y, Morimoto C: CD9 correlates with cancer stem cell potentials in human B-acute lymphoblastic leukemia cells. Biochem Biophys Res Commun. 2009, 382 (1): 57-62. 10.1016/j.bbrc.2009.02.123.
Coticchia CM, Revankar CM, Deb TB, Dickson RB, Johnson MD: Calmodulin modulates Akt activity in human breast cancer cell lines. Breast Cancer Res Treat. 2009, 115 (3): 545-560. 10.1007/s10549-008-0097-z.
Schmitt JM, Abell E, Wagner A, Davare MA: ERK activation and cell growth require CaM kinases in MCF-7 breast cancer cells. Mol Cell Biochem. 2009, 335 (1-2): 155-71. 10.1007/s11010-009-0252-9.
Berchtold MW, Egli R, Rhyner JA, Hameister H, Strehler EE: Localization of the human bona fide calmodulin genes CALM1, CALM2, and CALM3 to chromosomes 14q24-q31, 2p21.1-p21.3, and 19q13.2-q13.3. Genomics. 1993, 16 (2): 461-465. 10.1006/geno.1993.1211.
Fischer R, Koller M, Flura M, Mathews S, Strehler-Page MA, Krebs J, Penniston JT, Carafoli E, Strehler EE: Multiple divergent mRNAs code for a single human calmodulin. J Biol Chem. 1988, 263 (32): 17055-17062.
Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W: Relief of microRNA-Mediated Translational Repression in Human Cells Subjected to Stress. Cell. 2006, 125 (6): 1111-1124. 10.1016/j.cell.2006.04.031.
Vasudevan S, Steitz JA: AU-rich-element-mediated upregulation of translation by FXR1 and Argonaute 2. Cell. 2007, 128 (6): 1105-1118. 10.1016/j.cell.2007.01.038.
Leung AK, Sharp PA: microRNAs: a safeguard against turmoil?. Cell. 2007, 130 (4): 581-585. 10.1016/j.cell.2007.08.010.
Figueroa A, Cuadrado A, Fan J, Atasoy U, Muscat GE, Munoz-Canoves P, Gorospe M, Munoz A: Role of HuR in skeletal myogenesis through coordinate regulation of muscle differentiation genes. Mol Cell Biol. 2003, 23 (14): 4991-5004. 10.1128/MCB.23.14.4991-5004.2003.
Giessen van der K, Di-Marco S, Clair E, Gallouzi IE: RNAi-mediated HuR depletion leads to the inhibition of muscle cell differentiation. J Biol Chem. 2003, 278 (47): 47119-47128. 10.1074/jbc.M308889200.
Galban S, Kuwano Y, Pullmann R, Martindale JL, Kim HH, Lal A, Abdelmohsen K, Yang X, Dang Y, Liu JO, Lewis SM, Holcik M, Gorospe M: RNA-binding proteins HuR and PTB promote the translation of hypoxia-inducible factor 1alpha. Mol Cell Biol. 2008, 28 (1): 93-107. 10.1128/MCB.00973-07.
Esquela-Kerscher A, Slack FJ: Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006, 6 (4): 259-269. 10.1038/nrc1840.
Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Ménard S, Palazzo JP, Rosenberg A, Musiani P, Volinia S, Nenci I, Calin GA, Querzoli P, Negrini M, Croce CM: MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005, 65 (16): 7065-7070. 10.1158/0008-5472.CAN-05-1783.
Ma L, Teruya-Feldstein J, Weinberg RA: Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007, 449 (7163): 682-688. 10.1038/nature06174.
Ma L, Weinberg RA: Micromanagers of malignancy: role of microRNAs in regulating metastasis. Trends Genet. 2008, 24 (9): 448-456. 10.1016/j.tig.2008.06.004.
Tavazoie SF, Alarcon C, Oskarsson T, Padua D, Wang Q, Bos PD, Gerald WL, Massague J: Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008, 451 (7175): 147-152. 10.1038/nature06487.
Hostetter C, Licata LA, Witkiewicz A, Costantino CL, Yeo CJ, Brody JR, Keen JC: Cytoplasmic accumulation of the RNA binding protein HuR is central to tamoxifen resistance in estrogen receptor positive breast cancer cells. Cancer Biol Ther. 2008, 7 (9): 1496-506.
Maglott D, Ostell J, Pruitt KD, Tatusova T: Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2005, D54-58. 33 Database
Du P: lumiHumanAll.db: Illumina Human Expression BeadChips (include al versions: from version 1 to 3) annotation data. 1.2.0 Rpv.
Carlson M, Falcon S, Pages H, Li N: GO.db: A set of annotation maps describing the entire Gene Ontology. 2.2.0 Rpv.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/10/126/prepub
Acknowledgements
YK and MG were supported by the NIA-IRP, NIH. UA and JWD were supported by Department of Defense (Idea Award W81XWH-07-1-0406) as well as start up funds from University of Missouri. We wish to thank Dr. Michael McManus (UCSF, California) for his guidance with the lentiviral system.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
UA supervised the study. RC planned the experiments, performed microarrays and wrote the paper. MMG generated and performed analyses on cell lines as well as quantitative RT-PCR experiments. JWD performed statistical analyses on microarray experiments. JDM assisted in analyses of cell lines. YK and MG planned and performed biotin pull-down experiments. All authors read and approved the final manuscript.
Electronic supplementary material
12885_2009_1925_MOESM1_ESM.XLS
Additional file 1: Figure S2. Table of complete GO analysis. Listing of HuR-associated genes with odds ratios and functional categories. (XLS 54 KB)
12885_2009_1925_MOESM2_ESM.XLS
Additional file 2: Table S1. HuR targets five fold or greater. Listing of HuR-associated mRNAs in MB-231 and MCF-7 cell lines. (XLS 26 KB)
12885_2009_1925_MOESM3_ESM.PPT
Additional file 3: Figure S3. Total cellular levels of HuR are similar in MB-231 and MCF-7 cells. Nuclear and cytoplasmic separation was performed to measure levels of HuR in different compartments of MB-231 and MCF-7 cells. Total cellular HuR levels were very similar, whereas there was a small (10%) increase in HuR cytoplasmic levels in MB-231 cells as compared to MCF-7. Absence of β-tubulin staining demonstrates integrity of isolation as there should not be β-tubulin in the nuclear fraction. Bands were measured by densitometry and normalized to β-tubulin controls. (T = total cellular lysate; C = cytoplasmic lysate, N = nuclear lysate). (PPT 118 KB)
12885_2009_1925_MOESM4_ESM.PPT
Additional file 4: Figure S1. Relative baseline values of CALM1 , CALM2 , CALM3 , and CD-9 mRNAs in ER+ and ER- cells. Quantitative RT-PCR performed on mRNA extracted from cell lysates showing relative levels of CALM1, CALM2, CALM3, and CD-9 mRNAs in MB-231 and MCF-7 breast cancer cells. All values were normalized to GAPDH mRNA. All experiments were done in triplicate (n = 3) except for CALM3 (n = 2). (PPT 101 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Calaluce, R., Gubin, M.M., Davis, J.W. et al. The RNA binding protein HuR differentially regulates unique subsets of mRNAs in estrogen receptor negative and estrogen receptor positive breast cancer. BMC Cancer 10, 126 (2010). https://doi.org/10.1186/1471-2407-10-126
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-10-126