
Abstract
Background
Susceptibility to type 2 diabetes may be conferred by genetic variants having modest effects on risk. Genome-wide fixed marker arrays offer a novel approach to detect these variants.
Methods
We used the Affymetrix 100K SNP array in 1,087 Framingham Offspring Study family members to examine genetic associations with three diabetes-related quantitative glucose traits (fasting plasma glucose (FPG), hemoglobin A1c, 28-yr time-averaged FPG (tFPG)), three insulin traits (fasting insulin, HOMA-insulin resistance, and 0–120 min insulin sensitivity index); and with risk for diabetes. We used additive generalized estimating equations (GEE) and family-based association test (FBAT) models to test associations of SNP genotypes with sex-age-age2-adjusted residual trait values, and Cox survival models to test incident diabetes.
Results
We found 415 SNPs associated (at p < 0.001) with at least one of the six quantitative traits in GEE, 242 in FBAT (18 overlapped with GEE for 639 non-overlapping SNPs), and 128 associated with incident diabetes (31 overlapped with the 639) giving 736 non-overlapping SNPs. Of these 736 SNPs, 439 were within 60 kb of a known gene. Additionally, 53 SNPs (of which 42 had r2 < 0.80 with each other) had p < 0.01 for incident diabetes AND (all 3 glucose traits OR all 3 insulin traits, OR 2 glucose traits and 2 insulin traits); of these, 36 overlapped with the 736 other SNPs. Of 100K SNPs, one (rs7100927) was in moderate LD (r2 = 0.50) with TCF7L2 (rs7903146), and was associated with risk of diabetes (Cox p-value 0.007, additive hazard ratio for diabetes = 1.56) and with tFPG (GEE p-value 0.03). There were no common (MAF > 1%) 100K SNPs in LD (r2 > 0.05) with ABCC8 A1369S (rs757110), KCNJ11 E23K (rs5219), or SNPs in CAPN10 or HNFa. PPARG P12A (rs1801282) was not significantly associated with diabetes or related traits.
Conclusion
Framingham 100K SNP data is a resource for association tests of known and novel genes with diabetes and related traits posted at http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?id=phs000007. Framingham 100K data replicate the TCF7L2 association with diabetes.
Similar content being viewed by others

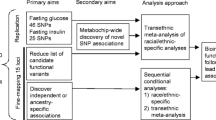

Background
Type 2 diabetes is a cause of poor health and early death that is spreading worldwide and exerting a fearsome human and economic toll [1, 2]. Prevention and control of diabetes requires a better understanding of its basic molecular causes. Type 2 diabetes is a heterogeneous disease arising from physiological dysfunction in the pancreas, skeletal muscle, liver, adipose and vascular tissue. Much of the heterogeneity of type 2 diabetes has a genetic basis. A full picture of the complex genetic architecture of diabetes has been elusive [3–7].
Among type 2 diabetes susceptibility genes few, if any, individual loci are expected to carry alleles of major effect explaining a substantial proportion of cases, although a few genes could have a substantial population effect but not give a strong genetic signal if the causal alleles were common and the increase in risk were modest [6, 7]. Such genes have proven hard to detect using linkage-based approaches, although recent rapid advances in genetic association methodologies have led to some successes. The P12A polymorphism in the gene encoding the peroxisome proliferator-activated receptor-g (PPARG) [7], the E23K polymorphism in the gene encoding the islet ATP-dependent potassium channel Kir6.2 (ABCC8-KCNJ11) [8–10] and common variants in the gene encoding the transcription factor 7-like 2 gene (TCF7L2) [11, 12] were all found using well-powered association mapping, and all have been reproducibly associated with diabetes in diverse samples at highly significant p-values.
Current gene discovery strategies have focused on coding regions, but regulatory variants also influence disease [11, 13, 14]. A comprehensive picture of diabetes genetics will require a wide and adequately dense search across coding and conserved non-coding genomic regions using an association analysis approach, where power is superior to linkage analysis when seeking common variants of modest effect [6]. Resources are now becoming available to perform such genome-wide association (GWA) studies of type 2 diabetes [15–18].
In this report we describe the Framingham Heart Study (FHS) Affymetrix 100K SNP genome-wide association (GWA) study resource for type 2 diabetes. This resource complements the several other large extant type 2 diabetes GWA studies in three major respects: it is population-based (not diabetes proband-based), studies two generations, and has decades of longitudinal, standardized, detailed follow-up. We describe results of a simple low p-value-based SNP selection strategy and an alternate novel SNP selection strategy that takes advantage of the unique FHS diabetes-related quantitative traits data. We use FHS 100K SNPs in an in silico replication analysis that tests the hypothesis that SNPs in LD with published causal variants in PPARG, ABCC8, TCF7L2, CAPN10, and HNFa are associated with diabetes and related quantitative traits.
Methods
Study subjects
The study sample is described in the Overview Methods section [19]. With respect to diabetes-related traits, Offspring subjects provided genotypes and diabetes-related traits to the analyses, and Offspring parents from the Original FHS Cohort contributed genotypes for linkage analysis and FBAT statistics. Of 1,345 FHS subjects with 100K SNP data, 1,087 were Offspring and of these 560 were women, the mean age at exam 5 was 52 years, and the mean age at last follow-up was 59 years. Every study subject provided written informed consent at every examination, including consent for genetic analyses, and the study was approved by Boston University's Institutional Review Board.
Genotyping and annotation
Affymetrix 100K SNP and Marshfield STR genotyping are described in the Overview Methods section [19]. Genotype annotation sources are described in the Overview Methods section [19].
Diabetes phenotyping
Diabetes and related quantitative traits have been ascertained at every FHS exam for every generation. Diabetes-related quantitative traits available in the FHS 100K resource are displayed in Table 1. FPG data for the analyses came from all 7 Offspring exams, but the remainder of the data came from exam 5 (1991–94), when subjects without diagnosed diabetes underwent a 75 gram oral glucose tolerance test, or exam 7 (1998–2001), the most recent exam. We defined diabetes as chart-review-confirmed diabetes, new or ongoing hypoglycemic treatment for diabetes at any exam, or a FPG > 125 mg/dl at two or more of the seven exams. Diabetes age-of-onset was defined as the subject's age at the exam at which diabetes was first identified. Among Offspring with diabetes, >99% have type 2 diabetes [4]. Of the 1,083 Offspring with 100K genotypes and known diabetes status, 91 had diabetes. The mean age of onset of was 58 yr; through exam 7, 9.3% of diabetic subjects had developed diabetes by age 40 yr, 33.0% by age 50, 68.1% by age 60, and 99.7% by age 80.
In this presentation we focus on six (three glucose and three insulin) primary Offspring diabetes-related quantitative traits. Glucose traits are fasting plasma glucose (FPG) and hemoglobin A1c (HbA1c) measured at exam 5, and up to 28 yr time-averaged FPG (tFPG) level obtained from the mean of up to seven serial exams. Glucose traits included all subjects, including those with diabetes regardless of treatment, as these were the most informative subjects with respect to hyperglycemia. Subjects with diabetes had the highest glucose values when subjects were ranked with respect to any glucose trait; those on treatment had the highest values. The three insulin traits are fasting insulin, homeostasis model-assessed insulin resistance (HOMA-IR), and Gutt's 0–120 min insulin sensitivity index (ISI_0-120) measured at exam 5. Subjects with insulin-treated diabetes were removed from all insulin trait analyses, as we had no information on insulin dose and so measured insulin values were confounded by insulin treatment [20–22]. We also analyzed incident diabetes from first exam through last follow-up. We previously have described FHS laboratory methods for these diabetes-related quantitative traits [4, 23–25]. In addition to glucose and insulin traits, levels of adiponectin and resistin are available in the FHS dbGaP resource. Plasma adiponectin and resistin concentrations were measured using a commercial ELISA (R&D Systems, Minneapolis, MN); inter- and intra-assays CVs were 5.3%–9.6% for adiponectin and 7.6%–10.5% for resistin.
SNP prioritization
We used two approaches to prioritize SNPs potentially associated with diabetes or diabetes related traits. In the first, we simply ordered SNPs from lowest to highest p-value for association with one or more of the six primary glucose and insulin traits. We also ordered SNPs or Marshfield STRS by highest to lowest LOD score for linkage to one or more of the six primary traits, and present LOD scores > 2.0. In an alternative SNP prioritization strategy, we selected SNPs associated with multiple-related traits. In this approach, we selected SNPs with consistent nominal associations (p < 0.01 in GEE or FBAT) with all three glucose traits OR all three insulin-related traits OR (two glucose and two insulin traits). Among these we used extent of LD to select a non-redundant set of SNPs; when several were perfect proxies for each other (r2 ≥ 0.8) only one SNP was selected, based on the highest genotyping call rate.
Statistical analysis
The general statistical methods for linkage and GWA analyses are described in the Overview Methods [19]. For diabetes-related quantitative traits we used additive GEE and FBAT models, testing associations between SNP genotypes and age-age2-sex-adjusted residual trait values. We kept 70,987 SNPs in the analyses that were on autosomes, had genotypic call rates ≥ 80%, HWE p ≥ 0.001 and MAF ≥ 10%.
We tested association of 100K SNPs with incident type 2 diabetes in two additional models using the same adjustment strategy. First, Martingale residuals were created to measure the age-of-onset of type 2 diabetes; residuals were analyzed with FBAT [26]. Individuals with lower values of this 'martingale residual' trait developed diabetes at younger ages, and those with the highest values had been observed for the longest time without development of diabetes [27]. Second, we used a Cox proportional hazard survival analysis with robust covariance estimates in order to find SNPs associated with development of diabetes over all seven exams [28].
Results
Diabetes-related quantitative traits available in the FHS 100K SNP resource are listed in Table 1 and posted on the NCBI web site [29]. Each trait is available as an age-age2-adjusted or age-age2-BMI-adjusted residuals from sex-specific models. In this analysis we only consider the age-age2-adjusted traits. Among these, the following were the primary traits used in this analysis: exam 5 fasting plasma glucose (FPG; n with data = 1,027; mean, SD 99, 24.7 mg/dl); exam 5 HbA1c (n = 623; 5.28, 0.9%); 28-year time averaged FPG (tFPG; n = 1,087; 98, 16.2 mg/dl); exam 5 fasting insulin (n = 982; 30.1, 16.4 uU/ml); exam 5 HOMA-IR (n = 980; 7.8, 7.3 units); and the 0–120 min insulin sensitivity index (ISI_0-120; n = 935; 26.1, 7.6 mg·l2/mmol·mU·min). Among 1,087 Offspring with 100K SNP data there were 91 cases of type 2 diabetes. Additional diabetes-related quantitative traits not used in this analysis but that are available in the FHS 100K SNP dbGaP resource include, at exam 7: FPG (n = 987; 103, 26 mg/dl); fasting insulin (n = 999; 15.8, 12.8 uU/ml); HOMA-IR (n = 969; 4.2, 4.1 units); HbA1c (n = 893; 5.59, 0.97%); resistin (n = 831; 14.5, 7.4 ng/dl); adiponectin (n = 828; 9.9, 6.2 ng/dl).
The six primary quantitative traits had significant associations with 415 SNPs in GEE models and 242 SNPs in FBAT models, using p-value < 0.001, and only considering SNPs with call rate ≥ 0.80, HWE p-value ≥ 0.001, and MAF ≥ 10%. Additionally, there were 91 significant associations with incident diabetes in the survival analyses and 42 significant associations with age-of-onset in FBAT, representing 128 non-overlapping SNPs. The 25 SNPs with lowest p-values in GEE or FBAT models, and LOD scores > 2.0 in linkage analyses, are displayed in Table 2. After accounting for the overlap between sets of significant associations, 736 non-overlapping SNPs were identified by the p-value approach for SNP prioritization.
The FHS has multiple measures of diabetes-related quantitative traits. We used a multiple-related trait approach in a strategy different from prioritizing SNPs based solely on small p-values. This approach yielded 203 SNPs associated with multiple traits. Of these, 53 were also associated with incident diabetes (p < 0.01 by GEE or FBAT). We defined redundant SNPs as those in LD with r2 >= 0.80 to select 168 non-redundant SNPs associated with multiple traits; 42 of these non-redundant SNPs also were associated with incident diabetes (Table 3). Examination of the multiple trait-based approach revealed 1) consistent associations of traits with SNPs that were in LD (providing reassurance that the signal was due to an association of traits with a particular genomic region rather than to technical error); 2) several putative associations of traits with SNPs in the same gene but not in perfect LD (suggesting that the association signal may be due to a functional role of that gene rather than a statistical fluctuation); and 3) associations of traits with SNPs in a variety of novel but plausible biological candidate genes.
We used the UCSC Genome Browser (http://genome.ucsc.edu/; accessed September 2006) to annotate SNP details [30, 31]. Of the 823 (736 + 203; 116 overlapped) SNPs identified by both prioritization methods without removing SNPs in LD (r2 >= 0.80), 304 (36.9%) were in genes, 173 (21%) were within 60 kb of a known gene and 5 (0.61%) were coding. For comparison, of the 70,987 SNPs included in this analysis, 25,916 (36.5%) were in genes, 14,333 (20.2%) were within 60 kb of a known gene and 421 (0.59%) were coding.
Some SNPs had p-values < 0.001 overlapping more than one analytical method. For instance, 18 SNPs were associated at p < 0.001 with at least one quantitative trait in both the GEE and the FBAT analyses. For incident diabetes, 5 SNPs were associated with diabetes survival in the Cox models and with age-of-onset in the FBAT analyses.
We used the FHS 100K array data to verify, in silico, replicated associations of reported diabetes candidate genes (Table 4). We found 7 SNPs in or near TCF7L2. One 100K SNP (rs7100927) was in moderate LD (r2 = 0.5) with TCF7L2-associated SNP rs7903146 and was nominally associated with a 56% increased relative risk of diabetes (p = 0.007) and with tFPG (GEE p = 0.03). We found 6 SNPs in or near ABCC8, but no SNPs in strong LD with ABCC8 A1369S (rs757110) or KCNJ11 E23K (rs5219), and thus could not replicate these associations. One 100K SNP (rs878208) ~25 kb upstream of ABCC8 showed nominal association with risk of diabetes, but it was not in LD with rs757110 in ABCC8 (r2 = 0.04). We found 15 SNPs in or near PPARG, but none were associated with diabetes. Four SNPs were associated (p < 0.05) with quantitative traits but were not in LD (r2 < 0.03) with PPARG P12A (rs1801282), the variant previously associated with type 2 diabetes [7]. We found no polymorphic (MAF > 1%) 100K SNPs in, near, or in LD with CAPN10 or HNFA.
We also assessed our approach for confirmation of 4 SNPs associated with FPG reported on the Boston University Department of Genetics and Genomics public site http://gmed.bu.edu/about/index.html that displays selected associations with FHS 100K data. We found no association (all p-values > 0.6) of incident diabetes or levels of FPG with SNPs rs10495355, rs9302082, rs10483948, or rs1148509.
Discussion and conclusion
In this paper we describe the characteristics and initial GWA results for type 2 diabetes and related quantitative traits in the FHS 100K SNP resource. Over 1000 men and women from a community-based sample have detailed linkage and association of diabetes-related phenotypes and 100K dense array SNP results available on the web. About 0.3%–0.6% of SNPs in the 100K array with MAF > 10% are associated at p < 0.001 with six diabetes-related quantitative traits or with incident type 2 diabetes. A similar proportion of SNPs in the array (0.21%) are associated with multiple related diabetes traits. These several hundred SNPs likely contain more false positive than true positive associations with diabetes and related traits, however, they offer logical next targets for the follow-up replication studies in independent samples necessary to resolve true diabetes risk genes. The FHS 100K data replicate the otherwise widely-replicated TCF7L2 association with diabetes [11, 12, 32–40] in an in silico analysis.
The FHS 100K SNP data resource has potential value to detect and replicate novel type 2 diabetes susceptibility genes. The 100K SNP array is limited by relatively sparse coverage in some regions, accounting on average for just 30%–40% of the human genome in whites [17, 41]. Association with the risk SNP in TCF7L2 is detectable at p < 0.05, but there are no SNPs in adequate LD with ABCC8 or PPARG to assess replication of causal SNPs in these accepted diabetes susceptibility genes. Thin coverage will be remedied to a large degree by the incipient availability in FHS of Affymetrix 500 k SNP array data as part of the planned FHS SHARe Study. (http://www.nhlbi.nih.gov/meetings/nhlbac/sept06sum.htm; accessed September 2006) Our analysis also demonstrates that true positive diabetes susceptibility gene signals are likely to be associated with modest p-values and will remain challenging to detect at the stringent p-values required for GWA studies. The enormous datasets generated by GWA scans have the potential to greatly advance understanding, or conversely to overwhelm the field with false leads. SNP prioritization strategies that leverage the complexity of the diabetes phenotype may offer some advantages over strictly p-value driven approaches. Replication, fine mapping, and functional studies are required to determine which approaches are most efficient and which SNPs are true positive diabetes risk factors. Integration with other GWA scans in similar cohorts will allow in silico replication of significant findings, increase power and reveal generalizability.
This report details the FHS contribution to publicly available diabetes-related genetic data. An important key to efficiently and economically achieving adequate power to detect association will be to integrate information from several GWA scans. While several cohorts have been assembled to perform GWA scans in type 2 diabetes, few possess the wealth of longitudinal, multigenerational phenotypic data available in Framingham. The FHS complements extant type 2 diabetes GWA studies. This report guides the way to harness the power of the FHS 100K SNP GWA resource to identify type 2 diabetes susceptibility genes.
Abbreviations
- FPG:
-
fasting plasma glucose
- FBAT:
-
family-based association test
- FHS:
-
Framingham Heart Study
- GEE:
-
generalized estimating equations
- GWA:
-
Genome-wide association
- HbA1c:
-
hemoglobin A1c
- HOMA-IR:
-
homeostasis model insulin resistance
- HWE:
-
Hardy Weinberg equilibrium
- IBD:
-
Identity-by-descent
- ISI_0-120:
-
0–120 min insulin sensitivity index
- LD:
-
Linkage disequilibrium
- LOD:
-
Log odds score
- MAF:
-
Minor allele frequency
- SNP:
-
Single nucleotide polymorphism
- TFPG:
-
28-yr time-averaged FPG.
References
King H, Aubert RE, Herman WH: Global burden of diabetes, 1995–2025: prevalence, numerical estimates, and projections. Diabetes Care. 1998, 21 (9): 1414-1431. 10.2337/diacare.21.9.1414.
Ritz E, Rychlik I, Locatelli F, Halimi S: End-stage renal failure in type 2 diabetes: A medical catastrophe of worldwide dimensions. American Journal of Kidney Diseases. 1999, 34 (5): 795-808.
Poulsen P, Kyvik KO, Vaag A, Beck-Nielsen H: Heritability of type II (non-insulin-dependent) diabetes mellitus and abnormal glucose tolerance – a population-based twin study. Diabetologia. 1999, 42 (2): 139-145. 10.1007/s001250051131.
Meigs JB, Cupples LA, Wilson PWF: Parental transmission of type 2 diabetes mellitus: the Framingham Offspring Study. Diabetes. 2000, 49: 2201-2207. 10.2337/diabetes.49.12.2201.
Florez JC, Hirschhorn J, Altshuler D: The inherited basis of diabetes mellitus: implications for the genetic analysis of complex traits. Annu Rev Genomics Hum Genet. 2003, 4: 257-291. 10.1146/annurev.genom.4.070802.110436.
Risch N, Merikangas K: The future of genetic studies of complex human diseases. Science. 1996, 273 (5281): 1516-1517. 10.1126/science.273.5281.1516.
Altshuler D, Hirschhorn JN, Klannemark M, Lindgren CM, Vohl MC, Nemesh J, Lane CR, Schaffner SF, Bolk S, Brewer C, Tuomi T, Gaudet D, Hudson TJ, Daly M, Groop L, Lander ES: The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet. 2000, 26 (1): 76-80. 10.1038/79216.
Gloyn AL, Weedon MN, Owen KR, Turner MJ, Knight BA, Hitman G, Walker M, Levy JC, Sampson M, Halford S, McCarthy MI, Hattersley AT, Frayling TM: Large-scale association studies of variants in genes encoding the pancreatic b-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes. 2003, 52 (2): 568-572. 10.2337/diabetes.52.2.568.
Florez JC, Burtt N, de Bakker PIW, Almgren P, Tuomi T, Holmkvist J, Gaudet D, Hudson TJ, Schaffner SF, Daly MJ, Hirschhorn JN, Groop L, Altshuler D: Haplotype structure and genotype-phenotype correlations of the sulfonylurea receptor and the islet ATP-sensitive potassium channel gene region. Diabetes. 2004, 53 (5): 1360-1368. 10.2337/diabetes.53.5.1360.
Barroso I, Luan J, Middelberg RPS, Harding A-H, Franks PW, Jakes RW, Clayton D, Schafer AJ, O'Rahilly S, Wareham NJ: Candidate gene association study in type 2 diabetes indicates a role for genes involved in β-cell function as well as insulin action. PLoS Biology. 2003, 1 (1): 41-55. 10.1371/journal.pbio.0000020.
Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, Helgason A, Stefansson H, Emilsson V, Helgadottir A, Styrkarsdottir U, Magnusson KP, Walters GB, Palsdottir E, Jonsdottir T, Gudmundsdottir T, Gylfason A, Saemundsdottir J, Wilensky RL, Reilly MP, Rader DJ, Bagger Y, Christiansen C, Gudnason V, Sigurdsson G, Thorsteinsdottir U, Gulcher JR, Kong A, Stefansson K: Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006, 38 (3): 320-323. 10.1038/ng1732.
Florez JC, Jablonski KA, Bayley N, Pollin TI, de Bakker PIW, Shuldiner AR, Knowler WC, Nathan DM, Altshuler D, Group tDPPR: TCF7L2 polymorphisms and progression to diabetes in the Diabetes Prevention Program. N Engl J Med. 2006, 355 (3): 241-250. 10.1056/NEJMoa062418.
Beysen D, Raes J, Leroy BP, Lucassen A, Yates JRW, Clayton-Smith J, Ilyina H, Brooks SS, Christin-Maitre S, Fellous M, Fryns JP, Kim JR, Lapunzina P, Lemyre E, Meire F, Messiaen LM, Oley C, Splitt M, Thomson J, Peer YVd, Veitia RA, De Paepe A, De Baere E: Deletions involving long-range conserved nongenic sequences upstream and downstream of FOXL2 as a novel disease-causing mechanism in blepharophimosis syndrome. Am J Hum Genet. 2005, 77 (2): 205-218. 10.1086/432083.
Drake JA, Bird C, Nemesh J, Thomas DJ, Newton-Cheh C, Reymond A, Excoffier L, Attar H, Antonarakis SE, Dermitzakis ET, Hirschhorn JN: Conserved noncoding sequences are selectively constrained and not mutation cold spots. 2006, 38 (2): 223-227.
The_International_HapMap_Consortium: A haplotype map of the human genome. Nature. 2005, 437: 1299-1320. 10.1038/nature04226.
Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D: The structure of haplotype blocks in the human genome. Science. 2002, 296 (5576): 2225-2229. 10.1126/science.1069424.
Pe'er I, de Bakker PIW, Maller J, Yelensky R, Altshuler D, Daly MJ: Evaluating and improving power in whole-genome association studies using fixed marker sets. 2006, 38 (6): 663-667.
Skol AD, Scott LJ, Abecasis GR, Boehnke M: Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet. 2006, 38 (2): 209-213. 10.1038/ng1706.
Cupples LA, Arruda H, Benjamin EJ, D'Agostino SRB, Demissie S, DeStefano AL, Dupuis J, Falls K, CS F, Gottlieb DJ, Govindaraju DR, Guo CY, Heard-Costa N, Hwang SJ, Kathiresan S, Kiel DP, Laramie JM, Larson MG, Levy D, Liu CY, Lunetta KL, Mailman MD, Manning AK, Meigs JB, Murabito JM, Newton-Cheh C, O'Connor GT, O'Donnell CJ, Pandey MA, Seshadri S, Vasan RS, Wang ZY, Wilk JB, Wolf PA, Yang Q, Atwood LD: The Framingham Heart Study 100K SNP genome-wide association study resource: Overview of 17 phenotype working group reports. BMC Med Genet. 2007, 8 (Suppl 1): S1-
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC: Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985, 28 (7): 412-419. 10.1007/BF00280883.
Gutt M, Davis CL, Spitzer SB, Llabre MM, Kumar M, Czarnecki EM, Schneiderman N, Skyler JS, Marks JB: Validation of the insulin sensitivity index (ISI(0,120)): comparison with other measures. Diabetes Res Clin Pract. 2000, 47 (3): 177-184. 10.1016/S0168-8227(99)00116-3.
Hanley AJ, Williams K, Gonzalez C, D'Agostino RB, Wagenknecht LE, Stern MP, Haffner SM: Prediction of type 2 diabetes using simple measures of insulin resistance: combined results from the San Antonio Heart Study, the Mexico City Diabetes Study, and the Insulin Resistance Atherosclerosis Study. Diabetes. 2003, 52 (2): 463-469. 10.2337/diabetes.52.2.463.
Meigs JB, Nathan DM, Wilson PWF, Cupples LA, Singer DE: Metabolic risk factors worsen continuously across the spectrum of nondiabetic glucose tolerance: the Framingham Offspring Study. Annals of Internal Medicine. 1998, 128: 524-533.
Meigs JB, D'Agostino RB, Nathan DM, Rifai N, Wilson PW: Longitudinal association of glycemia and microalbuminuria: the Framingham Offspring Study. Diabetes Care. 2002, 25 (6): 977-983. 10.2337/diacare.25.6.977.
Rutter MK, Meigs JB, Sullivan LM, D'Agostino RB, Wilson PW: Insulin Resistance, the Metabolic Syndrome, and Incident Cardiovascular Events in The Framingham Offspring Study. Diabetes. 2005, 54: 3252-3257. 10.2337/diabetes.54.11.3252.
Horvath S, Wei E, Xu X, Palmer LJ, Baur M: Family-based association test method: age of onset traits and covariates. Genet Epidemiol. 2001, 21 (Suppl 1): S403-408.
Hanson RL, Ehm MG, Pettitt DJ, Prochazka M, Thompson DB, Timberlake D, Foroud T, Kobes S, Baier L, Burns DK, Almasy L, Blangero J, Garvey WT, Bennett PH, Knowler WC: An autosomal genomic scan for loci linked to type II diabetes mellitus and body-mass index in Pima Indians. Am J Hum Genet. 1998, 63 (4): 1130-1138. 10.1086/302061.
Therneau TM, Grambsch PM: Modeling Survival Data: Extending the Cox Model. 2001, New York: Springer
National Center for Biotechnology Information dbGaP website. [http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?id=phs000007]
Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, Lu YT, Roskin KM, Schwartz M, Sugnet CW, Thomas DJ, Weber RJ, Haussler D, Kent WJ: The UCSC Genome Browser Database. Nucleic Acids Res. 2003, 31 (1): 51-54. 10.1093/nar/gkg129.
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D: The human genome browser at UCSC. Genome Res. 2002, 12 (6): 996-1006. 10.1101/gr.229102. Article published online before print in May 2002.
Groves CJ, Zeggini E, Minton J, Frayling TM, Weedon MN, Rayner NW, Hitman GA, Walker M, Wiltshire S, Hattersley AT, McCarthy MI: Association analysis of 6,736 U.K. subjects provides replication and confirms TCF7L2 as a type 2 diabetes susceptibility gene with a substantial effect on individual risk. Diabetes. 2006, 55 (9): 2640-2644. 10.2337/db06-0355.
Zhang C, Qi L, Hunter DJ, Meigs JB, Manson JE, van Dam RM, Hu FB: Variant of Transcription Factor 7-Like 2 (TCF7L2) Gene and the Risk of Type 2 Diabetes in Large Cohorts of U.S. Women and Men. Diabetes. 2006, 55 (9): 2645-2648. 10.2337/db06-0643.
Scott LJ, Bonnycastle LL, Willer CJ, Sprau AG, Jackson AU, Narisu N, Duren WL, Chines PS, Stringham HM, Erdos MR, Valle TT, Tuomilehto J, Bergman RN, Mohlke KL, Collins FS, Boehnke M: Association of transcription factor 7-like 2 (TCF7L2) variants with type 2 diabetes in a Finnish sample. Diabetes. 2006, 55 (9): 2649-2653. 10.2337/db06-0341.
Damcott CM, Pollin TI, Reinhart LJ, Ott SH, Shen H, Silver KD, Mitchell BD, Shuldiner AR: Polymorphisms in the transcription factor 7-like 2 (TCF7L2) gene are associated with type 2 diabetes in the Amish: Replication and evidence for a role in both insulin secretion and insulin resistance. Diabetes. 2006, 55 (9): 2654-2659. 10.2337/db06-0338.
Saxena R, Gianniny L, Burtt NP, Lyssenko V, Guiducci C, Sjögren M, Florez JC, Almgren P, Isomaa B, Orho-Melander M, Lindblad U, Daly MJ, Tuomi T, Hirschhorn JN, Groop L, Altshuler D: Common SNPs in TCF7L2 are reproducibly associated with type 2 diabetes and reduce the insulin response to glucose in non-diabetic individuals. Diabetes. 2006.
Cauchi S, Meyre D, Dina C, Choquet H, Samson C, Gallina S, Balkau B, Charpentier G, Pattou F, Stetsyuk V, Scharfmann R, Staels B, Fruhbeck G, Froguel P: Transcription factor TCF7L2 genetic study in the French population: expression in human beta-cells and adipose tissue and strong association with type 2 diabetes. Diabetes. 2006, 55 (10): 2903-2908. 10.2337/db06-0474.
van Vliet-Ostaptchouk JV, Shiri-Sverdlov R, Zhernakova A, Strengman E, van Haeften TW, Hofker MH, Wijmenga C: Association of variants of transcription factor 7-like 2 (TCF7L2) with susceptibility to type 2 diabetes in the Dutch Breda cohort. Diabetologia. 2006
Field SF, Howson JM, Smyth DJ, Walker NM, Dunger DB, Todd JA: Analysis of the type 2 diabetes gene, TCF7L2, in 13,795 type 1 diabetes cases and control subjects. Diabetologia. 2006
Humphries SE, Gable D, Cooper JA, Ireland H, Stephens JW, Hurel SJ, Li KW, Palmen J, Miller MA, Cappuccio FP, Elkeles R, Godsland I, Miller GJ, Talmud PJ: Common variants in the TCF7L2 gene and predisposition to type 2 diabetes in UK European Whites, Indian Asians and Afro-Caribbean men and women. J Mol Med. 2006
Nicolae DL, Wen X, Voight BF, Cox NJ: Coverage and characteristics of the Affymetrix GeneChip Human Mapping 100K SNP set. PLoS Genet. 2006, 2 (5): e67-10.1371/journal.pgen.0020067.
Acknowledgements and Disclosures
Supported by the by the National Heart, Lung, and Blood Institute's Framingham Heart Study (Contract No. N01-HC-25195), the Boston University Linux Cluster for Genetic Analysis (LinGA) funded by the NIH NCRR Shared Instrumentation grant (1S10RR163736-01A1), National Center for Research Resources (NCRR) General Clinical Research Center (GCRC) M01-RR-01066, and by an American Diabetes Association Career Development Award to Dr. Meigs. Dr. Meigs currently has research grants from GlaxoSmithKline, Wyeth and Sanofi-aventis, and serves on safety or advisory boards for GlaxoSmithKline, Merck, and Lilly. Dr. Florez is supported by the NIH Research Career Award K23 DK65978-03. The funding bodies had no role in the research design and conduct or the decision to publish this study.
This article has been published as part of BMC Medical Genetics Volume 8 Supplement 1, 2007: The Framingham Heart Study 100,000 single nucleotide polymorphisms resource. The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2350/8?issue=S1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
All authors participated in the design and conduct of the study and edited and approved the final manuscript. JM drafted the manuscript and coordinated the study. JM and CF contributed to FHS diabetes-related phenotyping. JD, AM, and and LAC coordinated the data management and conducted the statistical analyses. CL prepared traits for analyses. JF contributed the multiple-related traits method for SNP selection and the literature review for Table 4.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Meigs, J.B., Manning, A.K., Fox, C.S. et al. Genome-wide association with diabetes-related traits in the Framingham Heart Study. BMC Med Genet 8 (Suppl 1), S16 (2007). https://doi.org/10.1186/1471-2350-8-S1-S16
Published:
DOI: https://doi.org/10.1186/1471-2350-8-S1-S16