
Abstract
Background
Numerous studies suggest genetic influences on sleepiness and circadian rhythms. The Sleep Heart Health Study collected questionnaire data on sleep habits and sleepiness from 2848 Framingham Heart Study Offspring Cohort participants. More than 700 participants were genotyped using the Affymetrix 100K SNP GeneChip, providing a unique opportunity to assess genetic linkage and association of these traits.
Methods
Sleepiness (defined as the Epworth Sleepiness Scale score), usual bedtime and usual sleep duration were assessed by self-completion questionnaire. Standardized residual measures adjusted for age, sex and BMI were analyzed. Multipoint variance components linkage analysis was performed. Association of SNPs to sleep phenotypes was analyzed with both population-based and family-based association tests, with analysis limited to 70,987 autosomal SNPs with minor allele frequency ≥10%, call rate ≥80%, and no significant deviation from Hardy-Weinberg equilibrium (p ≥ 0.001).
Results
Heritability of sleepiness was 0.29, bedtime 0.22, and sleep duration 0.17. Both genotype and sleep phenotype data were available for 749 subjects. Linkage analysis revealed five linkage peaks of LOD >2: four to usual bedtime, one to sleep duration. These peaks include several candidate sleep-related genes, including CSNK2A2, encoding a known component of the circadian molecular clock, and PROK2, encoding a putative transmitter of the behavioral circadian rhythm from the suprachiasmatic nucleus. Association tests identified an association of usual bedtime with a non-synonymous coding SNP in NPSR1 that has been shown to encode a gain of function mutation of the neuropeptide S receptor, whose endogenous ligand is a potent promoter of wakefulness. Each copy of the minor allele of this SNP was associated with a 15 minute later mean bedtime. The lowest p value was for association of sleepiness with a SNP located in an intron of PDE4D, which encodes a cAMP-specific phosphodiesterase widely expressed in human brain. Full association results are posted at http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?id=phs000007.
Conclusion
This analysis confirms prior reports of significant heritability of sleepiness, usual bedtime, and usual sleep duration. Several genetic loci with suggestive linkage to these traits are identified, including linkage peaks containing circadian clock-related genes. Association tests identify NPSR1 and PDE4D as possible mediators of bedtime and sleepiness.
Similar content being viewed by others

Background
Daytime sleepiness is a common symptom, experienced at least 3 days per week by 29% of respondents in a recent poll of U.S. adults [1]. Sleepiness is a major cause of motor vehicle and occupational accidents, impaired social function, and reduced quality of life. Within individuals, the level of sleepiness is modulated by a combination of homeostatic (duration of wakefulness) and circadian (time of day) factors [2]. While behavioral factors and sleep disorders contribute to daytime sleepiness, there is great individual variability in the susceptibility to sleepiness in the context of disorders of sleep fragmentation [3] or sleep deprivation [4], which appears to be a stable individual trait. Evidence from several studies indicates that excessive sleepiness is heritable, with heritability estimates from recent twin studies in the range of 0.38–0.48 [5–7]. Persistent circadian rhythm disorders, such as advanced or delayed sleep phase syndrome, are relatively uncommon, estimated to affect <1% of the adult population [8]. However, individual differences in diurnal preference (morning types or "larks" versus evening types or "owls") have important implications for work scheduling and performance that are highly relevant in an economy in which almost one-fifth of employees are engaged in shift work [9]. Both twin and family studies suggest that diurnal preference is heritable, with heritability estimates of 0.23–0.47 for usual bedtime or more formal assessment of diurnal preference [10–12]. Usual sleep duration is an important determinant of daytime sleepiness; moreover, both short and long sleep duration have been associated in numerous epidemiologic studies with hypertension [13], diabetes mellitus [14, 15], coronary heart disease [16] and mortality [17–19], although the mechanisms underlying these associations are poorly understood. Significant heritability of usual sleep duration has been reported, with heritability estimates of 0.40–0.44 [10, 20].
While sleepiness, diurnal preference, and sleep duration have long been recognized as heritable traits, the genetic basis of this heritability is largely unknown. While it has been suggested that heritability of sleepiness may reflect genetic influences on sleep-disordered breathing [5], sleep drive is itself a highly regulated phenomenon and may be influenced by variations in the numerous genes involved in the circadian and homeostatic regulation of sleep and wakefulness. For example, a polymorphism in the gene encoding adenosine deaminase is reportedly associated with an increase in slow-wave sleep, a marker of homeostatic sleep drive [21]. Similarly, a polymorphism in HCRTR2, the gene encoding the orexin/hypocretin receptor 2, has been identified in 2 patients with idiopathic hypersomnolence but in no non-sleepy controls [22]. A polymorphism in the gene encoding this receptor is known to cause autosomal dominant canine narcolepsy [23, 24], although the role of polymorphisms in genes of the orexin/hypocretin system in human narcolepsy or daytime sleepiness in general remains uncertain. Polymorphisms in the human period 2 (PER2) and casein kinase 1d (CSNK1D) genes, known elements of the circadian molecular clock, are associated with autosomal dominant advanced sleep phase syndrome in isolated families [25, 26]. A polymorphism in the 3'-untranslated region of the CLOCK gene has been inconsistently reported in association with evening preference [27–29] and a length polymorphism in a tandem repeat region of the period 3 protein, containing either 4 or 5 repeats of an 18-amino acid motif, is reportedly associated with diurnal preference [30]. These examples notwithstanding, it appears that sleepiness and diurnal preference are polygenic traits. In a genome-wide linkage analysis of C57BL/6J × BALB/cJ hybrid mice, 14 loci were identified that were significantly linked to circadian phenotypes; only one of these loci was near a gene proposed to be part of the core mammalian circadian clock [31]. We are unaware of any published genome-wide linkage or association studies of daytime sleepiness, diurnal preference, or sleep duration in humans.
The present study takes advantage of sleep phenotype data collected by the Framingham Heart Study at the Offspring Cycle 6 Examination. These data include measures of daytime sleepiness, usual bedtime, usual sleep duration, and sleep-disordered breathing. The aims of this study were to replicate, in this unselected family-based sample, prior reports of heritability of these traits and to conduct genome-wide linkage and association studies of these traits. Although sleep phenotype data were collected from fewer than half of all Offspring participants, heritability of the sleep-related phenotypes was confirmed in this sample and preliminary linkage and association studies identified several loci of interest.
Methods
Subjects of this study are drawn from the 2848 Framingham Offspring Study participants who completed sleep habits questionnaires between 1995 and 1998 (Offspring Examination Cycle 6) for the Sleep Heart Health Study, a longitudinal study of the cardiovascular consequences of sleep-disordered breathing that has been described elsewhere [32]. Of these subjects, 891 members of 371 pedigrees had biological relatives with valid sleep phenotype data and thus contributed to the heritability analyses. Genome-wide SNP genotyping was performed in the "Family Plate Set" of 1345 members of the largest nuclear families participating in the Framingham Original and Offspring cohorts using an Affymetrix 100K SNP GeneChip, as described in the Framingham Heart Study 100K Project Overview [33]. A maximum of 738 members of 203 families contributed informative data to each of the genetic association analyses of sleep phenotypes.
Data on daytime sleepiness, usual bedtime, and usual sleep duration were obtained from a self-completion questionnaire either handed to the participant at the time of a regularly scheduled visit to the Framingham Study clinic or mailed to the participant. Sleepiness was defined as the score on the Epworth Sleepiness Scale, a widely used and well validated 8-item questionnaire that asks the likelihood of falling asleep in a variety of commonly encountered situations [34, 35]. Usual bedtime was obtained from the single question, "What time to you usually go to bed on weeknights (or work nights)?" Usual sleep duration was obtained from the single question, "How many hours of sleep do you usually get on weeknights (or work nights)?" with integer response options. The full Sleep Habits Questionnaire is available from the Sleep Heart Health Study website [36]. Data on work shift and retirement status were not available in this cohort; therefore, in order to exclude subjects in whom night shift work might lead to spurious estimates of circadian phenotype, the 0.5% of subjects reporting a usual bedtime between 5 AM and 6 PM were excluded from analyses of usual bedtime and usual sleep duration. Those whose usual bedtime differed by more than two hours between weekdays and weekends (1.3%) were excluded from analyses of usual bedtime, as behavioral factors were presumed to have a major influence on this measure. Similarly, those whose usual sleep duration differed by more than two hours between weekdays and weekends (4.2%) were excluded from analyses of sleepiness and usual sleep duration. As sleepiness, bedtime, and sleep duration may be influenced by age, sex and BMI [5, 10, 20], adjustment for these variables was made by linear regression. Standardized residuals of the adjusted sleep phenotype variables were used in genetic analyses. As further adjustment of sleepiness for usual sleep duration or self-reported symptoms of sleep-disordered breathing (snoring, nocturnal breathing pauses) reduced sample size but had little impact on the linkage and association results, analyses using these further adjustments are not presented. Standardized residuals were obtained using PROC REG in the SAS statistical software package (SAS version 9.1, SAS Institute, Cary, NC).
Heritability, linkage and genetic association analysis was performed as described in the Framingham Heart Study 100K Project Overview [33]. Briefly, multipoint linkage analysis was implemented using Merlin identity-by-descent estimates and variance component linkage in SOLAR with a subset of 10,588 SNPs supplementing 612 microsatellite markers from a previous genome scan. Association of SNPs to sleep phenotypes was studied with both population-based association tests using generalized estimating equations (GEE) and family-based association tests (FBAT). All association tests employed an additive model. Reported association test results are limited to SNPs located on autosomes and meeting the following quality control criteria: minor allele frequency of ≥10%; call rate ≥80%; and no significant deviation from Hardy-Weinberg equilibrium (p ≥ 0.001). Of the 70,987 SNPs meeting these criteria, 40,249 were located within 60 kb of a known or putative gene. Physical locations are based on National Center for Biotechnology Information build 35.
Of the 2848 Offspring Study participants who completed the sleep habits questionnaire, 699 also underwent overnight polysomnography; therefore, polysomnographic data on sleep-disordered breathing were available in a small subset of subjects included in the Family Plate Set (n = 219). There were too few subjects to permit linkage analysis and, as power was very low for association studies, these data are not considered in this manuscript but are posted on the web site. The correlation of sleepiness, bedtime, and sleep duration with polysomnographically measured apnea-hypopnea index was weak (correlation coefficients -0.07 to 0.10).
Results
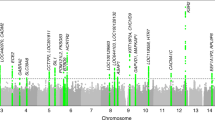
Sleep characteristics of the 749 subjects included in the Framingham Study Family Plate Set with available sleep phenotype data are shown in Table 1. Mean age was 55.8 (SD 9.4) years, mean BMI was 28.2 (SD 5.5) kg/m2, and 51.5% were women. Heritability of sleepiness was 0.29 (p < 0.001), of usual bedtime 0.22 (p < 0.01), and of usual sleep duration 0.17 (p = .02). The estimated heritability of sleepiness was not appreciably reduced by further adjustment for usual sleep duration and the presence of habitual snoring or witnessed apneas (0.28, p = 0.002). Linkage analysis to these three phenotypes identified no linkage peaks with LOD >3. Five linkage peaks with LOD >2 were identified, four to usual bedtime and one to usual sleep duration (Table 2c). The region of the strongest of these, a LOD score of 2.45 for usual bedtime with a linkage peak at 55.5 Mb on chromosome 16, includes the gene encoding casein kinase 2a2 (CSNK2A2, at 56.7 Mb), a known element of the Drosophila circadian clock. The linkage peak to usual sleep duration at 71.3 Mb on chromosome 3 includes the gene encoding prokineticin 2 (PROK2, at 71.9 Mb), which may be an important output molecule from the suprachiasmatic nucleus. Three additional linkage peaks to usual bedtime had maximum LOD scores of 1.9. One of these, located on chromosome 2, includes the interleukin-1 cytokine cluster. Another, located on chromosome 4 at 62.5 Mb, includes the CLOCK gene (at 56.0–56.1 Mb). A smaller linkage peak of LOD 1.5 was noted in this region (at 57.1 Mb) for sleepiness. Kurtosis was low for sleepiness and usual bedtime, at 0.2 and 1.1, respectively, although kurtosis of usual sleep duration was high at 3.0.
The five autosomal SNPs with the lowest p-value for association for each sleep phenotype by population-based and family-based association tests, and meeting quality control standards, are displayed in Tables 2a and 2b. Only one of these is located in a coding region: rs324981 in NPSR1, associated with usual bedtime, whose minor allele (frequency 0.44) is a non-synonymous mutation encoding an Asn107→Ile107 substitution in an exoloop lining the putative ligand-binding pocket of the neuropeptide S receptor [37]. The effect of this polymorphism is additive, with adjusted mean bedtime delayed by 14.9 minutes in heterozygotes and 29.5 minutes in homozygotes. Only eight SNPs were associated with any of the sleep phenotypes with p < 10-5. The SNP with the lowest p value for association to any of the sleep phenotypes is for the association of rs1823068 with sleepiness by population-based association testing (p = 2.5*10-8). This SNP is located in an intron of the gene encoding phosphodiesterase 4D (PDE4D). Other SNPs associated at p < 10-5 include one located in an intron of the gene encoding eyes absent 1 (EYA1) and two within an intron of the gene encoding myosin VIIA and Rab interacting protein (MYRIP) identified by population-based association tests, and two in or near the gene encoding opioid binding protein/cell adhesion molecule (OPCML) by family-based association tests. One was not located near a known gene.
The results of population-based and family-based association tests were modestly correlated. For example, when SNPs were ranked based on the p value for association with sleepiness, the Spearman correlation coefficient for rankings from population-based versus family-based tests was 0.22. Tables 2a and 2b show p-values for both approaches, allowing assessment of concordance across approaches for these SNPs. Only 3 SNPs were associated with sleepiness at p < 0.001 by both approaches; these were located in FHIT, VTA1, and LRP1B. Of the 5 SNPs meeting this criterion for usual bedtime, none was in or near a known gene, and no SNPs met this criterion for usual sleep duration. Similarly, little overlap was seen across the three phenotypes. No SNPs associated with sleepiness at p < 0.001 were associated with either usual bedtime or sleep duration at this nominal significance level for either population-based or family-based tests, and only 4 SNPs met this criterion for overlap between usual bedtime and sleep duration; three were located in known genes RYR2, JAZF1, and NDRG1.
Among potential candidate genes for sleep-related phenotypes, many were poorly represented on the Affymetrix 100K GeneChip. No SNP meeting quality control standards was located within PROK2, identified in the linkage analysis as a possible candidate for usual sleep duration. Only a single SNP was located in PER2 and none in CSNK1D or PER3, genes associated with familial advanced or delayed sleep phase syndromes. In contrast, 10 SNPs meeting quality control standards were typed within the CLOCK gene and 5 within CSNK2A2, none of which was significantly associated with any sleep phenotype by either the population-based or family-based approaches (lowest p value 0.12). The full results of the genetic association studies are available at the National Center for Biotechnology Information dbGaP website [38].
Discussion
In this family-based study, we have confirmed significant heritability of sleepiness, usual bedtime, and usual sleep duration that had been previously reported primarily from twin cohorts [5–7, 10–12, 20]. The heritability estimates in this study are lower than those reported in the literature from twin studies. This may reflect a greater contribution of environmental influences on these phenotypes in the present study that is detected as correlation between spouse-pairs. Alternatively, an underestimate of the shared environmental variance in twin studies may cause them to overstate the genetic contribution, as the estimated heritability of usual bedtime in this study is similar to that for diurnal preference in a previous family-based study [12]. We are unaware of any prior genome-wide linkage or association studies of these sleep-related phenotypes in humans. Because sleep phenotypes were available from only 56% of subjects included in the Framingham Family Plate Set, study power was limited and no linkage peaks reaching conventional levels of genome-wide significance were observed. Despite the greater heritability of sleepiness, most of the suggestive linkage peaks observed in this study were linked to usual bedtime and to a lesser extent to usual sleep duration. Several of these suggestive linkage peaks contain genes of potential importance to the circadian molecular clock. Two of these peaks are of particular interest. The linkage to usual bedtime on chromosome 16 was the strongest observed in this study (LOD = 2.45), with a peak close to the gene CSNK2A2. Its product, a catalytic subunit of casein kinase 2, has been shown to be an important component of the circadian molecular clock in Drosophila and other organisms [39]. Phosphorylation by casein kinase 2 promotes nuclear translocation of the PERIOD gene product, and mutations that impair catalytic activity or subunit multimerization cause a lengthening of the circadian period [40, 41]. Although casein kinase 2 has not previously been implicated in human circadian rhythm disorders, mutations in the human genes encoding casein kinase 1d and period 2 are associated with familial advanced sleep phase syndrome [25, 26]. The linkage to usual sleep duration on chromosome 3 (LOD = 2.17) has a peak close to the gene PROK2. Its product is the precursor of prokineticin 2, which is highly expressed in the suprachiasmatic nucleus, regulated by the circadian molecular clock, and believed to be an important output molecule from the suprachiasmatic nucleus, coordinating and transmitting the behavioral circadian rhythm to multiple brain regions [42, 43]. Although not previously implicated in human disorders, the total sleep duration of prokineticin null mice is reduced by 83.5 minutes per 24 hour period compared to their wild-type littermates [44]. The modest linkage to both bedtime and sleepiness near the CLOCK gene, a central component of the molecular circadian clock, is also intriguing. PROK2 could not be evaluated in association tests, as no SNPs were typed within this gene. As none of the 5 SNPs within CSNK2A2 or the 10 within CLOCK was significantly associated with usual bedtime, other genes in these regions or chance may be responsible for these linkage peaks.
Association tests identified several loci that merit follow-up in other cohorts. The most interesting of these is an association of usual bedtime with a non-synonymous coding SNP in NPSR1, which causes an Asn107→Ile107 substitution in the putative ligand-binding pocket of the neuropeptide S receptor [37]. This same variant has been linked to asthma in several Caucasian populations [45, 46] but has not been previously reported in association with any sleep or circadian phenotype. In mice, neuropeptide S is localized to a small area adjacent to the noradrenergic locus ceruleus and its intraventricular administration is a potent, transient stimulus to wakefulness [47]. The Asn107→Ile107 variant of the neuropeptide S receptor is a gain of function mutation, increasing sensitivity of the receptor to neuropeptide S [37]. Consistent with this effect on receptor function, mean bedtime is 15 minutes later for each copy of the gene encoding the Asn107→Ile107 variant.
Although not located in a coding region, the strong association of sleepiness with a SNP located in an intron of the gene encoding phosphodiesterase 4D also merits further study. Phosphodiesterase 4 is a cAMP-specific phosphodiesterase that has multiple splice variants, with PDE4D being widely expressed in human brain [48]. Mutations of PDE4D have been associated with stroke risk in several populations, possibly related to the role of PDE4 in modulating inflammatory processes, although the causal nature of the association remains controversial [49, 50]. While the nonselective phosphodiesterase inhibitors caffeine and theophylline have long been recognized to promote wakefulness, this is likely due to antagonism of dopamine receptors rather than phosphodiesterase inhibition [51]. However, variation in the effects of PDE4D on brain intracellular levels of cAMP or extracellular levels of adenosine might influence sleepiness, and the selective PDE4 inhibitor rolipram is a weak promoter of wakefulness in rats [52].
This study has a number of limitations. The sleep phenotypes were assessed by questionnaire only. While the Epworth Sleepiness Scale is a well-validated measure of usual sleepiness, the single questions regarding usual bedtime and sleep duration provide only crude measures of circadian phenotype. Moreover, as subjects did not have a clinical sleep evaluation, it was not possible to control for sleep apnea or other primary sleep disorders. A sleepiness phenotype further adjusted for self-reported usual sleep duration and frequent snoring or witnessed apneas was analyzed, however, and results are included in the web repository. This gave results very similar to those presented in this manuscript, although with somewhat lower power due to an additional 15% missing phenotype data. The present study has statistical limitations as well. The relatively small number of subjects included in the analysis limits the power to detect true linkage and association, while the large number of SNPs tested and apparent inflation of Type I error rates for the population-based association tests makes it likely that many of the observed associations are false positive findings. Kurtosis of the measure of sleep duration may have inflated the LOD scores from linkage analysis. Thus, all results of this study require replication in other populations. These statistical limitations are discussed in more detail in the Overview [33].
Notwithstanding these limitations, this study begins to apply the powerful methodologies of genetic epidemiology to the study of common sleep and circadian phenotypes, and identifies for further study several genes that have not previously been implicated in human sleep and circadian disorders. These findings require replication, which will be pursued in other cohorts and by a planned expansion of the SNP genotyping using a 500,000 SNP gene chip in a substantially larger sample of Framingham Study subjects. Collection of more detailed sleep phenotype data from a larger sample of Framingham Study subjects will increase the power to detect novel genes influencing sleep and circadian phenotypes.
Conclusion
This analysis confirms, in a family-based sample, prior reports of significant heritability of sleepiness, usual bedtime, and usual sleep duration. It identifies several genetic loci with suggestive linkage to these traits, including linkage peaks containing the circadian clock-related genes CSNK2A2, PROK2 and CLOCK. Among genes identified by association tests as possible mediators of sleep and circadian phenotypes, those most promising based on the strength of the associations and their known biological activity are NPSR1 and PDE4D, which may influence usual bedtime and daytime sleepiness, respectively. While these findings require replication in other samples, they provide evidence of the possible utility of genetic epidemiology approaches to understanding population variation in sleep and circadian phenotypes.
References
National Sleep Foundation: 2005 Sleep in America Poll. Washington, DC. 2005
Dijk DJ, Duffy JF, Czeisler CA: Circadian and sleep/wake dependent aspects of subjective alertness and cognitive performance. J Sleep Res. 1992, 1: 112-117.
Kapur VK, Baldwin CM, Resnick HE, Gottlieb DJ, Nieto FJ: Sleepiness in patients with moderate to severe sleep-disordered breathing. Sleep. 2005, 28: 472-477.
Van Dongen HP, Baynard MD, Maislin G, Dinges DF: Systematic interindividual differences in neurobehavioral impairment from sleep loss: evidence of trait-like differential vulnerability. Sleep. 2004, 27: 423-433.
Carmelli D, Bliwise DL, Swan GE, Reed T: A genetic analysis of the Epworth Sleepiness Scale in 1560 World War II male veteran twins in the NAS-NRC Twin Registry. J Sleep Res. 2001, 10: 53-58. 10.1046/j.1365-2869.2001.00241.x.
Desai AV, Cherkas LF, Spector TD, Williams AJ: Genetic influences in self-reported symptoms of obstructive sleep apnoea and restless legs: a twin study. Twin Res. 2004, 7: 589-595. 10.1375/1369052042663841.
Watson NF, Goldberg J, Arguelles L, Buchwald D: Genetic and environmental influences on insomnia, daytime sleepiness, and obesity in twins. Sleep. 2006, 29: 645-649.
Ando K, Kripke DF, Ancoli-Israel S: Estimated prevalence of delayed and advanced sleep phase syndromes. Sleep Res. 1995, 24: 509-
Drake CL, Roehrs T, Richardson G, Walsh JK, Roth T: Shift work sleep disorder: prevalence and consequences beyond that of symptomatic day workers. Sleep. 2004, 27: 1453-1462.
Heath AC, Kendler KS, Eaves LJ, Martin NG: Evidence for genetic influences on sleep disturbance and sleep pattern in twins. Sleep. 1990, 13: 318-335.
Vink JM, Groot AS, Kerkhof GA, Boomsma DI: Genetic analysis of morningness and eveningness. Chronobiol Int. 2001, 18: 809-822. 10.1081/CBI-100107516.
Klei L, Reitz P, Miller M, Wood J, Maendel S, Gross D, Waldner T, Eaton J, Monk TH, Nimgaonkar VL: Heritability of morningness-eveningness and self-report sleep measures in a family-based sample of 521 hutterites. Chronobiol Int. 2005, 22: 1041-1054. 10.1080/07420520500397959.
Gottlieb DJ, Redline S, Nieto FJ, Baldwin CM, Newman AB, Resnick HE, Punjabi NM: Association of usual sleep duration with hypertension: the Sleep Heart Health Study. Sleep. 2006, 29: 1009-1014.
Ayas NT, White DP, Al-Delaimy WK, Manson JE, Stampfer MJ, Speizer FE, Patel S, Hu FB: A prospective study of self-reported sleep duration and incident diabetes in women. Diabetes Care. 2003, 26: 380-384. 10.2337/diacare.26.2.380.
Gottlieb DJ, Punjabi NM, Newman AB, Resnick HE, Redline S, Baldwin CM, Nieto FJ: Association of sleep time with diabetes mellitus and impaired glucose tolerance. Arch Intern Med. 2005, 165: 863-867. 10.1001/archinte.165.8.863.
Ayas NT, White DP, Manson JE, Stampfer MJ, Speizer FE, Malhotra A, Hu FB: A prospective study of sleep duration and coronary heart disease in women. Arch Intern Med. 2003, 163: 205-209. 10.1001/archinte.163.2.205.
Hammond EC: Some Preliminary Findings on Physical Complaints from a Prospective Study of 1,064,004 Men and Women. Am J Public Health Nations Health. 1964, 54: 11-23.
Wingard DL, Berkman LF, Brand RJ: A multivariate analysis of health-related practices: a nine-year mortality follow-up of the Alameda County Study. Am J Epidemiol. 1982, 116: 765-775.
Kripke DF, Garfinkel L, Wingard DL, Klauber MR, Marler MR: Mortality associated with sleep duration and insomnia. Arch Gen Psychiatry. 2002, 59: 131-136. 10.1001/archpsyc.59.2.131.
Partinen M, Kaprio J, Koskenvuo M, Putkonen P, Langinvainio H: Genetic and environmental determination of human sleep. Sleep. 1983, 6: 179-185.
Retey JV, Adam M, Honegger E, Khatami R, Luhmann UF, Jung HH, Berger W, Landolt HP: A functional genetic variation of adenosine deaminase affects the duration and intensity of deep sleep in humans. Proc Natl Acad Sci USA. 2005, 102: 15676-15681. 10.1073/pnas.0505414102.
Thompson MD, Comings DE, bu-Ghazalah R, Jereseh Y, Lin L, Wade J, Sakurai T, Tokita S, Yoshida T, Tanaka H, Yanagisawa M, Burnham WM, Moldofsky H: Variants of the orexin2/hcrt2 receptor gene identified in patients with excessive daytime sleepiness and patients with Tourette's syndrome comorbidity. Am J Med Genet B Neuropsychiatr Genet. 2004, 129: 69-75. 10.1002/ajmg.b.30047.
Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, Qiu X, de Jong PJ, Nishino S, Mignot E: The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999, 98: 365-376. 10.1016/S0092-8674(00)81965-0.
Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, Fitch TE, Nakazato M, Hammer RE, Saper CB, Yanagisawa M: Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999, 98: 437-451. 10.1016/S0092-8674(00)81973-X.
Toh KL, Jones CR, He Y, Eide EJ, Hinz WA, Virshup DM, Ptacek LJ, Fu YH: An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science. 2001, 291: 1040-1043. 10.1126/science.1057499.
Xu Y, Padiath QS, Shapiro RE, Jones CR, Wu SC, Saigoh N, Saigoh K, Ptacek LJ, Fu YH: Functional consequences of a CKIdelta mutation causing familial advanced sleep phase syndrome. Nature. 2005, 434: 640-644. 10.1038/nature03453.
Katzenberg D, Young T, Finn L, Lin L, King DP, Takahashi JS, Mignot E: A CLOCK polymorphism associated with human diurnal preference. Sleep. 1998, 21: 569-576.
Robilliard DL, Archer SN, Arendt J, Lockley SW, Hack LM, English J, Leger D, Smits MG, Williams A, Skene DJ, von Schantz M: The 3111 Clock gene polymorphism is not associated with sleep and circadian rhythmicity in phenotypically characterized human subjects. J Sleep Res. 2002, 11: 305-312. 10.1046/j.1365-2869.2002.00320.x.
Mishima K, Tozawa T, Satoh K, Saitoh H, Mishima Y: The 3111T/C polymorphism of hClock is associated with evening preference and delayed sleep timing in a Japanese population sample. Am J Med Genet B Neuropsychiatr Genet. 2005, 133: 101-104.
Archer SN, Robilliard DL, Skene DJ, Smits M, Williams A, Arendt J, von Schantz M: A length polymorphism in the circadian clock gene Per3 is linked to delayed sleep phase syndrome and extreme diurnal preference. Sleep. 2003, 26: 413-415.
Shimomura K, Low-Zeddies SS, King DP, Steeves TD, Whiteley A, Kushla J, Zemenides PD, Lin A, Vitaterna MH, Churchill GA, Takahashi JS: Genome-wide epistatic interaction analysis reveals complex genetic determinants of circadian behavior in mice. Genome Res. 2001, 11: 959-980. 10.1101/gr.171601.
Quan SF, Howard BV, Iber C, Kiley JP, Nieto FJ, O'Connor GT, Rapoport DM, Redline S, Robbins J, Samet JM, Wahl PW: The Sleep Heart Health Study: design, rationale, and methods. Sleep. 1997, 20: 1077-1085.
Cupples LA, Benjamin EJ, D'Agostino RB, Demissie S, DeStefano AL, Dupuis J, Falls K, Fox CS, Gottlieb DJ, Govindaraju DR, Heard-Costa N, Hwang SJ, Kathiresan S, Kiel DP, Laramie JM, Larson MG, Levy D, Lunetta KL, Mailman MD, Manning AK, Meigs JB, Murabito JM, Newton-Cheh C, O'Connor GT, O'Donnell CJ, Pandey MA, Seshadri S, Vasan RS, Wilk JB, Wolf PA, Yang Q, Atwood LD: The Framingham Heart Study 100K SNP genome-wide association study resource: Overview of 17 phenotype working group reports. BMC Med Genet. 2007, 8 (Suppl 1): S1-
Johns MW: A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep. 1991, 14: 540-545.
Johns MW: Reliability and factor analysis of the Epworth Sleepiness Scale. Sleep. 1992, 15: 376-381.
Sleep Heart Health Study Sleep Habits Questionnaire. [http://www.jhucct.com/shhs/details/manual/forms/sh/shhshq2.pdf]
Reinscheid RK, Xu YL, Okamura N, Zeng J, Chung S, Pai R, Wang Z, Civelli O: Pharmacological characterization of human and murine neuropeptide s receptor variants. J Pharmacol Exp Ther. 2005, 315: 1338-1345. 10.1124/jpet.105.093427.
National Center for Biotechnology Information dbGaP website. [http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?id=phs000007]
Lin JM, Kilman VL, Keegan K, Paddock B, Emery-Le M, Rosbash M, Allada R: A role for casein kinase 2alpha in the Drosophila circadian clock. Nature. 2002, 420: 816-820. 10.1038/nature01235.
Lin JM, Schroeder A, Allada R: In vivo circadian function of casein kinase 2 phosphorylation sites in Drosophila PERIOD. J Neurosci. 2005, 25: 11175-11183. 10.1523/JNEUROSCI.2159-05.2005.
Akten B, Jauch E, Genova GK, Kim EY, Edery I, Raabe T, Jackson FR: A role for CK2 in the Drosophila circadian oscillator. Nat Neurosci. 2003, 6: 251-257. 10.1038/nn1007.
Cheng MY, Bullock CM, Li C, Lee AG, Bermak JC, Belluzzi J, Weaver DR, Leslie FM, Zhou QY: Prokineticin 2 transmits the behavioural circadian rhythm of the suprachiasmatic nucleus. Nature. 2002, 417: 405-410. 10.1038/417405a.
Li JD, Hu WP, Boehmer L, Cheng MY, Lee AG, Jilek A, Siegel JM, Zhou QY: Attenuated circadian rhythms in mice lacking the prokineticin 2 gene. J Neurosci. 2006, 26: 11615-11623. 10.1523/JNEUROSCI.3679-06.2006.
Hu WP, Li JD, Zhang C, Boehmer L, Siegel JM, Zhou QY: Altered circadian and homeostatic sleep regulation in prokineticin 2-deficient mice. Sleep. 2007, 30: 247-256.
Laitinen T, Polvi A, Rydman P, Vendelin J, Pulkkinen V, Salmikangas P, Makela S, Rehn M, Pirskanen A, Rautanen A, Zucchelli M, Gullsten H, Leino M, Alenius H, Petays T, Haahtela T, Laitinen A, Laprise C, Hudson TJ, Laitinen LA, Kere J: Characterization of a common susceptibility locus for asthma-related traits. Science. 2004, 304: 300-304. 10.1126/science.1090010.
Kormann MS, Carr D, Klopp N, Illig T, Leupold W, Fritzsch C, Weiland SK, von Mutius E, Kabesch M: G-Protein-coupled receptor polymorphisms are associated with asthma in a large German population. Am J Respir Crit Care Med. 2005, 171: 1358-1362. 10.1164/rccm.200410-1312OC.
Xu YL, Reinscheid RK, Huitron-Resendiz S, Clark SD, Wang Z, Lin SH, Brucher FA, Zeng J, Ly NK, Henriksen SJ, de Lecea L, Civelli O: Neuropeptide S: a neuropeptide promoting arousal and anxiolytic-like effects. Neuron. 2004, 43: 487-497. 10.1016/j.neuron.2004.08.005.
Perez-Torres S, Miro X, Palacios JM, Cortes R, Puigdomenech P, Mengod G: Phosphodiesterase type 4 isozymes expression in human brain examined by in situ hybridization histochemistry and [3H]rolipram binding autoradiography. Comparison with monkey and rat brain. J Chem Neuroanat. 2000, 20: 349-374. 10.1016/S0891-0618(00)00097-1.
Gretarsdottir S, Thorleifsson G, Reynisdottir ST, Manolescu A, Jonsdottir S, Jonsdottir T, Gudmundsdottir T, Bjarnadottir SM, Einarsson OB, Gudjonsdottir HM, Hawkins M, Gudmundsson G, Gudmundsdottir H, Andrason H, Gudmundsdottir AS, Sigurdardottir M, Chou TT, Nahmias J, Goss S, Sveinbjornsdottir S, Valdimarsson EM, Jakobsson F, Agnarsson U, Gudnason V, Thorgeirsson G, Fingerle J, Gurney M, Gudbjartsson D, Frigge ML, Kong A, Stefansson K, Gulcher JR: The gene encoding phosphodiesterase 4D confers risk of ischemic stroke. Nat Genet. 2003, 35: 131-138. 10.1038/ng1245.
Worrall BB, Mychaleckyj JC: PDE4D and stroke: a real advance or a case of the Emperor's new clothes?. Stroke. 2006, 37: 1955-1957. 10.1161/01.STR.0000234048.04053.39.
Nehlig A, Daval JL, Debry G: Caffeine and the central nervous system: mechanisms of action, biochemical, metabolic and psychostimulant effects. Brain Res Brain Res Rev. 1992, 17: 139-170. 10.1016/0165-0173(92)90012-B.
Lelkes Z, Alfoldi P, Erdos A, Benedek G: Rolipram, an antidepressant that increases the availability of cAMP, transiently enhances wakefulness in rats. Pharmacol Biochem Behav. 1998, 60: 835-839. 10.1016/S0091-3057(98)00038-0.
Acknowledgements
The authors wish to acknowledge the contribution of the Framingham Study participants to this research. This research was supported by the National Heart, Lung and Blood Institute through contract N01-HC 25195 (Framingham Heart Study) and cooperative agreement U01 HL53941 (Sleep Heart Health Study). A portion of the research was conducted using the Boston University Linux Cluster for Genetic Analysis (LinGA) funded by the NIH NCRR (National Center for Research Resources) Shared Instrumentation grant (1S10RR163736-01A1). Dr. Wilk is supported by a young clinical scientist award from the Flight Attendant Medical Research Institute.
This article has been published as part of BMC Medical Genetics Volume 8 Supplement 1, 2007: The Framingham Heart Study 100,000 single nucleotide polymorphisms resource. The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2350/8?issue=S1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
All authors participated in the conception and design of the study. DJG and GTO participated in phenotype data collection. DJG and JBW participated in the statistical analysis. DJG drafted the manuscript, with critical feedback from GTO and JBW. All authors read and approved the final manuscript.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Gottlieb, D.J., O'Connor, G.T. & Wilk, J.B. Genome-wide association of sleep and circadian phenotypes. BMC Med Genet 8 (Suppl 1), S9 (2007). https://doi.org/10.1186/1471-2350-8-S1-S9
Published:
DOI: https://doi.org/10.1186/1471-2350-8-S1-S9