
Abstract
Background
Fibroblast growth factor 23 (FGF23) is an important hormone in the regulation of phosphate metabolism. It is unclear whether FGF23 is associated with carotid artery calcification (CAAC) in predialysis patients. The present study aimed to clarify the relationship between FGF23 and CAAC in patients with chronic kidney disease (CKD) who were not on dialysis.
Methods
One-hundred ninety-five predialysis CKD patients were enrolled in this cross-sectional study. CAAC was assessed using multidetector computed tomography, and the prevalence of CAAC was examined. Intact FGF23 was measured in each patient. The risk factors for CAAC were evaluated using a logistic regression model.
Results
We found CAAC in 66% of the patients. The prevalence of CAAC significantly increased across CKD stages: it was 37% in CKD stages 1–2, 58% in stage 3; 75% in stage 4, and 77% in stage 5 (p < 0.01). In multivariate analysis, smoking, diabetes mellitus and log FGF23 were each identified as risk factors for CAAC. The study population was divided in quartiles of FGF23 levels. Compared with the lowest FGF23 quartile, each subsequent quartile had a progressively higher odds ratio (OR) for CAAC, adjusted for confounders (ORs [95% confidence interval] of 2.34 [0.78 to 7.31], 5.28 [1.56 to 19.5], and 13.6 [2.92 to 74.6] for the second, third, and fourth quartiles, respectively.
Conclusions
The prevalence of CAAC is increased with the decline in the kidney function. FGF23 is independently related to CAAC in patients with CKD who are not on dialysis.
Similar content being viewed by others


Background
Vascular calcification is highly prevalent in patients with chronic kidney disease (CKD), including end-stage renal disease (ESRD) [1–3]. Arterial calcification in CKD patients occurs at the intimal as well as medial layers of the vasculature. Vascular intimal calcification, as well as vascular medial calcification, is associated with decreased survival in CKD [4]. We previously reported that there is a high prevalence of carotid artery calcification (CAAC) at the initiation of hemodialysis in ESRD patients [3] and that CAAC at the same time is an independent risk factor for cardiovascular events after the initiation of hemodialysis [5]. Therefore, the fact that cardiovascular disease is the most common cause of mortality in patients with CKD, including ESRD, appears to be due in part to the presence of excess vascular calcification.
Fibroblast growth factor 23 (FGF23) is a 251-amino-acid protein secreted by osteocytes in adults [6], and it is an important regulator that maintains serum phosphorus levels within the normal range in patients with CKD by increasing urinary phosphate excretion and decreasing dietary phosphorus absorption through the inhibition of 1,25-dihydroxyvitamin D (1,25(OH)2D) synthesis [7]. In patients with CKD, FGF23 levels are thought to increase as an appropriate compensatory mechanism to maintain a normal phosphate balance, in parallel with a declining capacity for renal phosphate excretion [8, 9]. Notably, FGF23 is increased early in the course of kidney disease, well before the development of hyperphosphatemia and high FGF23 levels are thus considered to be among the earliest markers of disordered phosphorus metabolism in CKD [8, 10]. The role of FGF23 in mortality and in CKD progression in patients with predialysis and in prevalent dialysis patients has been documented [11–15]; higher FGF23 levels are associated with higher mortality and a more rapid decline in kidney function.
The relationship between FGF23and vascular calcification is controversial. In subjects with normal kidney function, FGF23 is not associated with coronary artery calcification [16], while there is a positive association of FGF23with abdominal aortic calcification in older males [17]. Several studies have attempted to determine the relationship between FGF23 and vascular calcification in predialysis and prevalent dialysis patients, but the relationship is ambiguous. In hemodialysis patients, some studies have reported an independent positive correlation between FGF23 and peripheral and aortic calcification [18, 19], but another study found that FGF23 was negatively correlated with peripheral calcification [20]. In predialysis CKD patients, FGF23 was not found to be associated with coronary artery calcification in a multivariate analysis [21]. A recent study found that serum FGF23 was associated with vascular calcification in patients at various CKD stages; however, 32% of the CKD patients examined were at stage 5D [22]. Based on these findings, it remains uncertain whether FGF23 is associated with vascular calcification in CKD patients who are not on dialysis. Therefore, the present study aimed to determine whether FGF23 levels are associated with CAAC in CKD patients who are not on dialysis.
Methods
In this cross-sectional study, 195 patients with CKD who were not on dialysis, and who were admitted to our hospital for evaluation and education regarding their CKD, were enrolled. None of the patients had been administered active vitamin D3 or phosphate binders before admission. All of the patients provided written informed consent for the protocol, which was approved by the Ethics Committee of National Kyushu Medical Center Hospital.
After admission, all of the patients were provided a diet containing low salt and protein, and blood samples (for serum creatinine (SCr), C-reactive protein, hemoglobin, serum corrected calcium, serum phosphorus, intact parathyroid hormone [PTH], 1,25(OH)2D, and intact FGF23 levels) were obtained in the early morning after an overnight fast. Daily proteinuria was also measured. Fasting serum was stored at −80°C. Intact FGF23 levels were measured using an FGF23 ELISA kit (Kainos Laboratories Int, Tokyo, Japan). The primary and secondary incubations were performed at room temperature for 2 hours and 1 hour, respectively, and the chromogenic reaction was carried out at room temperature for 30 min. The assay for intact FGF23 has a lower limit of detection of 3 pg/mL and intraassay and interassay coefficients of variation (CV) of less than 10%. Intact PTH levels were measured by electrochemiluminescence immunoassay (ECLIA) (Elecsys PTH, Roche Diagnostics GmbH, Mannheim, Germany). The incubation was performed at 37°C for 18 min. The detection limit of the assay was 1.20 pg/mL. The intraassay CV was less than 2.8% and interassay CV was less than 3.4%. A radioimmunoassay was used to measure 1,25(OH)2D (Immunodiagnostic Systems Limited, Boldon, UK). The incubation was performed using the following three-step protocol: (1) incubation overnight at 2-8°C, (2) incubation at room temperature for 1 hour, and (3) incubation at room temperature for 30min. The lower detection limit was 2.1 pg/mL and the intra and interassay CVs were less than 15%. SCr, C-reactive protein, hemoglobin, serum calcium, serum phosphorus and proteinuria levels were measured by standard laboratory methods. The estimated glomerular filtration rate (eGFR) (mL/min/1.73m2) was calculated by using the Modification of the Diet in the Renal Disease equation for Japanese patients: 194 × SCr-1.094 × age-0.287 × 0.739 (if female) [23].
All of the enrolled patients were interviewed and underwent a clinical examination at presentation. Their medical history and outpatient records were also evaluated in detail. Demographic information (age and sex) and atherosclerotic risk factors (hypertension, history of smoking, dyslipidemia, and diabetes mellitus) at presentation were recorded for each patient. Cigarette smoking was evaluated as current or past. Body mass index was calculated as the weight in kilograms divided by height in meters squared.
Carotid computed tomography (CT) images were performed to detect calcification of common, internal or external carotid arteries, using a 16-row multidetector CT (MDCT) scanner (Multispeed Ultra 16x; GE Medical Systems, Milwaukee, WI) with 22.9 mm/s table speed, 512 × 512 pixel matrix, 0.625-mm slice thickness, 0.6-mm reconstruction index, 120 kV peak tube energy and autoregulation of tube currents (Figure 1). The mean radiation dose of CT scans was 3.8 ± 3.0 (SD) (mSV). Because there is currently no scoring system to quantify CAAC, unlike the Agatston scoring system for evaluating calcium mass of coronary arteries, the presence or absence of CAAC based on the visual assessment of CT scans was examined by a double-check system of two radiologists who were blinded to the clinical information. However, intra- and interrater agreement for the detection of CAAC were not assessed.

Carotid artery calcification detected by multi-detector computed tomography was seen at the origin of the bilateral internal and external carotid arteries (arrows).
Statistical analysis
Continuous data are expressed as either the means ± SD or the median (interquartile range) depending on their distribution, and categorical data are expressed as numbers (with %). Non-normally distributed variables (FGF23, intact PTH, and 1,25(OH)2D) were also log-transformed to approximately achieve a normal distribution and were then used in subsequent statistical analysis. The significance of differences between group with and that without CAAC was examined using the chi-square test for categorical data, the Wilcoxon rank sum test for nonparametric data, and the unpaired Student’s t-test for parametric data.
Demographic, traditional and non-traditional risk factors, serological data and the prevalence of CAAC were compared across quartiles of FGF23 or CKD stages using the chi-square test for categorical data, ANOVA for continuous variables that were approximately normally distributed, and the Kruskal-Wallis test for skewed continuous variables analysis. Dunnett’s test was then used after ANOVA and the Kruskal-Wallis test. A logistic regression model was applied to determine the traditional and nontraditional risk factors associated with CAAC. Covariates associated with CAAC which were significant in age- and sex-adjusted logistic regression analysis were selected as risk factors in the multivariate analysis. We also examined whether higher quartiles of FGF23 were associated with CAAC in a logistic regression analysis. Quartiles of FGF23 levels were chosen as the risk factors for CAAC, with the lowest quartile serving as the reference category. The interaction effect of FGF23 and other variables on the outcome was estimated by adding interaction terms between FGF23 categories assigned serial numerical codes and the status of other variables (below and above the median) to the relevant model. The odds ratio and the 95% confidence interval were calculated for each variable. Data were analyzed using the JMP9 statistical package (SAS Institute, Cary, NC, USA). A p value below 0.05 indicated a significant difference.
Results
The mean age of the 195 patients (129 men and 66 women) in this study was 68.8 ± 11.3 years (range, 40–92 years). The primary causes of renal disease were chronic glomerulonephritis in 48 (24.6%) patients, diabetic nephropathy in 57 (29.2%), hypertensive nephrosclerosis in 60 (30.8%), other defined causes in 25 (12.8%), and unknown in 5 (2.6%). The clinical characteristics of the patients with and those without CAAC are summarized in Table 1. CAAC was found in 128 patients (66%). The mean age of the patients with CAAC was significantly higher than that of the patients without CAAC. The rates of hypertension, diabetes mellitus, smoking, and dyslipidemia in patients with CAAC were significantly higher compared with those in patients without CAAC. There were no significant differences in body mass index, proteinuria, C-reactive protein, and serum phosphate levels between the two groups. The values of hemoglobin, eGFR, and log 1,25(OH)2D in patients with CAAC were significantly lower than those in patients without CAAC, whereas PTH and FGF23 levels in patients with CAAC were significantly higher than those in patients without CAAC.
The clinical characteristics of the patients according to CKD stages are summarized in Table 2. Proteinuria in CKD stage 5 (n = 53) was significantly higher compared with that in CKD stages-1 and-2 (together, n = 27). There was no significant difference in C-reactive protein levels across CKD stages. Serum calcium levels were significantly lower and phosphate levels were significantly higher in CKD stage 5 patients compared with those in CKD stages 1–2 patients. The values of 1.25(OH)2D were significantly lower and PTH values were significantly higher in CKD stage 4 patients (n = 60) compared with those in CKD stages 1–2 patients. In contrast, significantly higher FGF23 levels in CKD stage 3 patients were already present compared with those in CKD stages 1–2 patients. In addition, the prevalence of CAAC was significantly increased across the CKD stages.
Table 3 shows the results of the logistic regression analysis for having CAAC. Adjusted for age and sex, hypertension, diabetes mellitus, smoking, proteinuria, calcium-phosphorus product, and log FGF23 were significantly associated with CAAC. Multivariate analysis showed that diabetes mellitus, smoking, and log FGF23 remained as independent determinants for CAAC.
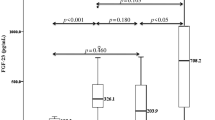
The study population was divided into quartiles of FGF23 levels (Table 4). With increasing quartiles of FGF23, mean serum phosphate and calcium-phosphorus product levels were higher, and mean hemoglobin and eGFR were lower. In addition, the prevalence of CAAC significantly increased with ascending quartiles of FGF23. Table 5 shows the odds ratios for having CAAC across quartiles of FGF23 levels obtained with the logistic regression model. In a crude model, both the third and fourth FGF23 quartiles were associated with CAAC. After being adjusted for demographic, traditional, and nontraditional risk factors (model4), the third and fourth FGF23 quartiles remained independently associated with CAAC. In addition, the effect of FGF23 and that of other variables, such as proteinuria, serum phosphorus, serum calcium, intact PTH, and eGFR, on the risk for CAAC were independent of each other (p for interaction; 0.61, 0.46, 0.11, 0.69, and 0.50, respectively).
Discussion
The principal finding in the present study was that FGF23 was independently associated with CAAC in CKD patients who were not on dialysis. The prevalence of CAAC was significantly increased as the kidney function declined. FGF23levels were significantly higher in CKD stage 3 patients than in CKD stages 1–2 patients. In contrast, serum phosphate levels were significantly higher only in CKD stage 5patients compared with CKD stages 1–2 patients, and were not related to CAAC. These results suggest that measurement of FGF23 is more important than measurement of serum phosphorus for the detection of risk factors associated with CAAC.
In ESRD patients, hyperphosphatemia is thought to be associated with vascular calcification [24–26]. In contrast, the association of serum phosphorus levels with vascular calcification in predialysis CKD patients has remained uncertain. Some studies have shown no significant correlation between serum phosphorus and vascular calcification [1, 27], while it has been demonstrated that higher serum phosphate levels, although still within the normal range, are associated with a greater prevalence of vascular and valvular calcification in subjects with moderate CKD [28]. Most patients develop hyperphosphatemia only at CKD stages 4 and 5 [29, 30], despite the progressive elevation of serum PTH and FGF23 levels as a defense mechanism to prevent an increase in serum phosphate levels. The present study also showed that serum phosphate levels were significantly high only in CKD stage 5 patients compared with CKD stages 1–2 patients, whereas significantly higher FGF23 levels were already observed in CKD stage 3 patients compared with CKD stages 1–2 patients. These findings are in agreement with previous studies [8, 10] reporting that an increase in FGF23 levels precedes an increase in serum phosphate levels in CKD patients, and they may support the hypothesis that elevated FGF23 concentrations are an early abnormality of disordered phosphorus metabolism in CKD patients [6].
Vascular calcification appears early in the course of CKD (i.e., well before ESRD is reached) [31]. One study reported that a high prevalence (64%) of coronary artery calcification was found in CKD patients with a median creatinine clearance of 37 mL/min/1.73m2 and a median serum phosphorus of 3.8 mg/dL [32]. In the present study, a high prevalence of CAAC was already present in CKD stage 3 patients, who also had a mean serum phosphate level of 3.4 mg/dL. Moreover, concerning the recent findings that high normal serum phosphate levels are associated with vascular calcification in predialysis CKD patients [28], it is possible that even lower serum phosphate levels contribute to vascular calcification. In addition, given the present finding that FGF23 levels, but not serum phosphate levels, were identified as an independent factor for CAAC, FGF23 measurement appears to be more important for detecting CAAC-associated risk factors in CKD patients who are not undergoing dialysis.
Although there is the possibility that elevated FGF23 levels affect bone metabolism or vascular calcification directly and/or indirectly, the precise mechanism of such an effect remains unknown. The potential finding of FGF23 levels as a biomarker of CAAC in the present study cannot lead to the conclusion of a real role of FGF23 in the pathogenesis of vascular calcification.
The main physiological stimuli of FGF23 secretion are increased dietary phosphorus intake and increased 1,25(OH)2D levels [33]. Accordingly, in the present study, we studied patients who had not been administered active vitamin D3 or phosphate binders, to more precisely determine whether FGF23 levels are associated with vascular calcification. Burnett et al. demonstrated that dietary phosphate loading increases the fractional excretion of phosphate and circulating FGF23 in 66 healthy males and females, suggesting that dietary phosphate is an important regulator of circulating FGF23 levels in humans [34]. However, an association of phosphate loading with changes in FGF23 levels has not been fully documented in CKD patients. In a small, randomized controlled trial of CKD patients, Isakova et al. recently reported that CKD patients receiving a higher phosphorus diet plus placebo for 2 weeks demonstrated a significant increase in FGF23 levels, with no significant changes over time in serum phosphate levels, suggesting an important effect of dietary phosphorus intake on FGF23 levels rather than serum phosphate levels [35]. However, it remains unknown whether high dietary phosphate loading contributes to the development of vascular calcification in CKD patients.
Moderately uremic mice fed a high phosphate diet are not hyperphosphatemic but have a significant rise in serum FGF23 levels that are significantly correlated with arterial medial calcification [36]. In the present study, phosphate intake or loading was not assessed in a quantitative manner over prolonged time periods before the patients’ admission. Accordingly, it remains unknown whether elevated FGF23 levels in some of our study population might reflect a reaction to phosphate loading, and the effect of phosphate loading on vascular calcification could not be assessed.
A previous study demonstrated that in normophosphatemic CKD patients there was a significant decrease in FGF23 levels, but not serum phosphorus levels, in patients treated with the non-calcium containing phosphate binder, sevelamer, for 6 weeks, whereas there was no decrease in FGF23 in calcium acetate-treated patients [37]. However, it is unclear whether a decrease in FGF23 by non-calcium containing phosphate binders is related to a simultaneous decrease in vascular calcification in CKD patients not on dialysis. Therefore, long, randomized-intervention studies are required to clarify the effect of non-calcium containing phosphate binders on changes in both FGF23 and vascular calcification in CKD.
The current study has some limitations. First, the patient cohort was relatively small. Second, the multidetector CT assessment depended on the presence or absence of CAAC, but calcium content was not scored. If the CAAC score is quantified, then the associated risk factors may be more precisely documented, and the relationship between CAAC scores and FGF23 levels should be defined. Third, serum klotho levels were not examined in the present study. It has been reported that klotho deficiency contributes to soft-tissue calcification in CKD [38]. FGF23 generally requires its cofactor, klotho, for its activity. High levels of FGF23, as well as deficient renal klotho expression and function, are found in CKD patients. It is possible that, in the absence of klotho, increased FGF23 levels exert pro-calcific effects via non-specific low affinity binding to its receptors [39–41]. A recent study showed that in human arteries, restoration of klotho expression by vitamin D receptor activators could unmask anticalcific effect of FGF23 [42]. Further investigations are warranted to determine whether high FGF23 levels, independent of klotho deficiency, contribute to the development of vascular calcification in CKD patients.
Conclusions
In CKD patients, the prevalence of CAAC is significantly increased across the CKD stages. Our findings also demonstrate that FGF23 is independently associated with CAAC. In the future, additional investigations are warranted to further clarify the precise mechanisms by which FGF23 contributes to the development of vascular calcification.
References
Russo D, Palmiero G, De Blasio AP, Balletta MM, Andreucci VE: Coronary artery calcification in patients with CRF not undergoing dialysis. Am J Kidney Dis. 2004, 44: 1024-1030. 10.1053/j.ajkd.2004.07.022.
Sigrist M, Bungay P, Taal MW, McIntyre CW: Vascular calcification and cardiovascular function in chronic kidney disease. Nephrol Dial Transplant. 2006, 21: 707-714. 10.1093/ndt/gfi236.
Sumida Y, Nakayama M, Nagata M, Nakashita S, Suehiro T, Kaizu Y, Ikeda H, Izumaru K: Carotid artery calcification and atherosclerosis at the initiation of hemodialysis in patients with end-stage renal disease. Clin Nephrol. 2010, 73: 360-369.
El-Abbadi M, Giachelli CM: Arteriosclerosis, calcium phosphate deposition and cardiovascular disease in uremia: current concepts at the bench. Curr Opin Nephrol Hypertens. 2005, 14: 519-524. 10.1097/01.mnh.0000168335.29381.23.
Nakayama M, Ura Y, Nagata M, Okada Y, Sumida Y, Nishida K, Ikeda H, Kaizu Y: Carotid artery calcification at the initiation of hemodialysis is a risk factor for cardiovascular events in patients with end-stage renal disease: a cohort study. BMC Nephrol. 2011, 12: 56-10.1186/1471-2369-12-56.
Stubbs J, Liu S, Quarles LD: Role of fibroblast growth factor 23 in phosphate homeostasis and pathogenesis of disordered mineral metabolism in chronic kidney disease. Semin Dial. 2007, 20: 302-308. 10.1111/j.1525-139X.2007.00308.x.
Liu S, Quarles LD: How fibroblast growth factor 23 works. J Am Soc Nephrol. 2007, 18: 1637-1647. 10.1681/ASN.2007010068.
Gutierrez O, Isakova T, Rhee E, Shah A, Holmes J, Collerone G, Juppner H, Wolf M: Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J Am Soc Nephrol. 2005, 16: 2205-2215. 10.1681/ASN.2005010052.
Evenepoel P, Meijers B, Viaene L, Bammens B, Claes K, Kuypers D, Vanderschueren D, Vanrenterghem Y: Fibroblast growth factor-23 in early chronic kidney disease: additional support in favor of a phosphate-centric paradigm for the pathogenesis of secondary hyperparathyroidism. Clin J Am Soc Nephrol. 2010, 5: 1268-1276. 10.2215/CJN.08241109.
Isakova T, Wahl P, Vargas GS, Gutiérrez OM, Scialla J, Xie H, Appleby D, Nessel L, Bellovich K, Chen J, Hamm L, Gadegbeku C, Horwitz E, Townsend RR, Anderson CA, Lash JP, Hsu CY, Leonard MB, Wolf M: Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011, 79: 1370-1378. 10.1038/ki.2011.47.
Gutiérrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, Smith K, Lee H, Thadhani R, Jüppner H, Wolf M: Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008, 359: 584-592. 10.1056/NEJMoa0706130.
Jean G, Terrat JC, Vanel T, Hurot JM, Lorriaux C, Mayor B, Chazot C: High levels of serum fibroblast growth factor (FGF)-23 are associated with increased mortality in long haemodialysis patients. Nephrol Dial Transplant. 2009, 24: 2792-2796. 10.1093/ndt/gfp191.
Isakova T, Xie H, Yang W, Xie D, Anderson AH, Scialla J, Wahl P, Gutiérrez OM, Steigerwalt S, He J, Schwartz S, Lo J, Ojo A, Sondheimer J, Hsu CY, Lash J, Leonard M, Kusek JW, Feldman HI, Wolf M, Chronic Renal Insufficiency Cohort (CRIC) Study Group: Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA. 2011, 305: 2432-2439. 10.1001/jama.2011.826.
Kendrick J, Cheung AK, Kaufman JS, Greene T, Roberts WL, Smits G, Chonchol MK, HOST Investigators: FGF-23 associates with death, cardiovascular events, and initiation of chronic dialysis. J Am Soc Nephrol. 2011, 22: 1913-1922. 10.1681/ASN.2010121224.
Fliser D, Kollerits B, Neyer U, Ankerst DP, Lhotta K, Lingenhel A, Ritz E, Kronenberg F, MMKD Study Group, Kuen E, König P, Kraatz G, Mann JF, Müller GA, Köhler H, Riegler P; MMKD Study Group: Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: the Mild to Moderate Kidney Disease (MMKD) Study. J Am Soc Nephrol. 2007, 18: 2600-2608. 10.1681/ASN.2006080936.
Roos M, Lutz J, Salmhofer H, Luppa P, Knauss A, Braun S, Martinof S, Schömig A, Heemann U, Kastrati A, Hausleiter J: Relation between plasma fibroblast growth factor-23, serum fetuin-A levels and coronary artery calcification evaluated by multislice computed tomography in patients with normal kidney function. Clin Endocrinol (Oxf). 2008, 68: 660-665. 10.1111/j.1365-2265.2007.03074.x.
Schoppet M, Hofbauer LC, Brinskelle-Schmal N, Varennes A, Goudable J, Richard M, Hawa G, Chapurlat R, Szulc P: Serum level of the phosphaturic factor FGF23 is associated with abdominal aortic calcification in men: the STRAMBO study. J Clin Endocrinol Metab. 2012, 97: E575-E583. 10.1210/jc.2011-2836.
Jean G, Bresson E, Terrat JC, Vanel T, Hurot JM, Lorriaux C, Mayor B, Chazot C: Peripheral vascular calcification in long-haemodialysis patients: associated factors and survival consequences. Nephrol Dial Transplant. 2009, 24: 948-955.
Nasrallah MM, El-Shehaby AR, Salem MM, Osman NA, El Sheikh E, Sharaf El Din UA: Fibroblast growth factor-23 (FGF-23) is independently correlated to aortic calcification in haemodialysis patients. Nephrol Dial Transplant. 2010, 25: 2679-2685. 10.1093/ndt/gfq089.
Inaba M, Okuno S, Imanishi Y, Yamada S, Shioi A, Yamakawa T, Ishimura E, Nishizawa Y: Role of fibroblast growth factor-23 in peripheral vascular calcification in non-diabetic and diabetic hemodialysis patients. Osteoporos Int. 2006, 17: 1506-1513. 10.1007/s00198-006-0154-6.
Gutiérrez OM, Januzzi JL, Isakova T, Laliberte K, Smith K, Collerone G, Sarwar A, Hoffmann U, Coglianese E, Christenson R, Wang TJ, De Filippi C, Wolf M: Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation. 2009, 119: 2545-2552. 10.1161/CIRCULATIONAHA.108.844506.
Desjardins L, Liabeuf S, Renard C, Lenglet A, Lemke HD, Choukroun G, Drueke TB, Massy ZA, The European Uremic Toxin (EUTox) Work Group: FGF23 is independently associated with vascular calcification but not bone mineral density in patients at various CKD stages. Osteoporos Int. 2012, 23: 2017-2025. 10.1007/s00198-011-1838-0.
Matsuo S, Imai E, Horio M, Yasuda Y, Tomita K, Nitta K, Yamagata K, Tomino Y, Yokoyama H, Hishida A: Collaborators developing the Japanese equation for estimated GFR: Revised equations for estimated GFR from serum creatinine in Japan. Am J Kidney Dis. 2009, 53: 982-992. 10.1053/j.ajkd.2008.12.034.
Goodman WG, Goldin J, Kuizon BD, Yoon C, Gales B, Sider D, Wang Y, Chung J, Emerick A, Greaser L, Elashoff RM, Salusky IB: Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N Engl J Med. 2000, 342: 1478-1483. 10.1056/NEJM200005183422003.
Raggi P, Boulay A, Chasan-Taber S, Amin N, Dillon M, Burke SK, Chertow GM: Cardiac calcification in adult hemodialysis patients. A link between end-stage renal disease and cardiovascular disease?. J Am Coll Cardiol. 2002, 39: 695-701. 10.1016/S0735-1097(01)01781-8.
London GM, Guérin AP, Marchais SJ, Métivier F, Pannier B, Adda H: Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant. 2003, 18: 1731-1740. 10.1093/ndt/gfg414.
Toussaint ND, Lau KK, Strauss BJ, Polkinghorne KR, Kerr PG: Associations between vascular calcification, arterial stiffness and bone mineral density in chronic kidney disease. Nephrol Dial Transplant. 2008, 23: 586-593.
Adeney KL, Siscovick DS, Ix JH, Seliger SL, Shlipak MG, Jenny NS, Kestenbaum BR: Association of serum phosphate with vascular and valvular calcification in moderate CKD. J Am Soc Nephrol. 2009, 20: 381-387. 10.1681/ASN.2008040349.
Levin A, Bakris GL, Molitch M, Smulders M, Tian J, Williams LA, Andress DL: Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney Int. 2007, 71: 31-38. 10.1038/sj.ki.5002009.
Moranne O, Froissart M, Rossert J, Gauci C, Boffa JJ, Haymann JP, M’rad MB, Jacquot C, Houillier P, Stengel B, Fouqueray B, NephroTest Study Group: Timing of onset of CKD-related metabolic complications. J Am Soc Nephrol. 2009, 20: 164-171. 10.1681/ASN.2008020159.
Temmar M, Liabeuf S, Renard C, Czernichow S, Esper NE, Shahapuni I, Presne C, Makdassi R, Andrejak M, Tribouilloy C, Galan P, Safar ME, Choukroun G, Massy Z: Pulse wave velocity and vascular calcification at different stages of chronic kidney disease. J Hypertens. 2010, 28: 163-169. 10.1097/HJH.0b013e328331b81e.
Tomiyama C, Higa A, Dalboni MA, Cendoroglo M, Draibe SA, Cuppari L, Carvalho AB, Neto EM, Canziani ME: The impact of traditional and non-traditional risk factors on coronary calcification in pre-dialysis patients. Nephrol Dial Transplant. 2006, 21: 2464-2471. 10.1093/ndt/gfl291.
Ferrari SL, Bonjour JP, Rizzoli R: Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab. 2005, 90: 1519-1524.
Burnett SM, Gunawardene SC, Bringhurst FR, Jüppner H, Lee H, Finkelstein JS: Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. J Bone Miner Res. 2006, 2: 1187-1196.
Isakova T, Gutiérrez OM, Smith K, Epstein M, Keating LK, Jüppner H, Wolf M: Pilot study of dietary phosphorus restriction and phosphorus binders to target fibroblast growth factor 23 in patients with chronic kidney disease. Nephrol Dial Transplant. 2011, 26: 584-591. 10.1093/ndt/gfq419.
El-Abbadi MM, Pai AS, Leaf EM, Yang HY, Bartley BA, Quan KK, Ingalls CM, Liao HW, Giachelli CM: Phosphate feeding induces arterial medial calcification in uremic mice: role of serum phosphorus, fibroblast growth factor-23, and osteopontin. Kidney Int. 2009, 75: 1297-1307. 10.1038/ki.2009.83.
Oliveira RB, Cancela AL, Graciolli FG, Dos Reis LM, Draibe SA, Cuppari L, Carvalho AB, Jorgetti V, Canziani ME, Moysés RM: Early control of PTH and FGF23 in normophosphatemic CKD patients: a new target in CKD-MBD therapy?. Clin J Am Soc Nephrol. 2010, 5: 286-291. 10.2215/CJN.05420709.
Hu MC, Shi M, Zhang J, Quiñones H, Griffith C, Kuro-o M, Moe OW: Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol. 2011, 22: 124-136. 10.1681/ASN.2009121311.
Razzaque MS: The FGF23-Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol. 2009, 5: 611-619. 10.1038/nrendo.2009.196.
Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T: Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006, 444: 770-774. 10.1038/nature05315.
Koh N, Fujimori T, Nishiguchi S, Tamori A, Shiomi S, Nakatani T, Sugimura K, Kishimoto T, Kinoshita S, Kuroki T, Nabeshima Y: Severely reduced production of klotho in human chronic renal failure kidney. Biochem Biophys Res Commun. 2001, 280: 1015-1020. 10.1006/bbrc.2000.4226.
Lim K, Lu TS, Molostvov G, Lee C, Lam FT, Zehnder D, Hsiao LL: Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation. 2012, 125: 2243-2255. 10.1161/CIRCULATIONAHA.111.053405.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2369/14/22/prepub
Acknowledgements
We would like to express our gratitude to Dr S Yamada (Departments of Medicine and Clinical Science, Kyushu University, Fukuoka, Japan) for his pertinent comments on this manuscript, to the 195 participants of our study, and to radiologists for their assessment of carotid artery calcification by CT.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MN1 was involved in the study design, sample collection, analysis and interpretation of the data and in the writing of the report. YK acquired the data and participated in the interpretation of the data. MN2 participated in the analysis and interpretation of the data. YU, HI, SS and KK participated in the sample collection. All authors have read and approved the final version of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Nakayama, M., Kaizu, Y., Nagata, M. et al. Fibroblast growth factor 23 is associated with carotid artery calcification in chronic kidney disease patients not undergoing dialysis: a cross-sectional study. BMC Nephrol 14, 22 (2013). https://doi.org/10.1186/1471-2369-14-22
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2369-14-22