
Abstract
Genetic and biochemical studies support the apolipoprotein E (APOE) ε4 allele as a major risk factor for late-onset Alzheimer's disease (AD), though ~50% of AD patients do not carry the allele. APOE transports cholesterol for luteinizing hormone (LH)-regulated steroidogenesis, and both LH and neurosteroids have been implicated in the etiology of AD. Since polymorphisms of LH beta-subunit (LHB) and its receptor (LHCGR) have not been tested for their association with AD, we scored AD and age-matched control samples for APOE genotype and 14 polymorphisms of LHB and LHCGR. Thirteen gene-gene interactions between the loci of LHB, LHCGR, and APOE were associated with AD. The most strongly supported of these interactions was between an LHCGR intronic polymorphism (rs4073366; lhcgr2) and APOE in males, which was detected using all three interaction analyses: linkage disequilibrium, multi-dimensionality reduction, and logistic regression. While the APOE ε4 allele carried significant risk of AD in males [p = 0.007, odds ratio (OR) = 3.08(95%confidence interval: 1.37, 6.91)], ε4-positive males carrying 1 or 2 C-alleles at lhcgr2 exhibited significantly decreased risk of AD [OR = 0.06(0.01, 0.38); p = 0.003]. This suggests that the lhcgr2 C-allele or a closely linked locus greatly reduces the risk of AD in males carrying an APOE ε4 allele. The reversal of risk embodied in this interaction powerfully supports the importance of considering the role gene-gene interactions play in the etiology of complex biological diseases and demonstrates the importance of using multiple analytic methods to detect well-supported gene-gene interactions.
Similar content being viewed by others

Background
Alzheimer's disease (AD) is a progressive neurodegenerative disorder characterized by neuronal and synaptic loss, neurofibrillary tangles in neuronal cytoplasm, and deposition of β-amyloid (Aβ) in extracellular, neuritic plaques. To date, only four genes have been unambiguously associated with AD, of which only one, Apolipoprotein E (APOE), is associated with the common, late-onset form of AD [1]. The APOE4 allele (ε4) was first identified as a risk factor for late-onset AD in the early 1990s [2, 3], and corroborated as such by a number of subsequent studies [4]. However, the risk for AD imparted by one or two ε4 alleles is only partially penetrant: ~50% of AD patients do not carry an ε4 allele [5]. Application of quantitative genetics methodology in fact supports the presence of 4 as yet unidentified AD-associated loci in the human genome, each expected to affect age of onset (AoO) as much or more than APOE [6]. Additional genetic risk factors for AD, therefore, remain to be found. Yet, a majority of studies have failed to find any evidence for association of their genetic target(s) with AD (e.g., recently, Chapuis et al. [7] and [8]), and large-scale meta-analyses, which combine the datasets of numerous studies, often negate or call into question any putative associations inferred from individual datasets [9].
The disproportionate number of women who suffer from AD has long suggested that an aspect of reproductive physiology lies at the origin of AD pathogenesis. Recently, this idea was supported by the discovery that polymorphisms of the estrogen receptors alpha and beta were associated with AD, further implicating estradiol signaling in the pathogenesis of AD [10, 11]. Several converging lines of evidence make another member of the hypothalamic-pituitary-gonadal axis, luteinizing hormone (LH), a worthwhile candidate for genetic study: (1) LH is elevated in AD patients [12–14]; (2) LH crosses the blood-brain barrier [15]; (3) in the brain, LH/chorionic gonadotropin receptors (LHCGR) are most concentrated in the hippocampus [16]; (4) increased concentration of LH has been shown to increase Aβ secretion in a neuronal cell line while suppression of serum LH decreases brain Aβ in mice [17]; and, (5) reduced serum LH has been shown to decrease cognitive loss and Aβ deposition in AβPP transgenic mice [18]. Interestingly, through its regulation of steroidogenic enzymes, LH mediates neurosteroid production from cholesterol [19]; both animal and human clinical studies strongly support the crucial neuroprotective functions of steroids in the brain [20, 21]. Since APOE is a cholesterol transport protein [22] involved in the transport of cholesterol into neurons [23] for neurosteroid synthesis, a functional link exists between APOE and LH signaling.
Numerous polymorphisms of LH beta-subunit (LHB) and LHCGR have been documented (for comprehensive reviews, see [24] and [25]). While the majority of mutations underlying these polymorphisms are associated with rare reproductive disorders, a few are relatively more common and worthy of exploring for their association with AD. Two non-synonymous single nucleotide polymorphisms (SNPs) in LHB are collectively referred to as variant LH (vLH) [26]. In a study of 40 Japanese women, vLH carriers exhibited greater LH secretion in response to GnRH stimulation [27]. In breast cancer patients, an LQ-insert in exon 1 of LHCGR was associated with a significantly earlier age of onset and worse survival rate [27]. Exon 10 of LHCGR is required for binding of LH [28] and is the location of 2 relatively common non-synonymous SNPs [29]. The functional consequences of the mutations underlying the other LHB and LHCGR polymorphisms scored in our study, however, are largely unknown. Therefore, in this study we examined polymorphmic sites of LH β-subunit (LHB) and LHCGR, as well as gene-gene interactions between LHB, LHCGR, and APOE for association with AD. Our results suggest that a specific LHCGR allele significantly decreases the risk of AD in individuals carrying an APOE ε4 allele.
Results
APOE genotype and 14 previously reported polymorphisms of LHB and LHCGR were scored (Table 1). The A/G polymorphism in exon 2 of LHB (rs5030775) was invariant and, therefore, not included in any analyses. The 2 SNPs comprising vLH (rs1800447) covaried without fail, as did the LHB polymorphisms rs2387588 and rs4287687; in both instances, only one of the polymorphisms was subjected to analysis. Table 1 lists the names assigned to the remaining 11 polymorphisms of LHB and LHCGR.
Analysis of single-locus, main effects
HWE and AoO
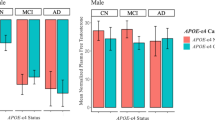
The only locus demonstrating significant divergence from HWE at the modified FDR level was lhcgr3 in the control (C) group (AD: p = 0.052, C: p = 0.008; α = 0.0082). This result is apparently driven by samples of the female control group (Cf; p = 0.034), as the male control group (Cm) is not even marginally divergent from HWE (p = 0.149). No main or interactive effects identified in our analyses included lhcgr3. Only one genotype model was significantly associated with age of onset (AoO) at the modified FDR level. In the ADm group, AoO was affected by lhcgr2 genotype (Figure 1). Males with 1 or 2 C-alleles of lhcgr2 had a mean AoO of 81.06 years, while males with no C-allele had a mean AoO of 78.33 (n = 50, p = 0.0047; α = 0.0077) – i.e., the C-allele significantly delays AoO in males.

Age of onset is significantly affected by lhcgr2 genotype in males. AoO in lhcgr2 heterozygous and homozygous males (n = 50, p = 0.001).
χ 2-tests for single-locus associations
Whether stratified by gender or not, no significant associations between LHB or LHCGR loci and AD were identified. As expected, the frequency of the APOE ε4 allele was much greater in AD than in C samples: 0.35 and 0.09, respectively. ε4 was also found at a higher frequency in AD females (ADf; 0.39) than in AD males (ADm; 0.32). Compared with ADm, a noticeably greater number of ε4 alleles were found in ADf heterozygotes (ε2/ε4 and ε3/ε4: 0.35 in ADm, 0.62 in ADf) than in homozygotes (ε4/ε4: 0.14 in ADm, 0.08 in ADf). A significant association, at modified FDR levels, between the APOE ε4 allele and AD was detected in AD vs. C (p < 0.0001; α = 0.0082), ADm vs. Cm (p < 0.001; α = 0.0077), and ADf vs. C female (Cf; p < 0.0001; α = 0.0081) comparisons. Both the 'ε4 dosage' and 'ε4 positive' models of APOE genotype were associated with AD at marginally to highly significant levels in AD vs. C (p < 0.0001; α = 0.0082), ADm vs. Cm (ε4 dosage: p = 0.003; ε4 positive: p = 0.007; α = 0.0077), and ADf vs. Cf (p < 0.0001; α = 0.0081). The estimated OR associated with ε4 was considerably higher in females ['ε4 dosage': 18.53 (6.18, 55.61); 'ε4 positive': 20.53 (6.80, 62.01)] than males ['ε4 dosage': 2.81 (1.36, 5.82); 'ε4 positive': 2.66 (1.14, 6.20).
Analysis of gene-gene interactions
LD analysis
In the ADm group but not the Cm group, significant multi-locus LD at the modified FDR level was detected between APOE and lhcgr2 (p = 0.003; α = 0.0077; Table 2), while a number of pairs of loci exhibited marginally significant LD (p < 0.05; Table 2). Significant LD at the modified FDR level was also detected between APOE and lhcgr2 in Cf (p = 0.007; α = 0.0077; Table 2). A number of LHCGR loci in a number of different AD and control groups were found to be in significant LD with several LHB loci, especially lhb1 (Table 2).
APOE and LHB are closely linked to one another (chromosomal region 19q13), separated by only 4.1 megabases. We took great care to ensure that any associations with AD observed in LHB were not the result of linkage with APOE. The only instance of significant LD between an LHB locus and APOE alone was found in the ADf and total AD groups (lhb3, p < 0.0001 for both groups; α = 0.0081 and α = 0.0082, respectively). It is difficult to interpret this result as an indication of LD that is simply due to physical proximity of the loci, since none of the other LHB loci exhibited even marginally significant LD with APOE. lhb3 was not identified as a main effect, nor as a component of any other significant interactions.
MDR analysis
MDR models were deemed significant when they met the a priori significance criteria described in Methods. For the AD vs. C comparison, one multi-locus combination was significantly associated with AD: lhcgr1/lhcgr2/APOE was selected as the best model in 6 of 10 cross-validation (CV) runs and produced a training accuracy of >0.5 in 9 of 10 CV runs. Two multi-locus models exhibited significant association with AD in the ADm vs. Cm comparison: lhcgr2/APOE (8 of 10 CVs, >0.5 training accuracy in 7 of 10 CVs) and lhcgr2/lhcgr5/APOE (7 of 10 CVs, >0.5 training accuracy in 9 of 10 CVs). One multi-locus model, lhb5/APOE (5 out of 10 CVs, >0.5 training accuracy in 10 of 10 CVs), was selected as significantly associated with AD upon comparison of the ADf and Cf datasets. No significant gene-gene interactions were detected using APOE-free datasets.
LR analysis
LR analysis of the interactions suggested by LD and MDR analyses supported two interactions as marginally significant (lhcgr5/lhb2, both co-dominant, p = 0.05; lhcgr5/lhb4, both co-dominant, p = 0.041) and one interaction (lhcgr2/APOE) as significant at the modified FDR level. The most significant combination of lhcgr2 and APOE genotype models was the ε4-positive model of APOE and the C-dominant model of lhcgr2 (p = 0.003). As illustrated in Figure 2, the OR associated with this interaction [0.06 (0.01, 0.38)], represents a marked decrease in risk of AD for males who possess 1 or 2 'C-alleles' for lhcgr2 and are ε4-positive. Upon identification of this interaction we sequenced lhcgr2 and APOE in 10 additional male (5C and 5 AD) and 30 additional female (6C and 24 AD) samples. Use of the augmented datasets in LR analysis did not change the p-value or OR associated with the male-specific interaction and the lhcgr2/APOE interaction was still insignificant amongst females (p = 0.871).

Risk associated with ε4 allele is negated in male, lhcgr2 heterozygotes. Contingency table illustrating the relative frequencies of lhcgr2/APOE genotypes in males and females (black = AD; white = Control).
Identification of a novel, missense mutation in LHCGR
Two males, one control and one AD, were heterozygous for a previously unreported C->T (Arg->Stop) missense mutation at the first position of codon 479 (exon 11) of LHCGR (Figure 3).

Novel missense mutation in exon 11 of LHCGR. (A) Chromatogram from one of two samples heterozygous for a novel C->T (Arg->Stop) missense mutation in codon 479 (exon 11) of LHCGR. (B) Chromatogram from a sample homozygous for 'C' at the same site.
Discussion
In general, our results suggest that putative associations should be treated with caution if they do not receive consistent support from biologically or statistically distinct analyses or are discovered using only one analytic method. Results of multiple analyses have the potential to strengthen support for disease association, point to alternate explanations of anomalous allele or genotype frequencies, or disabuse one of the notion that a particular polymorphism, unrelated to a disease of interest, plays a central role in its etiology.
Multi-analytic approach to detection of gene-gene interaction
Studies that aim to identify genetic interactions are best served by the multi-analytic approach to data analysis practiced here and, for family-based data, by [30]. In the absence of biochemical evidence, putative genetic interactions are often difficult to accept intellectually. Multiple lines of statistical support, which represent the identification of a significant genetic interaction using different statistical and biological approaches, increase statistical and intellectual confidence in the biological reality of an interaction. In this study, LR and MDR provided distinct statistical/computational methods for the detection of significant genetic interactions, while LD analysis provided a distinct biological approach to the problem of interaction detection. A majority of AD-associated interactions reported here lack intra-study corroboration, as they were only identified by one analytical method (Table 3). As such, these interactions are not well supported. On the other hand, lhcgr2/APOE (the only interaction supported by all 3 analytic methods at the modified FDR level) is a better candidate for biochemical study. In addition, more support for lhcgr2/APOE is derived from the association of lhcgr2 with differences in AoO. The different analyses are not, of course, statistically independent of one another, as the same dataset is being analyzed in each case. Nevertheless, because they are not independent, failure to identify the same interaction using distinct, robust analytical methods seriously impugns the biological reality of a putative interaction.
APOE, LH signaling, gender-specific effects, and AD
Polymorphisms of other HPG-axis proteins (estrogen receptors α and β) are associated with increased susceptibility to AD in women [10, 11]. In this respect, prophylactic and therapeutic use of natural estrogen (17β-estradiol) has been consistently demonstrated to delay disease progression in women [11]. As LH signaling is directly involved with reproduction, produces gender-specific physiological and anatomical endpoints, and has been associated with AD, LH and its receptor also present good candidates for gender-specific associations with disease. The male-specific nature of the significant lhcgr2/APOE interaction identified in our analyses (Table 3), and its relation to APOE genotype, is important. Gender is thought to interact with APOE genotype [31, 32], and our data support the hypothesis that the ε4 allele is more strongly associated with female than male AD: 49% of ADm and 70% of ADf were ε4 positive. If ε4 does provide less explanatory power in males, it is logical to suggest that male-specific risk factors for AD do exist. Indeed, one recent study identified an association between number of CAG repeats in the androgen receptor and AD [33]. Should further sampling corroborate the male-specific association with AD of lhcgr2/APOE (Table 3), it will become imperative to elucidate the biochemical basis of this gender bias (see discussion of the lhcgr2 site below). While gender-specific hormonal fluctuations, namely the rise in LH serum levels following menopause, have been suggested to account for the disproportionately greater number of females who acquire AD [34, 35], the idea that common differences in the actual sequence and structure of LHβ and its receptor might only affect males is intriguing. We can exclude issues related to genetic or trait heterogeneity as explaining our results since all scored loci exhibit HWE in males and the dataset is consistent with past sampling of APOE genotype in males. ε2, ε3, and ε4 allele frequencies among affected males in our study were 0.03, 0.66, and 0.32, respectively, which are not obviously different from those reported in a meta-analysis of 5107 case-control Caucasian AD subjects (4): 0.039 (ε2), 0.594 (ε3), and 0.367 (ε4). 49% of ADm and 70% of ADf were ε4-positive in our study, which is strikingly similar to the 46.6% ε4-postive males and 72% ε4-positive females reported in a previous paper suggesting interaction between gender and APOE genotype (36). Additionally, neither mean age (83.34 +/- 5.14 yrs in males, 83.34 +/- 5.58 yrs in females; p = 1.00) nor mean AoO (79.18 +/- 3.47 yrs in ADm and 80.26 +/- 5.07 yrs in ADf; p = 0.22) were significantly different in males and females. Documented instances of alcohol and drug use, cardiovascular disease, and stroke were equally rare in males and females of our cohort.
Despite strong support for the association between lhcgr2/APOE and AD, the details of the interaction are paradoxical. While the ε4 allele carried significant risk of AD in males of our dataset (p = 0.007), males who carried 1 or 2 C-alleles at the lhcgr2 locus and were ε4 positive had a significantly reduced risk of AD (odds ratio: 0.04; 95% confidence level: 0.01, 0.32; p = 0.002). As both increased LH levels [17] and the APOE ε4 allele [36, 37] are associated with increased Aβ deposition, and neurosteroid production, it is reasonable to suggest that LH signaling and APOE genotype interact to modify an individual's susceptibility to AD. The significant decrease in risk of AD observed in ε4-positive males with 1 or 2 lhcgr2 C-alleles lends support to the possibility that lhcgr2-dependent alternative splicing of LHCGR pre-mRNA leads to isoforms of LHCGR that are functionally distinct (see below), or that lhcgr2 is part of an intron-derived microRNA (miRNA) capable of regulating APOE mRNA translation (see below). Despite the absence of empirical evidence to support the existence of unreported LHCGR isoforms or miRNAs derived from LHCGR introns, our data do support a complex, gender-specific interaction between LHCGR and APOE. Of note, LH elevates APOE secretion from cultured interstitial cells, thereby increasing the availability of cholesterol for sex hormone production [38]. LH also increases low-density-lipoprotein receptor-related protein expression in granulose cells [39]. If such processes occur in the brain, then the protective effects of an LHCGR-APOE interaction in males may be mediated via increased neurosteroid production and the male-specific nature might be explained by differential protective effects of androgens and estrogens.
Intronic polymorphisms and lhcgr2 as a cryptic splice site or intron-derived miRNA
The most significant interaction associated with AD in this study includes lhcgr2, a polymorphism located in intron 1 of LHCGR. Intronic polymorphisms are frequently implicated in increased disease susceptibility [11, 40, 41] and intronic mutations have the potential to alter protein sequence dramatically. An intronic mutation can cause the disarrangement of an existing splice site or introduce a cryptic splice site, resulting in the addition/removal of hundreds of amino acids or premature termination of translation. A second mechanism by which intronic mutations might affect disease etiology is through their effect on the miRNAs that may be derived from the mutated intron. ~10–30% of spliced intronic material is exported to the cytoplasm, where it has the potential to act as an miRNA and alter protein expression [42]. It follows that mutation of intronic sequence could decrease or increase the complementarity of an intron-derived miRNA to its target mRNA, thereby causing a relative decrease or increase in expression of the encoded protein. Indeed the 15 bases immediately upstream of lhcgr2 are complementary to APOE mRNA (Figure 4A), indicating this polymorphism may cap an intron-derived miRNA.

lhcgr2 shares similarity with consensus 3' acceptor splice sites and ApoE mRNA. (A) lhcgr2 as a potential miRNA that regulates the expression of ApoE. The reversed LHCGR sequence is complimentary to a fragment of APOE mRNA at 13 out of 16 sites. (B) Pairwise sequence alignment of a portion of LHR intron 1 in human and mouse (Mus musculus). The human sequence includes lhr2 (rs4073366) and bears sequence features characteristic of 3' acceptor splice sites.
Examination of the sequence surrounding lhcgr2 and alignment of human and mouse (Mus musculus) LHCGR intron 1 indicate lhcgr2 may be located within a cryptic 3' acceptor splice site (Figure 4B). Acceptor splice sites are characterized by two conserved sequence patterns: a pyrimidine-rich sequence, known as the polypyrimidine tract, and the proximate terminal 'AG' of the intron [43]. Although the length of the polypyrimidine tract and its distance from the end of the intron are variable, the terminal 'AG' is invariant [43, 44]. Polymorphism lhcgr2 is located within a pyrimidine-rich region of intron 1 (Figure 4B). The lhcgr2 'C-allele' increases the CT-content of the surrounding 19 nucleotides to 79%, and, more locally, a 'C' at lhcgr2 forms a contiguous sequence of 7 Cs and Ts. Four bases downstream of this pyrimidine-rich tract is a 3' acceptor site consensus sequence, CAGG. Absence of a homologous sequence in mouse LHCGR intron 1 may indicate that retention of this sequence in Homo sapiens is due to its potential use in alternative splicing. To investigate the degree to which a 'C' at lhcgr2 increases the similarity of the local sequence to human acceptor sites in general, lhcgr2 'C' and 'G' alleles were entered in the online splice site prediction programs GENIO/splice and SpliceScan [45]. Both programs identified lhcgr2 as a potential acceptor splice site and, based on the programs' output scores, indicated that a 'C' at the site does increase its similarity to stereotyped 3' acceptor splice sites.
Linkage disequilibrium between lhcgr1 and multiple LHB loci
LD between lhcgr1 and LHB loci was commonly detected in several AD and C datasets (Table 2), including 6 instances in the 'All male' dataset. The frequent identification of LD between lhcgr1 and LHB loci indicates that specific LHB haplotypes are found most frequently in individuals that are hetero- or homozygous for the LQ-insertion at lhcgr1. We questioned whether our data supported the idea that the LQ-insertion at lhcgr1 is, in general, in LD with LHB loci. To examine this possibility, we compared the frequency of genotypes at all 6 LHB loci between individuals with no LQ-inserts and those with 1 or 2 LQ-inserts (Figure 5). In both the group of all males and the group of all samples, χ2-tests revealed that the genotype counts of a majority of LHB loci were markedly different from one another in the LQ-insert and no-LQ-insert strata (Figure 5). This suggests: (1) a non-random force, such as selection, is driving the non-random association of LQ-insert alleles with specific LHB haplotypes in the North American Caucasian population – i.e., certain variants of LHB may be better suited to the LQ-insert variant of LHCGR; (2) given the extent of LD between lhcgr1 and LHB loci in C and AD samples, instances of significant LD between lhcgr1 and LHB loci in AD samples are not indicative of AD-associated interactions.

Significant linkage disequilibrium between lhcgr1 and multiple LHB loci. In the schematic of LHB, exons are represented as black boxes and the positions of the 7 LHB polymorphisms scored are indicated by vertical lines. lhb3 is composed of two SNPs, which are in complete LD with each other. For each polymorphism, genotype frequencies are represented in the form of two pie charts, where the left chart represents genotype frequencies among individuals lacking an LQ-insert allele in exon 1 of LHCGR (lhcgr1) and the right chart represents genotype frequencies among individuals possessing 1 or 2 LQ-insert alleles. Frequencies above the graphs correspond to AD males, while frequencies below the graph correspond to all individuals sampled in this study. * = significant difference (p < 0.05), ** = highly significant difference (p < 0.01).
Methods
Case-control setup
The National Cell Repository for Alzheimer's Disease (NCRAD; University of Indiana, Bloomington, IN) provided total DNA samples from 200 control (negative for AD and other neurodegenerative diseases, 50 male and 50 female) and late-onset AD patients (negative for other neurodegenerative diseases, 50 male and 50 female). All samples were obtained from North American Caucasian subjects. All samples were derived from individuals >= 75 years of age, and all AD samples were acquired from individuals whose AoO was = 75. Mean age of the control group was 84.73 w 4.61 years, while mean age of the AD group was 81.95 ± 5.69 years. Among the AD samples, AoO was 79.18 ± 3.47 years for males and 80.26 ± 5.07 years for females. DNA was obtained from 40 additional samples (10 male samples – 5 control, 5 AD and 30 female samples – 6 control, 24 AD) to test for significant interaction between APOE and an LHCGR locus (lhcgr2, see below). Direct sequencing of APOE, LHB (promoter, signal, and coding regions), and LHCGR (exons 1, 10, and 11) was performed using the primer pairs listed in Table 4. Cycle sequencing products were run on an ABI 3730 XL DNA analyzer at the University of Wisconsin Biotechnology Center (Madison, WI) and the resultant chromatograms were analyzed with FinchTV v1.4 (Geospiza, Seattle, WA). This study was carried out with IRB approval from the Health Sciences Institutional Review Board of the University of Wisconsin.
Data analysis
Similar to the analytic paradigm suggested by [30] for family-based data, we chose to analyze our case-control dataset using an array of analytic methods, testing for interactive as well as main effects, and treating the convergence of results from distinct analyses as the best evidence of association. Allele and genotype counts were used in the following analyses: (1) χ2 tests of allele and genotype counts to test for main effects of individual polymorphisms; (2) tests of each locus for Hardy-Weinberg Equilibrium (HWE); (3) tests of combinations of two or three loci for linkage disequilibrium (LD); (4) tests for gene-gene interactions using multifactor dimensionality reduction (MDR); (5) tests for interactions using logistic regression (LR), and; (6) tests for association of polymorphisms with age of onset using one-way ANOVA. Additionally, to control for heterogeneity we stratified the dataset according to gender and applied the same 6 analyses. Finally, for each bi-allele locus, four genotype models were analyzed in tests for interactive effects: co-dominant, allele 1 dominant, allele 2 dominant, and over-dominant. This schema enabled us to: (1) address the possibility of heterozygote advantage, and; (2) test both alleles for dominance, as we had no a priori knowledge of which allele might carry risk.
For each sample, genotype and demographic data were entered into a MySQL relational database, enabling the quick identification of samples meeting an array of criteria. APOE genotype and 14 previously reported polymorphisms of LHB and LHCGR were scored. For each polymorphism, allele and genotype frequencies of the AD and control groups were calculated. Additionally, both groups were stratified by gender and gender-specific allele and genotype frequencies were calculated. Four separate genotype models were used in tests for main and interactive effects of bi-allele loci. For example, the following models would be used for a locus that varied between alleles B and b: (1) co-dominant (BB vs. Bb vs. bb); (2) B dominant [(BB + Bb) vs. bb]; (3) b dominant [BB vs. (Bb + bb)], and; (4) over-dominant [(BB + bb) vs. Bb]. For the tri-allele APOE, an 'ε4 dosage' model (genotypes grouped by the number of ε4 alleles) and an 'ε4 positive' model (ε4 allele present or not) were used in analyses.
The program Genetic Data Analysis (GDA) [46] was used to test each polymorphic locus for HWE. Minitab [47] was used to test for the association of individual polymorphisms with AD (χ2 tests of allele and genotype counts) and AoO in the AD groups (ANOVA). In all tests for LD, genotypes were preserved in order to prevent significant deviations from HWE at a single locus from contributing to the measure of LD. We considered a number of theoretical issues when designing our analytical approach to detect gene-gene interactions (see Supplementary Information) and ultimately chose the combination of LD, MDR and LR analyses. Pairwise tests for LD were performed using the program PyPop, where the p-value reported here is derived from the difference between the likelihood of the inferred haplotype frequencies and the likelihood of the data if the two loci are assumed to be in linkage equilibrium [48]. We reported the D' measure of LD, as this is an intuitive metric that represents the estimated proportion of maximum possible LD exhibited by the sample data. MDR was performed using MDR Software [49], which output the best 1-, 2-, 3-, and 4-factor models for a given dataset. 10-fold cross-validation was used. Given the weight APOE carries as a single factor, MDR was also run using APOE-free datasets in order to detect any interactions that did not include APOE. An interaction model was considered significant if it was selected as the best model in 5 or more of the CV runs and exhibited a testing accuracy of >0.5 in 7 or more CV runs. Pairs of loci exhibiting significant LD (p <= 0.02) and significant multi-locus models discovered using MDR were input as disease models in LR analyses performed in [47]. This form of LR model selection was necessary, as a lack of several multi-locus combinations made backward model selection impossible and the lack of significant main effects in most loci studied made forward model selection impractical.
To account for multiple tests, testwise α levels were corrected using modified FDR (see Supplementary Information). Because a multi-locus combination was only tested with LR if LD and/or MDR analyses were suggestive of its association with AD, only a subset of the total array of possible LR tests were actually performed and the total, male, and female datasets were subjected to a different total number of tests: 182, 252, and 190 tests, resulting in modified FDR α levels of 0.0082, 0.0077, and 0.0081, respectively.
Conclusion
We report the discovery of a genetic interaction between APOE and LHCGR alleles that is associated with a significantly decreased risk for AD in males. The biochemical basis for this interaction is uncertain, although alternative mRNA splicing and intron-derived miRNA regulation are hypothesized as distinct possibilities. Our results emphasize the importance of testing for gene-gene interactions in studies of complex disease. We suggest that the best evidence for epistasis is obtained when multiple analyses, distinct in their biological or statistical basis, converge on a positive result.
Appendix
Methodological strategies for the detection of gene-gene interactions
We searched for genetic association of single loci with AD using standard χ2 and HWE tests and considered that subsequent tests for gene-gene interactions may identify interactions whose loci may or may not produce significant main effects on their own. There are a number of analytical issues to consider when searching for multiple interacting genetic (or environmental) factors associated with a disease of complex etiology. For one, genetic or trait heterogeneity among the samples (e.g., AD samples from males or females, or, with or without hypertension) has the potential to confound data analysis. Stratification of the dataset is the most straightforward method used to control for heterogeneity. For example, one might examine the effects of APOE heterogeneity by splitting control and AD groups into ε4 positive and negative strata and asking: Is the frequency of an SNP of interest significantly higher in ε4-positive AD samples than in ε4-positive controls, but not in the comparable ε4-negative comparison? If so, the data suggest an interactive effect between the SNP and ε4. Significantly, a non-stratified comparison of AD and control groups in such a case might lead a researcher to conclude the SNP has no association with AD, or, conversely, that the SNP is associated with AD in ε4 positive and negative individuals. Though more mathematical methods to control for heterogeneity exist, the majority of them are not applicable to case-control data.
In studying the genetics of a complex disease, it is important to consider the possibility that gene-gene or gene-environment interactions produce interactive effects that provide significant explanatory power, even in the absence of single factor, main effects [53]. A number of methods allow researchers to test for gene-gene interactions using case-control data. A traditional method is logistic regression (LR), which, given a dataset, models the probability of a discrete outcome (in our case, AD or not) on n factors and their interactions, each qualified by a coefficient estimated using Maximum Likelihood Estimation. Attractively, LR produces an Odds Ratio (OR), which provides the researcher with an intuitive measure of how a particular array of genetic and/or environmental factors affects the likelihood of developing the disease. The major shortcoming of LR is the so-called 'curse of dimensionality', which refers to poor coefficient estimation resulting from too few or no examples of various multi-factor combinations in the dataset if sample size is too small or number of factors too large [54].
An alternative to LR analysis for the detection of interactions is multifactor dimensionality reduction (MDR) [55], which is advantageous for several reasons. A chief, practical advantage is that the researcher can easily test disease models that include interaction terms whose components lack significant main effects. This is critical, since it is increasingly apparent that genetic interactions, in the absence of main effects, frequently contribute to the susceptibility of an individual to complex diseases like AD [56]. Also important, MDR is not "cursed" by dimensionality: the method is robust even when the input dataset lacks examples of various, multi-factor combinations. Finally, unlike LR, MDR analysis automatically measures the predictive accuracy and validity of a selected model through partitioning of the original dataset into training and testing subsets [57, 58].
Another approach to gene-gene interaction detection is to calculate linkage disequilibrium (LD) amongst combinations of the loci under investigation. Significant LD among 2 or more loci indicates non-random segregation of the loci in question, which implies that at least one multi-allele combination at these loci is overrepresented. Logically, any multi-allele combination enriched in the case but not the control group is considered to contribute increased susceptibility to the disease, and is reflected by a measurement of significant LD in the case group only [59]. Williams et al. [60] used this approach in a study of polymorphisms associated with hypertension, discovering 16 combinations of 7 loci in 5 genes that exhibited significant LD in the case group only. Significantly, none of these loci were associated with a main effect on hypertension, indicating LD analysis has the ability to detect potentially significant gene-gene interactions in the absence of main effects.
References
Bertram L, Tanzi RE: The current status of Alzheimer's disease genetics: what do we tell the patients?. Pharmacol Res. 2004, 50: 385-396. 10.1016/j.phrs.2003.11.018.
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA: Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993, 261: 921-923. 10.1126/science.8346443.
Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ, et al: Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurology. 1993, 43: 1467-1472.
Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM: Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. Jama. 1997, 278: 1349-1356. 10.1001/jama.278.16.1349.
Roses A, Devlin B, Conneally P, Small G, Saunders A, Pritchard M: Measuring the genetic contribution of APOE in late-onset Alzheimer disease. Am J Hum Genet. 1995, 57: A202-
Daw EW, Payami H, Nemens EJ, Nochlin D, Bird TD, Schellenberg GD, Wijsman EM: The number of trait loci in late-onset Alzheimer disease. Am J Hum Genet. 2000, 66: 196-204. 10.1086/302710.
Chapuis J, Tian J, Shi J, Bensemain F, Cottel D, Lendon C, Amouyel P, Mann D, Lambert JC: Association study of the vascular endothelial growth factor gene with the risk of developing Alzheimer's disease. Neurobiol Aging. 2006, 27: 1212-1215. 10.1016/j.neurobiolaging.2005.07.013.
Ozturk A, DeKosky ST, Kamboh MI: Lack of association of 5 SNPs in the vicinity of the insulin-degrading enzyme (IDE) gene with late-onset Alzheimer's disease. Neurosci Lett. 2006, 406: 265-269. 10.1016/j.neulet.2006.07.054.
Pritchard A, Harris J, Pritchard CW, St Clair D, Lemmon H, Lambert JC, Chartier-Harlin MC, Hayes A, Thaker U, Iwatsubo T, Mann DM, Lendon C: Association study and meta-analysis of low-density lipoprotein receptor related protein in Alzheimer's disease. Neurosci Lett. 2005, 382: 221-226. 10.1016/j.neulet.2005.03.016.
Monastero R, Cefalu AB, Camarda C, Noto D, Camarda LK, Caldarella R, Imbornone E, Averna MR, Camarda R: Association of estrogen receptor alpha gene with Alzheimer's disease: a case-control study. J Alzheimers Dis. 2006, 9: 273-278.
Pirskanen M, Hiltunen M, Mannermaa A, Helisalmi S, Lehtovirta M, Hanninen T, Soininen H: Estrogen receptor beta gene variants are associated with increased risk of Alzheimer's disease in women. Eur J Hum Genet. 2005, 13: 1000-1006. 10.1038/sj.ejhg.5201447.
Bowen RL, Isley JP, Atkinson RL: An association of elevated serum gonadotropin concentrations and Alzheimer disease?. J Neuroendocrinol. 2000, 12: 351-354. 10.1046/j.1365-2826.2000.00461.x.
Short RA, Bowen RL, O'Brien PC, Graff-Radford NR: Elevated gonadotropin levels in patients with Alzheimer disease. Mayo Clin Proc. 2001, 76: 906-909.
Hogervorst E, Bandelow S, Combrinck M, Smith AD: Low free testosterone is an independent risk factor for Alzheimer's disease. Exp Gerontol. 2004, 39: 1633-1639. 10.1016/j.exger.2004.06.019.
Lukacs H, Hiatt ES, Lei ZM, Rao CV: Peripheral and intracerebroventricular administration of human chorionic gonadotropin alters several hippocampus-associated behaviors in cycling female rats. Horm Behav. 1995, 29: 42-58. 10.1006/hbeh.1995.1004.
Lei ZM, Rao CV: Novel presence of luteinizing hormone/human chorionic gonadotropin (hCG) receptors and the down-regulating action of hCG on gonadotropin-releasing hormone gene expression in immortalized hypothalamic GT1-7 neurons. Mol Endocrinol. 1994, 8: 1111-1121. 10.1210/me.8.8.1111.
Bowen RL, Verdile G, Liu T, Parlow AF, Perry G, Smith MA, Martins RN, Atwood CS: Luteinizing hormone, a reproductive regulator that modulates the processing of amyloid-beta precursor protein and amyloid-beta deposition. J Biol Chem. 2004, 279: 20539-20545. 10.1074/jbc.M311993200.
Casadesus G, Atwood CS, Bowen RL, Smith MA: Luteinizing hormone modulates cognition and amyloid-beta deposition in Alzheimer APP tansgenic mice. Biochem Biophys Acta. 2006
Liu T, Wimalasena J, Bowen RL, Atwood CS: Luteinizing hormone receptor mediates neuronal pregnenolone production via up-regulation of steroidogenic acute regulatory protein expression. J Neurochem. 2007, 100: 1329-1339. 10.1111/j.1471-4159.2006.04307.x.
Weill-Engerer S, David JP, Sazdovitch V, Liere P, Eychenne B, Pianos A, Schumacher M, Delacourte A, Baulieu EE, Akwa Y: Neurosteroid quantification in human brain regions: comparison between Alzheimer's and nondemented patients. J Clin Endocrinol Metab. 2002, 87: 5138-5143. 10.1210/jc.2002-020878.
Simpkins JW, Yang SH, Wen Y, Singh M: Estrogens, progestins, menopause and neurodegeneration: basic and clinical studies. Cell Mol Life Sci. 2005, 62: 271-280. 10.1007/s00018-004-4382-2.
Mahley RW: Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988, 240: 622-630. 10.1126/science.3283935.
Andersen OM, Willnow TE: Lipoprotein receptors in Alzheimer's disease. Trends Neurosci. 2006, 29: 687-694. 10.1016/j.tins.2006.09.002.
Themmen APN, Huhtaniemi IT: Mutations of gonadotropins and gonadotropin receptors: elucidating the physiology and pathophysiology of pituitary-gonadal function. Endocr Rev. 2000, 21: 551-583. 10.1210/er.21.5.551.
Huhtaniemi IT, Themmen AP: Mutations in human gonadotropin and gonadotropin-receptor genes. Endocrine. 2005, 26: 207-217. 10.1385/ENDO:26:3:207.
Furui K, Suganuma N, Tsukahara S, Asada Y, Kikkawa F, Tanaka M, Ozawa T, Tomoda Y: Identification of two point mutations in the gene coding luteinizing hormone (LH) beta-subunit, associated with immunologically anomalous LH variants. J Clin Endocrinol Metab. 1994, 78: 107-113. 10.1210/jc.78.1.107.
Takahashi K, Kurioka H, Ozaki T, Kanasaki H, Miyazaki K, Karino K: Pituitary response to luteinizing hormone-releasing hormone in women with variant luteinizing hormone. Eur J Endocrinol. 2000, 143: 375-381. 10.1530/eje.0.1430375.
Muller T, Gromoll J, Simoni M: Absence of exon 10 of the human luteinizing hormone (LH) receptor impairs LH, but not human chorionic gonadotropin action. J Clin Endocrinol Metab. 2003, 88: 2242-2249. 10.1210/jc.2002-021946.
Richter-Unruh A, Martens JW, Verhoef-Post M, Wessels HT, Kors WA, Sinnecker GH, Boehmer A, Drop SL, Toledo SP, Brunner HG, Themmen AP: Leydig cell hypoplasia: cases with new mutations, new polymorphisms and cases without mutations in the luteinizing hormone receptor gene. Clin Endocrinol (Oxf). 2002, 56: 103-112. 10.1046/j.0300-0664.2001.01437.x.
Ashley-Koch AE, Mei H, Jaworski J, Ma DQ, Ritchie MD, Menold MM, Delong GR, Abramson RK, Wright HH, Hussman JP, Cuccaro ML, Gilbert JR, Martin ER, Pericak-Vance MA: An analysis paradigm for investigating multi-locus effects in complex disease: examination of three GABA receptor subunit genes on 15q11-q13 as risk factors for autistic disorder. Ann Hum Genet. 2006, 70: 281-292. 10.1111/j.1469-1809.2006.00253.x.
Ghebremedhin E, Schultz C, Thal DR, Rub U, Ohm TG, Braak E, Braak H: Gender and age modify the association between APOE and AD-related neuropathology. Neurology. 2001, 56: 1696-1701.
Payami H, Zareparsi S, Montee KR, Sexton GJ, Kaye JA, Bird TD, Yu CE, Wijsman EM, Heston LL, Litt M, Schellenberg GD: Gender difference in apolipoprotein E-associated risk for familial Alzheimer disease: a possible clue to the higher incidence of Alzheimer disease in women. Am J Hum Genet. 1996, 58: 803-811.
Lehmann DJ, Butler HT, Warden DR, Combrinck M, King E, Nicoll JA, Budge MM, de Jager CA, Hogervorst E, Esiri MM, Ragoussis J, Smith AD: Association of the androgen receptor CAG repeat polymorphism with Alzheimer's disease in men. Neurosci Lett. 2003, 340: 87-90. 10.1016/S0304-3940(03)00069-7.
Smith MA, Perry G, Atwood CS, Bowen RL: Estrogen replacement and risk of Alzheimer disease. Jama. 2003, 289: 1100; author reply 1101-2.
Meethal SV, Smith MA, Bowen RL, Atwood CS: The gonadotropin connection in Alzheimer's disease. Endocrine. 2005, 26: 317-326. 10.1385/ENDO:26:3:317.
Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, Schmechel D, Saunders AM, Goldgaber D, Roses AD: Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993, 90: 8098-8102. 10.1073/pnas.90.17.8098.
Fazekas F, Enzinger C, Ropele S, Schmidt H, Schmidt R, Strasser-Fuchs S: The impact of our genes: consequences of the apolipoprotein E polymorphism in Alzheimer disease and multiple sclerosis. J Neurol Sci. 2006, 245: 35-39. 10.1016/j.jns.2005.08.018.
Zhang G, Curtiss LK, Wade RL, Dyer CA: An apolipoprotein E synthetic peptide selectively modulates the transcription of the gene for rat ovarian theca and interstitial cell P450 17alpha-hydroxylase, C17-20 lyase. J Lipid Res. 1998, 39: 2406-2414.
Foster JD, Strauss JF, Paavola LG: Cellular events involved in hormonal control of receptor-mediated endocytosis: regulation occurs at multiple sites in the low density lipoprotein pathway, including steps beyond the receptor. Endocrinology. 1993, 132: 337-350. 10.1210/en.132.1.337.
Milne RL, Ribas G, Gonzalez-Neira A, Fagerholm R, Salas A, Gonzalez E, Dopazo J, Nevanlinna H, Robledo M, Benitez J: ERCC4 Associated with Breast Cancer Risk: A Two-Stage Case-Control Study Using High-throughput Genotyping. Cancer Res. 2006, 66: 9420-9427. 10.1158/0008-5472.CAN-06-1418.
Yu Y, Panhuysen C, Kranzler HR, Hesselbrock V, Rounsaville B, Weiss R, Brady K, Farrer LA, Gelernter J: Intronic variants in the dopa decarboxylase (DDC) gene are associated with smoking behavior in European-Americans and African-Americans. Hum Mol Genet. 2006, 15: 2192-2199. 10.1093/hmg/ddl144.
Lin SL, Miller JD, Ying SY: Intronic MicroRNA (miRNA). J Biomed Biotechnol. 2006, 2006: 26818-
Wessagowit V, Nalla VK, Rogan PK, McGrath JA: Normal and abnormal mechanisms of gene splicing and relevance to inherited skin diseases. J Dermatol Sci. 2005, 40: 73-84. 10.1016/j.jdermsci.2005.05.006.
Matlin AJ, Clark F, Smith CW: Understanding alternative splicing: towards a cellular code. Nat Rev Mol Cell Biol. 2005, 6: 386-398. 10.1038/nrm1645.
Churbanov A, Rogozin IB, Deogun JS, Ali H: Method of predicting splice sites based on signal interactions. Biol Direct. 2006, 1: 10-10.1186/1745-6150-1-10.
Lewis P, Zaykin D: Computer program for the analysis of allelic data. Version 1.0(d16c). 2007, [http://hydrodictyon.eeb.uconn.edu/people/plewis/software.php]
Minitab: Minitab v 14.0 State College, PA.
Lancaster A, Nelson MP, Meyer D, Thomson G, Single RM: PyPop: a software framework for population genomics: analyzing large-scale multi-locus genotype data. Pac Symp Biocomput. 2003, 514-525.
Hahn LW, Ritchie MD, Moore JH: Multifactor dimensionality reduction software for detecting gene-gene and gene-environment interactions. Bioinformatics. 2003, 19: 376-382. 10.1093/bioinformatics/btf869.
Thornton-Wells TA, Moore JH, Haines JL: Genetics, statistics and human disease: analytical retooling for complexity. Trends Genet. 2004, 20: 640-647. 10.1016/j.tig.2004.09.007.
Peduzzi P, Concato J, Kemper E, Holford TR, Feinstein AR: A simulation study of the number of events per variable in logistic regression analysis. J Clin Epidemiol. 1996, 49: 1373-1379. 10.1016/S0895-4356(96)00236-3.
Ritchie MD, Hahn LW, Roodi N, Bailey LR, Dupont WD, Parl FF, Moore JH: Multifactor-dimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer. Am J Hum Genet. 2001, 69: 138-147. 10.1086/321276.
Moore JH: The ubiquitous nature of epistasis in determining susceptibility to common human diseases. Hum Hered. 2003, 56: 73-82. 10.1159/000073735.
Coffey CS, Hebert PR, Ritchie MD, Krumholz HM, Gaziano JM, Ridker PM, Brown NJ, Vaughan DE, Moore JH: An application of conditional logistic regression and multifactor dimensionality reduction for detecting gene-gene interactions on risk of myocardial infarction: the importance of model validation. BMC Bioinformatics. 2004, 5: 49-10.1186/1471-2105-5-49.
Motsinger AA, Ritchie MD: The effect of reduction in cross-validation intervals on the performance of multifactor dimensionality reduction. Genet Epidemiol. 2006, 30: 546-555. 10.1002/gepi.20166.
Moore JH, Williams SM: New strategies for identifying gene-gene interactions in hypertension. Ann Med. 2002, 34: 88-95. 10.1080/07853890252953473.
Williams SM, Addy JH, Phillips JA, Dai M, Kpodonu J, Afful J, Jackson H, Joseph K, Eason F, Murray MM, Epperson P, Aduonum A, Wong LJ, Jose PA, Felder RA: Combinations of variations in multiple genes are associated with hypertension. Hypertension. 2000, 36: 2-6.
Jiang M, Lamminen T, Pakarinen P, Hellman J, Manna P, Herrera RJ, Huhtaniemi I: A novel Ala(-3)Thr mutation in the signal peptide of human luteinizing hormone beta-subunit: potentiation of the inositol phosphate signalling pathway and attenuation of the adenylate cyclase pathway by recombinant variant hormone. Mol Hum Reprod. 2002, 8: 201-212. 10.1093/molehr/8.3.201.
Atger M, Misrahi M, Sar S, Le Flem L, Dessen P, Milgrom E: Structure of the human luteinizing hormone-choriogonadotropin receptor gene: unusual promoter and 5' non-coding regions. Mol Cell Endocrinol. 1995, 111: 113-123. 10.1016/0303-7207(95)03557-N.
Kremer H, Martens JW, van Reen M, Verhoef-Post M, Wit JM, Otten BJ, Drop SL, Delemarre-van de Waal HA, Pombo-Arias M, De Luca F, Potau N, Buckler JM, Jansen M, Parks JS, Latif HA, Moll GW, Epping W, Saggese G, Mariman EC, Themmen AP, Brunner HG: A limited repertoire of mutations of the luteinizing hormone (LH) receptor gene in familial and sporadic patients with male LH-independent precocious puberty. J Clin Endocrinol Metab. 1999, 84: 1136-1140. 10.1210/jc.84.3.1136.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/9/37/prepub
Acknowledgements
This research was supported in part by the Office of Research and Development, Department of Veteran Affairs and the Alzheimer's Association. We thank our colleagues Miguel J. Gallego, Andrea C. Wilson, Prashob Porayette and Jacob Basson for their valuable comments. This is Geriatrics Research, Education and Clinical Core VA paper number 2008-10.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
RJH designed the study, collected the samples, performed the genotyping, analyzed the data, performed the statistical analyses and drafted the manuscript. MRA performed genotyping and data analysis. SVM and RLB helped to draft the manuscript. CEG, SCJ and SA provided DNA samples from confirmed AD and control individuals that they characterized. CSA conceived the study, participated in its design and coordination and helped to draft the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Haasl, R.J., Ahmadi, M.R., Meethal, S.V. et al. A luteinizing hormone receptor intronic variant is significantly associated with decreased risk of Alzheimer's disease in males carrying an apolipoprotein E ε4 allele. BMC Med Genet 9, 37 (2008). https://doi.org/10.1186/1471-2350-9-37
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-9-37