
Abstract
Background
Charcot-Marie-Tooth neuropathies are a group of genetically heterogeneous diseases of the peripheral nervous system. Mutations in the MFN2 gene have been reported as the primary cause of Charcot-Marie-Tooth disease type 2A.
Methods
Patients with the clinical diagnosis of Charcot-Marie-Tooth type 2 were screened using single strand conformation polymorphism (SSCP). All DNA samples showing band shifts in the SSCP analysis were amplified from genomic DNA and cycle sequenced.
Results
We analyzed a total of 73 unrelated patients with a clinical diagnosis of CMT 2. Overall, novel mutations were detected in 6 patients. c.380G>T (G127V), c.1128G>A (M376I), c.1040A>T (E347V), c.1403G>A (R468H), c.2113G>A (V705I), and c.2258_2259insT (L753fs).
Conclusion
We confirmed a significant role of mutations in MFN2 in the pathogenesis of Charcot-Marie-Tooth disease type 2.
Similar content being viewed by others
Background
Charcot-Marie-Tooth neuropathies (CMT), also named as hereditary motor and sensory neuropathies (HMSN) are a group of genetically heterogeneous diseases of the peripheral nervous system. CMT has been classified into two major subgroups. A severe reduction of nerve conduction velocities (NCV) with NCV < 38 m/s is found in the demyelinating CMT type 1. The axonal type (CMT type 2) is characterized by preferential degeneration of the axon showing amplitude reductions in nerve conduction studies but only mildly reduced NCV. Both CMT1 and CMT2 have been recognized to be genetically heterogeneous. Several genetic loci have been defined for both clinical types. CMT2A was located to chromosome 1p35–p36 [1–3]. Although a mutation in KIF1B was published in a large CMT2A family, no further mutations were detected in other families with CMT2A [4, 5]. Züchner et al. identified the MFN2 gene by means of linkage studies and reported mutations in MFN2 in seven large families with linkage to the CMT2A locus [5]. Recently, other groups confirmed the MFN2 gene as the primary cause of CMT2A [6–8]. We screened 73 unrelated patients with the clinical diagnosis of CMT2 and identified six new disease-causing mutations.
Methods
Subjects
We analyzed samples of 73 unrelated patients sent to our diagnostic laboratory with the request of genetic analysis for Charcot-Marie-Tooth disease. Linkage analysis was not possible in these individuals. Inclusion criteria for the MFN2 analysis were a clinical classification of CMT2, nerve conduction velocity (NCV) > 38 m/s, or histological findings of axonal degeneration. EDTA blood samples were taken from patients after informed consent. DNA was extracted from blood leukocytes by standard methods. All control samples used to check the distribution of sequence alterations mentioned in the results sections were taken from an anonymous collective of ethnically matched samples.
Mutation analysis of MFN2 gene
For mutation analysis in MFN2, primers were designed to amplify all exons including flanking intronic regions. Primer sequences are available on request. Exon 7 and 8, and exon 10 and 11, respectively, were each amplified in one amplicon including the intermediate intron. PCRs were performed in 96-well microtiter plates (Thermowell Costar Corning, NY) using a thermocycler (Biometra, Goettingen, Germany). Each well contained 50 ng DNA in 10 μl reaction volume, GC buffer (Genecraft, Münster, Germany), 10 pMol of forward and reverse primer, 1 U Taq Polymerase (Genecraft, Münster, Germany), 2 mMol of each dNTP, and a MgCl2 concentration of 1 mM. For SSCP analysis, 0.06 μl of [α32 P] dCTP (10 mCi/ml) was included in the PCR. PCR conditions included initial denaturation (2 min at 94°C), two initial cycles at 94°C (15 s) and 6°C and 3°C above the main annealing temperature (30 s) followed by 30 s at 72°C, and 28–32 cycles of 94°C (15 s), annealing temperature (30 s) and final elongation step at 72°C (30 s). Annealing temperature was 62°C except for exons 7/8, 10/11, 15, 16, 17, and 8 (58°C). PCR products were digested with suitable restriction enzymes depending on the fragment size to optimize the mutation detection rate by SSCP analysis. In the SSCP analysis the denatured PCR products were separated by polyacrylamide (PAA) gel electrophoresis using two different conditions: 30% PAA (acrylamide/bisacrylamide: 19/1) gel, 1xTBE, and either 10% glycerol or 5% glycerol/1 M urea. Electrophoresis was carried out at 55 W for 3–4 h at 4°C. Gels were evaluated by autoradiography or exposure to a phosphoimager screen, using the corresponding software. Exon 13 was screened using SSCP and DHPLC. DNA samples showing band shifts in the SSCP analysis were amplified from genomic DNA and cycle sequenced by standard protocols using the Megabace 1000 (Amersham Bioscience, Freiburg, Germany). If possible, restriction analysis with a suitable restriction enzyme was applied to confirm the mutation and to analyse control samples. Enzymes were ordered from New England Biolabs if not indicated otherwise.
Results
We analyzed a total of 73 unrelated patients with a clinical diagnosis of CMT 2. Overall, novel mutations were detected in 6 patients. The clinical features and the neurographical data of these patients are summarized (tables 1 and 2). Nerve biopsy has not been performed in any of these 6 patients.
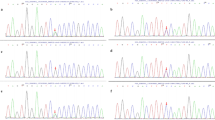
The heterozygous point mutation c.380 G>T in exon 5 of MFN2 (fig. 1a) was detected in a 58 year old female patient (patient 1), predicting the exchange of Glycine to Valine at position 127 of the MFN2 protein (G127V). The mutation leads to a loss of a recognition site for the restriction enzyme Msc I, which was used to exclude this mutation in 97 control persons (194 control chromosomes). Clinically the patient shows a predominant axonal neuropathy with normal motor nerve conduction velocity (mNCV) in electrophysiological studies of the median nerve. The family history is compatible with autosomal-dominant inheritance, but other family members were not available for genetic analysis.

Electropherograms showing novel mutations in the MFN2 gene detected in this analysis. Numbering of nucleotides is according to the open reading frame of the cDNA sequence as deposited in GenBank (GenBank accession no. BC017061).
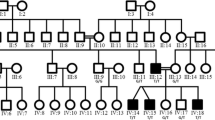
Patient 2, a 34 year old male, is heterozygous for the point mutation c.1128G>A in exon 11, predicting the exchange of Methionine to Isoleucine at position 376 (M376I) (fig. 1b). This mutation is detectable by a loss of a NlaIII restriction site, and was excluded by restriction analysis in 190 control chromosomes. The patient has distal sensory loss in his legs, absent Achilles' tendon reflexes, pes cavus and severely reduced amplitudes of the compound motor nerve action potentials of the peroneal and tibial nerves, but preserved motor nerve conduction velocities. His mother, one brother and one sister reportedly have similar symptoms of peripheral neuropathy but denied further neurophysiological testing or genetic analysis.
The point mutation c.1040A>T in exon 11, leading to the exchange of Glutamine to Valine at position 347 (E347V) of the protein, was detected in a 32 year old male (fig. 1c, patient 3). The mutation was not observed by SSCP analysis in over 200 control chromosomes, detection by restriction analysis was not possible. The parents of the patient were reported to be healthy and unrelated, there is no history of peripheral neuropathies in the family. The patient has distal atrophy of his legs since childhood. He also shows high-grade paresis of peroneus muscles in both legs, severe pain in both legs and distal sensory loss. Electrophysiological studies indicate an axonal neuropathy with mildly decreased mNCV.
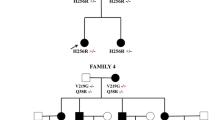
Patient 4, a 27 year old female, presented the mutation c.1403G>A in exon 14 (fig. 1d). The mutation leads to the exchange of Arginine to Histidine (R468H). Clinically the patient shows distal weakness and atrophy of the legs, distal sensory loss and decreased mNCV. Her father has Parkinson disease and distal neuropathy. Her paternal grandfather had pes cavus. Her sister and her mother both do not present any symptoms of neuropathy. Mutation analysis in the parents by restriction with AciI and by sequencing proved the same mutation in her father, indicating cosegregation. However, the mutation was also detected in one of 260 control chromosomes.
A 44 year old female patient (patient 5) with symptoms of peripheral neuropathy since the age of six was found to carry a point mutation c.2113G>A in exon 18 (fig. 1e) leading to the exchange of Valine to Isoleucine at position 705 (V705I). No mutation was found in 212 control chromosomes by restriction digest with HinI (Fermentas). Her deceased father and her brother were reported to have similar symptoms, but family members were not available for genetic analysis. Patient 5 shows distal weakness and atrophy of the lower and upper extremities. She has drop feet and steppage gait, absent Achilles' tendon reflexes, distal sensory loss of the lower extremities and decreased sensitivity to vibration of both ankles.
Patient 6 carries a frameshift mutation (c.2258_2259insT (L753fs)) in exon 19 (fig. 1f). This frameshift mutation causes a change of the last four amino acids of the open reading frame and leads to an extension of five new amino acids at the end of the protein. By restriction analysis of exon 19 with the enzyme PstI, this mutation was not observed in 200 control chromosomes. Molecular genetic analysis was carried out for this patient at the age of 73 years. There is no family history of peripheral neuropathy but he was analyzed under suspect of CMT2 because of symmetric distal weakness and increased distal paresis of the legs.
Discussion
We detected MFN2 mutations in 6 patients out of 73 patients with a clinical diagnosis of axonal CMT2, indicating a rate of 8% similar to the findings around 10% up to 20% observed by other groups. None of these mutations has been previously published, and (except the mutation c.1403G→ A in patient 4) none of these has been found in unrelated controls.
Our data confirm the findings of dominant mutations in the MFN2 gene to be associated with CMT2A [5]. Recently, mutations in MFN2 have also been linked to two rare forms of hereditary neuropathy, namely HMSN V (with pyramidal signs) [8] and HMSN VI (with optic atrophy) [9]. Our patients with mutations in MFN2 have axonal polyneuropathy without additional symptoms. Severity of symptoms is relatively mild and the rate of clinical progression slow.
Although there are several hints linking peripheral neuropathies to the mitochondrial network, the function of mitofusins in peripheral nerves and also the role of MFN2 mutations in the pathogenesis of peripheral neuropathy is only partly understood.
Mitochondria are active organelles forming a single dynamic network whose continuity is maintained by a balance of fission and fusion events [10]. In both yeast and drosophila, mitochondrial fusion is controlled by the nuclear-encoded mitochondrial transmembrane GTPase fuzzy onions (Fzo) [11–13]. In humans and mice MFN2 and MFN1, two human homologues of Fzo exist and both are essential for embryonic development and mitochondrial fusion [13]. MFN2 is the second HMSN disease gene directly involved in the maintenance of the mitochondrial network. In contrast to mitofusins, overexpression of ganglioside-induced differentiation associated protein 1 (GDAP1) is interfering with mitochondrial fusion and induces fragmentation of mitochondria, and recessive mutations in GDAP1 have been found in patients with CMT4A [14, 15].
MFN2 is localized to the outer mitochondrial membrane. The protein contains two coiled-coil regions flanking a transmembrane segment and a GTPase domain. Functional coiled-coil regions are essential for tethering of mitochondria before fusion [16–18]. An intact GTPase domain is indispensable for the function of mitofusins [12, 17, 19]. Overall, more than 25 different mutations in MFN2 have been observed up to now in patients with CMT2A, but also HMSN V and HMSN VI. There is currently no explanation for the divergence of phenotypes associated with mutations in MFN2 [5]. Many mutations are predicted to affect the GTPase domain of the protein, but an equal proportion has been detected for the region linking the GTPase domain and the first coiled coil region (see fig. 2). The mutations described up to now seem to cluster in several hotspot regions (aa94, aa236–251, aa273–284, aa357–364), but we have not found any recurrent mutation.

Structure of the MFN2 protein, showing functional domains and published mutations. Mutations described in this paper are underlined. Mutations associated with HMSN V and HMSN VI are marked with + and *, respectively.
Five out of six mutations described in this paper are missense mutations, like the majority of mutations detected in MFN2 up to now [5–8]. Of the five missense mutations, one (G127V) affects the GTPase domain, and two mutations change the region linking the GTPase domain and the first coiled coil region (E347V and M376I). The two other missense mutations (R468H and V705I) are predicted to cause amino acid substitutions between the transmembrane domain and the coiled coil regions (fig. 2).
The missense mutation c.1403G→ A (R468H) is the first described in the region between the transmembrane domain and the C-terminal coiled coil region, and was detected in patient 4 and her symptomatic father, indicating cosegregation of mutation and disease. The same substitution was detected in one out of 130 anonymous control samples given by blood donors of different ages assumed to be healthy. Since mild symptoms of CMT2 may have been missed in a younger blood donor added to the control collective, this does not exclude the substition R468H as pathogenic, but it may also represent a rare sequence variant. Molecular biological studies may be necessary to finally determine the nature of this substitution.
Another novel mutation (c.1040A>T) found in patient 3 in our series causes the change of the polar glutamine to the non-polar valine at position 347. This amino acid exchange occurred in a highly conserved region of MFN2. This region is one of the indispensable segment of the protein that might provides binding sites for the assembly [18].
Only one stop mutation, R418X, has been described recently in a patient with CMT6 [9]. Interestingly, the frameshift mutation c.2258_2259insT in our patient 6 would result in a protein only differing by four amino acids and an extension of further five new amino acids at the C-terminus of the protein. This mutation may affect the C-terminal coiled-coil domain at the end of the fzo-mitofusin domain. Analysis of MFN2 deletion constructs revealed that GTPase-dependent interaction between the N-terminal and C-terminal tails of MFN2 through their coiled-coil domains and a highly conserved domain in the most N-terminal region is essential for mitochondrial fusion [18]. On the other hand it has been suggested that the C-terminus of MFN2 contains determinants required for targeting of the protein to the mitochondrial membrane [20].
Further studies of promoter domains in the MFN2 gene will help to clarify if these regions are involved in the development of CMT2.
Conclusion
We confirmed a significant role of mutations in MFN2 in the pathogenesis of Charcot-Marie-Tooth disease type 2.
Abbreviations
- CMT:
-
Charcot-Marie-Tooth
- CMT2A:
-
Charcot-Marie-Tooth type 2A
- GDAP1:
-
ganglioside-induced differentiation associated protein 1
- KIF1B:
-
kinesin family member protein 1B
- MFN2:
-
mitofusin 2
- mNCV:
-
motor nerve conduction velocity
- SSCP:
-
single strand conformation polymorphism
References
Ben Othmane K, Middleton LT, Loprest LJ, Wilkinson KM, Lennon F, Rozear MP, Stajich JM, Gaskell PC, Roses AD, Pericak-Vance MA, et al: Localization of a gene (CMT2A) for autosomal dominant Charcot-Marie-Tooth disease type 2 to chromosome 1p and evidence of genetic heterogeneity. Genomics. 1993, 17 (2): 370-375. 10.1006/geno.1993.1334.
Harding AE, Thomas PK: The clinical features of hereditary motor and sensory neuropathy types I and II. Brain. 1980, 103 (2): 259-280.
Kuhlenbäumer G, Young P, Hunermund G, Ringelstein B, Stogbauer F: Clinical features and molecular genetics of hereditary peripheral neuropathies. J Neurol. 2002, 249 (12): 1629-1650. 10.1007/s00415-002-0946-3.
Zhao C, Takita J, Tanaka Y, Setou M, Nakagawa T, Takeda S, Yang HW, Terada S, Nakata T, Takei Y, Saito M, Tsuji S, Hayashi Y, Hirokawa N: Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell. 2001, 105 (5): 587-597. 10.1016/S0092-8674(01)00363-4.
Züchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgrafov O, Jonghe PD, Takahashi Y, Tsuji S, Pericak-Vance MA, Quattrone A, Battaloglu E, Polyakov AV, Timmerman V, Schröder JM, Vance JM: Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004, 36 (5): 449-451. 10.1038/ng1341.
Kijima K, Numakura C, Izumino H, Umetsu K, Nezu A, Shiiki T, Ogawa M, Ishizaki Y, Kitamura T, Shozawa Y, Hayasaka K: Mitochondrial GTPase mitofusin 2 mutation in Charcot-Marie-Tooth neuropathy type 2A. Hum Genet. 2005, 116 (1-2): 23-27. 10.1007/s00439-004-1199-2.
Lawson VH, Graham BV, Flanigan KM: Clinical and electrophysiologic features of CMT2A with mutations in the mitofusin 2 gene. Neurology. 2005, 65 (2): 197-204.
Zhu D, Kennerson ML, Walizada G, Züchner S, Vance JM, Nicholson GA: Charcot-Marie-Tooth with pyramidal signs is genetically heterogeneous: families with and without MFN2 mutations. Neurology. 2005, 65 (3): 496-497.
Züchner S, De Jonghe P, Jordanova A, Claeys KG, Guergueltcheva V, Cherninkova S, Hamilton SR, Van Stavern G, Krajewski KM, Stajich J, Tournev I, Verhoeven K, Langerhorst CT, de Visser M, Baas F, Bird T, Timmerman V, Shy M, Vance JM: Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann Neurol. 2006, 59 (2): 276-281. 10.1002/ana.20797.
Nunnari J, Marshall WF, Straight A, Murray A, Sedat JW, Walter P: Mitochondrial transmission during mating in Saccharomyces cerevisiae is determined by mitochondrial fusion and fission and the intramitochondrial segregation of mitochondrial DNA. Mol Biol Cell. 1997, 8 (7): 1233-1242.
Hermann GJ, Shaw JM: Mitochondrial dynamics in yeast. Annu Rev Cell Dev Biol. 1998, 14: 265-303. 10.1146/annurev.cellbio.14.1.265.
Hales KG, Fuller MT: Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell. 1997, 90 (1): 121-129. 10.1016/S0092-8674(00)80319-0.
Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC: Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003, 160 (2): 189-200. 10.1083/jcb.200211046.
Nelis E, Erdem S, Van Den Bergh PY, Belpaire-Dethiou MC, Ceuterick C, Van Gerwen V, Cuesta A, Pedrola L, Palau F, Gabreels-Festen AA, Verellen C, Tan E, Demirci M, Van Broeckhoven C, De Jonghe P, Topaloglu H, Timmerman V: Mutations in GDAP1: autosomal recessive CMT with demyelination and axonopathy. Neurology. 2002, 59 (12): 1865-1872.
Niemann A, Ruegg M, La Padula V, Schenone A, Suter U: Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: new implications for Charcot-Marie-Tooth disease. J Cell Biol. 2005, 170 (7): 1067-1078. 10.1083/jcb.200507087.
Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC: Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004, 305 (5685): 858-862. 10.1126/science.1099793.
Santel A, Fuller MT: Control of mitochondrial morphology by a human mitofusin. J Cell Sci. 2001, 114 (Pt 5): 867-874.
Honda S, Aihara T, Hontani M, Okubo K, Hirose S: Mutational analysis of action of mitochondrial fusion factor mitofusin-2. J Cell Sci. 2005, 118 (Pt 14): 3153-3161. 10.1242/jcs.02449.
Eura Y, Ishihara N, Yokota S, Mihara K: Two mitofusin proteins, mammalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J Biochem (Tokyo). 2003, 134 (3): 333-344.
Rojo M, Legros F, Chateau D, Lombes A: Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci. 2002, 115 (Pt 8): 1663-1674.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/7/53/prepub
Acknowledgements
We thank the patients and their families for their cooperation
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
KE carried out the molecular genetic studies, KE and MM participated in the sequence alignment and drafted the manuscript. MH participated and assisted in molecular genetic studies and the sequence alignment. MM conceived of the study, and participated in the design. JTE participated in its design and coordination and helped to draft the manuscript. MV, GH and JG made major contributions to the clinical characterisation of the patients. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Engelfried, K., Vorgerd, M., Hagedorn, M. et al. Charcot-Marie-Tooth neuropathy type 2A: novel mutations in the mitofusin 2 gene (MFN2). BMC Med Genet 7, 53 (2006). https://doi.org/10.1186/1471-2350-7-53
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-7-53