
Abstract
Background
Grebe-type chondrodysplasia (GCD) is a rare autosomal recessive syndrome characterized by severe acromesomelic limb shortness with non-functional knob like fingers resembling toes. Mutations in the cartilage-derived morphogenetic protein 1 (CDMP1) gene cause Grebe-type chondrodysplasia.
Methods
Genotyping of six members of a Pakistani family with Grebe-type chondrodysplasia, including two affected and four unaffected individuals, was carried out by using polymorphic microsatellite markers, which are closely linked to CDMP1 locus on chromosome 20q11.22. To screen for a mutation in CDMP1 gene, all of its coding exons and splice junction sites were PCR amplified from genomic DNA of affected and unaffected individuals of the family and sequenced directly in an ABI Prism 310 automated DNA sequencer.
Results
Genotyping results showed linkage of the family to CDMP1 locus. Sequence analysis of the CDMP1 gene identified a novel four bases insertion mutation (1114insGAGT) in exon 2 of the gene causing frameshift and premature termination of the polypeptide.
Conclusion
We describe a 4 bp novel insertion mutation in CDMP1 gene in a Pakistani family with Grebe-type chondrodysplasia. Our findings extend the body of evidence that supports the importance of CDMP1 in the development of limbs.
Similar content being viewed by others


Background
Grebe-type chondrodysplasia is a rare autosomal recessive syndrome characterized by severe acromesomelia, dwarfism, severe micromelia with increasing severity in a proximo-distal gradient and deformation of upper and lower limbs [1]. Radiologically, it is characterized by extremely short limbs, the legs being more severely affected than arms. The hands are extremely short with toe-like fingers, and the feet are in the valgus position [2]. Short and deformed middle long bones, fusion of carpal bones and several metacarpal and metatarsal, and absence of proximal and middle phalanges have been observed in the affected individuals. Other features include obesity and delayed mental development, but facial appearance and intelligence are normal with no vertebral abnormalities. Grebe-type chondrodysplasia is caused by mutations in the cartilage derived bone morphogenetic protein1 (CDMP1), located on chromosome 20q11.22. The CDMP1 gene is predominantly expressed in cartilaginous tissues of the developing long bones and the more distal elements of the appendicular skeleton that develop from the budding limb [3].
The CDMP1 has a murine homologue growth and differentiation factor-5 (Gdf5) and its absence leads to brachypodism in mice [4] and a number of skeletal malformation syndrome in humans including brachydactyly type A2 [5, 6], brachydactyly type C (BDC) [7–10], fibular hypoplasia and complex brachydactyly (Du Pan syndrome) [11], Grebe-type chondrodysplasia [2, 12–14], Hunter-Thompson type acromesomelic dysplasia [15], angel-shaped phalangeal dysplasia [16], multiple synostoses syndrome type 1 [17] and proximal symphalangism (SYM1) [5, 10]. Recently, +104T/C (rs143383), a single nucleotide polymorphism in the untranslated region of the CDMP1 gene, was found to show significant association with osteoarthritis in Asian and European populations [18, 19].
In the present study, we have ascertained a Pakistani family demonstrating autosomal recessive form of Grebe-type chondrodysplasia. DNA sequence analysis identified a novel four base insertion in CDMP1 gene.
Methods
Research subjects
For the present study a Pakistani family with autosomal recessive Grebe-type chondrodysplasia (Fig. 1) was investigated. Prior to start of the study approval was obtained from the Quaid-i-Azam University Institutional Review Board. Informed consent was obtained from all subjects participating in the study. All affected and normal individuals underwent examination at Department of Diagnostic Imaging, Pakistan Institute of Medical Sciences, Islamabad. Blood samples were obtained from two affected (IV-1, IV-4) and four unaffected members (III-I, III-2, IV-3, IV-5) of the family.

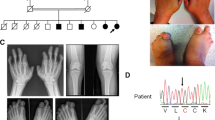
Pedigree of a Pakistani family with Grebe type chondrodysplasia. Open squares and circles represent unaffected males and females, respectively. Filled squares and circles represent affected individuals. Double lines between symbols are representative of consanguineous unions.
DNA extraction and genotyping
Genomic DNA was isolated from peripheral blood following a standard protocol and diluted to 40 ng/ul for amplification by polymerase chain reaction (PCR). The amplification of microsatellite markers was performed according to standard procedure in a total volume of 25 μl, containing 40 ng genomic DNA, 20 pmol of each primer, 200 μM of each deoxyribonucleoside triphosphates (dNTP), 1 unit of Taq DNA polymerase and 2.5 μl reaction buffer (MBI Fermentas, York, UK). The thermal cycling conditions used included 95°C for 1 min, followed by 35 cycles of 95°C for 1 min, 57°C for 1 min, 72°C for 1 min, and final extension at 72°C for 10 minutes. PCR products were resolved on 8% non-denaturing polyacrylamide gel, stained with ethidium bromide and genotypes were assigned by visual inspection.
Due to the autosomal recessive mode of inheritance and clinical features of the patients compatible with Grebe-type chondrodysplasia, the family was tested for linkage using microsatellite markers (D20S843, D20S195, D20S909, D20S865, D20S847, D20S834, D20S884) linked to CDMP1 gene on chromosome 20q11.22.
Mutation detection
To search for an underlying mutation in CDMP1 gene, exons and splice junctions of the gene were amplified by PCR from genomic DNA using primers designed from intronic sequences of the gene. After PCR-amplified products were purified with commercially available kits (Marligen Biosciences, USA), sequencing was performed using DTCS-Quick start sequencing Kit (Beckman Coulter, USA) together with CQ8800 DNA sequencer (Beckman Coulter, USA). To amplify 440 bp PCR fragment encompassing part of exon 2 of the CDMP1 gene the following primers were used.
5'-GCTGGGAGGTGTTCGACATCT-3' (Exon 2, Sense)
5'-GCACTCCTGGAATCACAGAGG-3' (Exon 2, Antisense)
Results
Clinical findings
In the affected individuals of the family, presented here, abnormal findings were limited to the limbs (Fig 2). In the upper limbs, both arms were short, with forearms relatively shorter and bowed. The hands were very short and the fingers were replaced by globular appendages. In the lower limbs, the proximal segments were short and the middle segments were relatively shorter. In the feet the toes were small and overriding with limited movement. Polydactyly was observed in both hands.

Clinical features of the Grebe-type chondrodysplasia. A 10 year's old affected member (IV-4) of the family (a), feet (b) and hands with hexadyctyly (c, d) of the same individual.
Radiographic examination of the upper limbs revealed that radius was malformed and the radial head was displaced upward out of the elbow joint. Pruning of the ulna was noted with sharp distal edges (Fig 3). Carpal bones were distorted and hypoplastic, and metacarpals were completely missing. Distal phalanges were present however proximal and middle phalanges were absent. In the lower limb femoral and tibial bones were short with the absence of tibial plateau. Fibulas were completely absent. Thoracic spines, bones and skull appeared normal.

Radiographs of the 10 year's old affected boy (IV-4) with Grebe type chondrodysplasia. Radiographs of hand (a), arm (b), pelvis (c) and foot (d).
Genotyping
To identify the underlying autosomal recessive Grebe-type chondrodysplasia in the family, genotyping was performed with microsatellite markers closely linked to CDMP1 gene on chromosome 20q11.22. The affected members of the family were homozygous for four markers (D20S843, D20S195, D20S865, D20S847), thus suggesting linkage of the family to CDMP1 gene.
Mutation analysis
The entire coding portion and intron-exon boundaries of CDMP1 gene was sequenced in 6 individuals including two affected (IV-1 and IV-4) of the family. Sequence analysis of exon 2 of the CDMP1 gene from affected individual revealed a 4-bp insertion starting at nucleotide position 1114 (1114insGAGT) (Fig. 4a) resulting in immediate stop codon. This insertion was present in the heterozygous state in obligate carriers III-1 and III-2 (Fig. 4b). The mutation was not identified in normal individuals of the family (Fig. 4c). To ensure that the mutation does not represent a neutral polymorphism in this population, a panel of 40 unrelated unaffected ethnically matched control individuals was screened for the mutation and it was not identified outside the family.

Sequence analysis of the CDMP1 gene mutation. DNA sequence of exon 2 of CDMP1 gene from (a) an affected individual (b) a heterozygous carrier and (c) a control individual. The bar in panel (a) represents the sequence that was inserted in the homozygous state in the affected individuals.
Discussion
In the present investigation we have described a second Pakistani family with Grebe-type chondrodysplasia. The phenotype of the present family shares clinical and radiographic features with three families with Grebe-type chondrodysplasia reported earlier [2, 12, 13]. The Pakistani family, presented here, displayed severe shortening of both upper and lower limbs. Hexadactyly was observed in both hands of the affected individuals. Radiographic examination of the affected individuals showed severely deformed tibiae, ulnae and radii. Fibulae were completely absent in the affected individuals. Physically apparent clinical defects were not seen in any of the obligate carriers of the family. Lack of availability of skeletal radiographs of the obligate carriers makes it difficult to exclude subtle skeletal deformities which were not detected by physical examination.
To date, eighteen pathogenic mutations in the CDMP1 gene have been identified including three in patients affected with Grebe type Chondrodysplasia [2, 12, 13]. Cartilage-derived morphogenetic protein-1 (CDMP1), a secreted protein, is a member of the bone morphogenetic protein (BMP) family and the transforming growth factor-beta (TGF-β) superfamily [3, 20, 21]. The CDMP1 precursor polypeptide contains 501 amino acids with a RRKRR polybasic proteolytic processing site at amino acids 377–381 and seven highly conserved cystiene at its C-terminus. It forms homodimers or heterodimers with other BMP partners linked by a single inter-chain disulfide bond. These dimers undergo proteolysis, producing an active mature CDMP1 dimer that is secreted from the cell [20]. The CDMP1 is a ligand of BMP receptors (Types 1 and 2) resulting in an activation of BMPR1B by transphosphorylation through BMPR2. The transmembrane serine/threonine kinase receptor BMPR1b subsequently activates the SMAD-dependent pathway and/or the p38 MAP kinase pathway. Both signal cascades regulate the transcription of specific target genes that are involved in bone formation [22–24].
In the family with Grebe type Chondrodysplasia, presented here, sequence analysis of the CDMP1 gene detected a homozygous 4-bp insertion mutation (1114insGAGT) in both the affected individuals. This mutation is predicted to produce a truncated protein consisting of 371 amino acids and lacking an active domain, which is expected to abolish the subsequent signaling pathways in CDMP1 target cells.
The CDMP1/GDF5 is expressed predominantly in developing limbs at joint interzones, and is known to play an important role in joint formation [2, 3]. The expression pattern of GDF5 at joint interzones suggested a possible role for CDMP1/GDF5 in establishing boundaries between skeletal elements [25]. As suggested by Francis et al. [26] that the interzones can be regarded as a signaling center, regulating chondrocytes proliferation and differentiation and orchestrating joint formation. Absence of CDMP1/GDF5 results in abnormal joint formation and ligament defects.
Conclusion
We have reported the fourth mutation in CDMP1 gene, which results in autosomal recessive Grebe-type chondrodysplasia. The study presented here confirms that CDMP1 plays an important role in the regulation of the development skeleton, and its absence does not impair other development processes.
Consent
Written consent was provided by father of the patients for publishing photographs and other material.
Abbreviations
- CDMP1 :
-
cartilage-derived morphogenetic protein 1
- GDF5 :
-
growth and differentiation factor 5
- BMP:
-
bone morphogenetic protein
- BMPR:
-
bone morphogenetic protein receptor
- TGF:
-
transforming growth factor
- DTCS:
-
dye terminator cycle sequencing.
References
Grebe H: Die Achondrogenesis: ein einfach rezessives Erbmerkmal. Folia Hered Pathol (Milano). 1952, 2: 23-28.
Thomas JT, Kilpatrick MW, Lin K: Disruption of human limb morphogenesis by a dominant negative mutation in CDMP1. Nat Genet. 1997, 17: 58-64. 10.1038/ng0997-58.
Storm EE, Kingsley DM: GDF5 coordinates bone and joint formation during digit development. Dev Biol. 1999, 209: 11-27. 10.1006/dbio.1999.9241.
Storm EE, Huynh TV, Copeland NG, Jenkins NA, Kingsley DM, Lee SJ: Limb alterations in brachypodism mice due to mutations in a new member of the TGF beta-superfamily. Nature. 1994, 368: 639-643. 10.1038/368639a0.
Seemann P, Schwappacher R, Kjaer KW, Krakow D, Lehmann K, Dawson K, Stricker S, Pohl J, Ploger F, Staub E, Nickel J, Sebald W, Knaus P, Mundlos S: Activating and deactivating mutations in the receptor interaction site of GDF5 cause symphalangism or brachydactyly type A2. J Clin Invest. 2005, 115: 2373-2381. 10.1172/JCI25118.
Ploger F, Seemann P, Kegler MS, Lehmann K, Seidel J, Kjaer KW, Pohl J, Mundlos S: Brachydactyly type A2 associated with a defect in proGDF5 processing. Hum Mol Genet. 2008, 217: 1222-1233. 10.1093/hmg/ddn012.
Polinkovsky A, Robin NH, Thomas JT: Mutations in CDMP1 cause autosomal dominant brachydactyly type C. Nat Genet. 1997, 17: 18-19. 10.1038/ng0997-18.
Everman DB, Bartels CF, Yang Y, Yanamandra N, Goodman FR, Mendoza-Londono JR, Savarirayan R, White SM, Graham JM, Gale RP, Svarch E, Newman WG, Kleckers AR, Francomano CA, Govindaiah V, Singh L, Morrison S, Thomas JT, Warman ML: The mutational spectrum of brachydactyly type C. Am J Med Genet. 2002, 112: 291-296. 10.1002/ajmg.10777.
Savarirayan R, White SM, Goodman FR, Graham JM, Delatycki MB, Lachman RS, Rimoin DL, Everman DB, Warman ML: Broad phenotypic spectrum caused by an identical heterozygous CDMP-1 mutation in three unrelated families. Am J Med Genet A. 2003, 117: 136-142. 10.1002/ajmg.a.10924.
Yang W, Cao L, Liu W, Jiang L, Sun M, Zhang D, Wang S, Lo HYW, Luo Y, Zhang X: Novel point mutations in GDF5 associated with two distinct limb malformations in Chinese: brachydactyly type C and proximal symphalangism. J Hum Genet. 2008, 53: 368-374. 10.1007/s10038-008-0253-7.
Faiyaz-Ul-Haque M, Ahmad W, Zaidi SHE, Haque S, Teebi AS, Ahmad M, Cohn DH, Tsui LC: Mutation in the cartilage-derived morphogenetic protein-1 (CDMP1) gene in a kindred affected with fibular hypoplasia and complex brachyactyly (Du Pan syndrome). Clin Genet. 2002, 61: 454-458. 10.1034/j.1399-0004.2002.610610.x.
Faiyaz-Ul-Haque M, Ahmad W, Wahab A, Haque S, Azim AC, Zaidi SHE, Teebi AS, Ahmad M, Cohn DH, Siddique T, Tsui LC: Frameshift mutation in the cartilage-derived morphogenetic protein-1 (CDMP1) gene and severe acromesomelic chondrodysplasia resembling grebe type chondrodysplasia. Am J Med Genet. 2002, 111: 31-37. 10.1002/ajmg.10501.
Al-Yahyaee SA, Al-Kindi MN, Habbal O, Kumar DS: Clinical and molecular analysis of Grebe acromesomelic dysplasia in an Omani family. Am J Med Genet. 2003, 121: 9-14. 10.1002/ajmg.a.20256.
Stelzer C, Winterpacht A, Spranger J, Zabel B: Grebe dysplasia and the spectrum of CDMP1 mutations. Pediatr Pathol Mol Med. 2003, 22: 77-85. 10.1080/15227950307698.
Thomas JT, Lin K, Nandedkar M, Camarqo M, Cervenka J, Luyten FP: A human chondrodysplasia due to a mutation in TGF-β superfamily member. Nat Genet. 1996, 12: 315-317. 10.1038/ng0396-315.
Holder-Espinasse M, Escande F, Mayrargue E, Dieux-Coeslier A, Fron D, Doual-Bisser A, Boute-Benejean O, Robert Y, Porchet N, Manouvrier-Hanu S: Angel shaped phalangeal dysplasia, hip dysplasia, and positional teeth abnormalities are part of the brachydactyly C spectrum associated with CDMP-1 mutations. J Med Genet. 2004, 41: e78-10.1136/jmg.2003.013904.
Dawson K, Seeman P, Sebald E, King L, Edwards M, Williams J, Mundlos S, Krakow D: GDF5 is a second locus for multiple-synostosis syndrome. Am J Hum Genet. 2006, 78: 708-712. 10.1086/503204.
Miyamoto Y, Mabuchi A, Shi D, Kubo T, Takatori Y, Saito S, Fujioka M, Sudo A, Uchida A, Yamamoto S, Ozaki K, Takigawa M, Tanaka T, Nakamura Y, Jiang Q, Ikegawa S: A functional polymorphism in the 50 UTR of GDF5 is associated with susceptibility to osteoarthritis. Nat Genet. 2007, 39: 529-533. 10.1038/2005.
Southam L, Rodriguez-Lopez J, Wilkins JM, Pombo-Suarez M, Snelling S, Gomez-Reino JJ, Chapman K, Gonzalez A, Loughlin J: An SNP in the 50-UTR of GDF5 is associated with osteoarthritis susceptibility in Europeans and with in vivo differences in allelic expression in articular cartilage. Hum Mol Genet. 2007, 16: 2226-2232. 10.1093/hmg/ddm174.
Luyten FP: Cartilage-derived morphogenetic protein-1. Int J Biochem Cell Biol. 1997, 29: 1241-1244. 10.1016/S1357-2725(97)00025-3.
Chang SC, Hoang B, Thomas JT, Vukicevic S, Luyten FP, Ryba NJ, Kozak CA, Reddi AH, Moos M: Cartilage-derived morphogenetic proteins: new members of the transforming growth factor-β superfamily predominantly expressed in long bones during human embryonic development. J Biol Chem. 1994, 269: 28227-28234.
Gilboa L, Nohe A, Geissendorfer T, Sebald W, Henis YI, Knaus P: Bone morphogenetic protein receptor complexes on the surface of live cells: a new oligomerization mode for serine/threonine kinase receptors. Mol Biol Cell. 2000, 11: 1023-1035.
von Bubnoff A, Cho K: Intracellular BMP signaling regulation in vertebrates: pathway or network?. Dev Biol. 2001, 239: 1-14. 10.1006/dbio.2001.0388.
Nohe A, Hassel S, Ehrlich M: The mode of bone morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. J Biol Chem. 2002, 277: 5330-5338. 10.1074/jbc.M102750200.
Settle SH, Rountree RB, Sinha A, Thacker A, Higgins K, Kingsley DM: Multiple joint and skeletal patterning defects caused by single and double mutations in the mouse Gdf6 and Gdf5 genes. Dev Biol. 2003, 254: 116-130. 10.1016/S0012-1606(02)00022-2.
Francis-West PH, Abdelfattah A, Chen P, Allen C, Parish J, Ladher R, Allen S, MacPherson S, Luyten FP, Archer CW: Mechanisms of GDF-5 action during skeletal development. Development. 1999, 126: 1305-1315.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/9/102/prepub
Acknowledgements
We sincerely wish to thank the family members for their participation. The work presented here was funded by the Higher Education Commission (HEC), Islamabad, Pakistan. Sulman Basit is supported by indigenous PhD fellowship from HEC, Islamabad, Pakistan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SB participated in the design of the study, performed PCR, gene sequencing and manuscript writing. SKN, NW, GA, MA studied family, collected blood samples and extracted DNA. WA analyzed the data, participated in manuscript preparation and collected funds for the study. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Basit, S., Naqvi, S.KuH., Wasif, N. et al. A novel insertion mutation in the cartilage-derived morphogenetic protein-1 (CDMP1) gene underlies Grebe-type chondrodysplasia in a consanguineous Pakistani family. BMC Med Genet 9, 102 (2008). https://doi.org/10.1186/1471-2350-9-102
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-9-102