
Abstract
Background
Population heterogeneity may be a significant confounding factor hampering detection and verification of late onset Alzheimer's disease (LOAD) susceptibility genes. The Amish communities located in Indiana and Ohio are relatively isolated populations that may have increased power to detect disease susceptibility genes.
Methods
We recently performed a genome scan of dementia in this population that detected several potential loci. However, analyses of these data are complicated by the highly consanguineous nature of these Amish pedigrees. Therefore we applied the Combinatorial Mismatch Scanning (CMS) method that compares identity by state (IBS) (under the presumption of identity by descent (IBD)) sharing in distantly related individuals from such populations where standard linkage and association analyses are difficult to implement. CMS compares allele sharing between individuals in affected and unaffected groups from founder populations. Comparisons between cases and controls were done using two Fisher's exact tests, one testing for excess in IBS allele frequency and the other testing for excess in IBS genotype frequency for 407 microsatellite markers.
Results
In all, 13 dementia cases and 14 normal controls were identified who were not related at least through the grandparental generation. The examination of allele frequencies identified 24 markers (6%) nominally (p ≤ 0.05) associated with dementia; the most interesting (empiric p ≤ 0.005) markers were D3S1262, D5S211, and D19S1165. The examination of genotype frequencies identified 21 markers (5%) nominally (p ≤ 0.05) associated with dementia; the most significant markers were both located on chromosome 5 (D5S1480 and D5S211). Notably, one of these markers (D5S211) demonstrated differences (empiric p ≤ 0.005) under both tests.
Conclusion
Our results provide the initial groundwork for identifying genes involved in late-onset Alzheimer's disease within the Amish community. Genes identified within this isolated population will likely play a role in a subset of late-onset AD cases across more general populations. Regions highlighted by markers demonstrating suggestive allelic and/or genotypic differences will be the focus of more detailed examination to characterize their involvement in dementia.
Similar content being viewed by others
Background
With over 4 million individuals affected with Alzheimer's disease (AD) in the U.S., dementia of the Alzheimer's Type (DAT) is the leading cause of dementia in the elderly. These current estimates are projected to triple over the next 50 years as the population ages [1–3]. AD has a complex etiology with strong genetic and environmental determinants. Tremendous evidence suggests the involvement of at least three genes in early-onset autosomal dominant AD. Amyloid precursor protein (APP on chromosome 21) [4, 5], presenilin I (PSEN1 on chromosome 14) [6–9], and presenilin II (PSEN2 on chromosome 1) [10, 11] are all prominent early-onset Alzheimer's disease genes. Understanding of the more common late-onset Alzheimer disease (LOAD), is centered on the role of one universally accepted risk gene, the apolipoprotein E locus (APOE) [12]. The APOE ε4 allele (frequency approximately 16%) [13, 14] acts in a dose-related manner to increase risk for LOAD and decrease age-of-onset [15, 16]. Although its involvement is without question, APOE accounts for less than half of late-onset AD susceptibility [15]. Given the strong heritability of AD, other genetic factors are likely to be involved. Multiple linkage screens have been conducted to elucidate additional regions harboring susceptibility genes for late-onset AD [17–35]. While regions on chromosomes 9, 10 and 12 are most consistently identified, candidate genes within those regions have yet to be clearly implicated in AD. Meanwhile, numerous other regions have been implicated but have not been the focus of detailed study due to the prominence of 9, 10, and 12.
Though numerous promising LOAD candidate genes have been examined, the lack of replication across studies has made a definitive declaration of their involvement difficult (Reviewed in [36, 37]). Genetic heterogeneity is likely to be one of the underlying reasons for this lack of replication. Given this, one possible solution is to study populations likely to be more genetically homogeneous, thereby enriching for a more homogeneous set of risk alleles. The North American Amish population is a relatively isolated, genetically well-defined homogeneous population, well-suited for this type of study. Further detail regarding the establishment of the North American Amish population has been described elsewhere [38–43]. While there may be a number of LOAD susceptibility genes contributing to disease in the general population, the relatively homogeneous Amish population is likely to contain a smaller set of risk alleles.
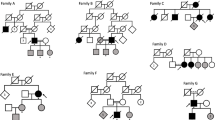
One challenge in performing linkage analysis in Amish pedigrees is to utilize the extensive pedigree information available while maintaining tractability of the computations. Due to their strong religious and cultural beliefs, the Amish very rarely marry outside of their communities, thereby promoting a genetically isolated population [38–41]. This in turn has led to an elevated degree of consanguinity, yielding family pedigrees that contain many loops that can often be traced back three or more generations. In fact, through use of the Anabaptist Genealogy Database (AGDB), we find that 93% of our overall study population of 460 individuals and more specifically 25/27 (93%) of the individuals used in this study can be traced back 10 generations to a single founding couple. Moreover, 100% of individuals within our entire Amish sample (460) belong to one very large extended pedigree when allowing parent-child and marriage links to be included [44]. Since LOAD cannot be ascertained until late in life, affected individuals are usually only available for genotyping in a single generation. Hence, by far, most of the individuals in the pedigree have unknown phenotype and genotype status. It should be noted that there are limited methodologies available to analyze disease gene linkage utilizing such large complex pedigrees. One such method is SimWalk2 which utilizes descent graph theory and Markov Chain Monte Carlo (MCMC) simulation to compute lod scores [45]. This is a computationally demanding process and because of the uncertainty of MCMC convergence, the accuracy of the scores obtained may be difficult to assess.
Combinatorial mismatch scanning (CMS) is an alternative technique to search for IBS sharing in distantly related individuals from isolated founder populations where standard linkage and association analyses are difficult to implement. While several other methods could be implemented, this approach was used because of its simplicity in examining existing data. This method was also chosen because at the onset of analysis, we lacked the more detailed knowledge of the inter-relatedness of our sample often required to perform similar, but more sophisticated approaches within large inbred pedigrees [46–48]. This strategy is designed to circumvent the confounding issue of genetic heterogeneity, by examining affected and unaffected persons from relatively small founder populations [49]. By genetically evaluating case and control individuals selected from such a population, whose common ancestor is no more closely related than grandparents, some prevailing problems in allelic association studies of complex disease within generally outbred populations can be avoided. Population stratification can lead to allelic association and be misinterpreted as linkage disequilibrium. In this approach, population stratification is less of an issue due to the relative isolation and common heritage of the study population. Another difficulty facing genetic studies within large outbred populations is that these populations are likely to exhibit locus heterogeneity. Within an isolated population, the probability that the risk allele of interest might have entered the gene pool only once or rarely, provides a great advantage. This in turn will likely facilitate the distinction between true and spurious association. Heath and colleagues highlight another advantage of examining isolated populations by alluding to the potential for detecting gene-gene interactions [49]. These epistatic interactions may play a substantial role in complex disease, effectively hampering the ability to detect association using single locus methods within heterogeneous populations. With reference to this problem, there are good reasons to believe that epistatic (gene-gene) interactions are ubiquitous in complex disease and may in fact be more important than single-gene effects [50].
Methods
Subjects and phenotypes
The Amish often have large sibships and extensive pedigree records that permit the accurate estimation of IBS gene sharing to be accurately evaluated. The estimated coefficient of inbreeding for the entire population is 0.0151, which is approximately equivalent to having second cousins as parents [51]. This effect has led present-day Amish to possess genes inherited identically from a common ancestor at rates higher than observed in the general population. By searching the Anabaptist Genealogy Database (AGDB) with the query software PedHunter, we have determined the level of relatedness of our sample more precisely [44, 52]. We calculated the average kinship coefficient for our overall ascertained Amish sample to be 0.019 ± 0.00053 (mean ± SEM). This measure demonstrates a significant difference from the average kinship coefficients calculated for the within cases group (0.011 ± 0.0013, mean ± SEM), the within controls group (0.0094 ± 0.0011, mean ± SEM), and the between cases and controls group (0.010 ± 0.00065, mean ± SEM). These calculations provide us with additional confidence that our cases and controls are more distantly related to each other relative to our overall sample population.
The subjects included in this study are a subset of individuals described in extensive detail elsewhere [43]. Briefly, individuals enrolled in the study each were assigned to one of three clinical impression categories; dementia (probable or possible Alzheimer's disease); unclear (includes mild cognitive impairment (MCI)); or unaffected (cognitively normal). Participants were administered the Mini-Mental State Exam (MMSE) [53], with possible scores ranging from 0 to 30. All individuals scoring 27 or greater were classified as cognitively normal/unaffected. Those scoring 23 or less were classified as cognitively impaired and labeled as probable dementia. Those who scored 24–26 had additional neuropsychological testing including the Dementia Rating Scale (DRS) [54], the Boston Naming Test (BNT) [55], and a reading subtest from the Wide Range Achievement Test-Revised (WRAT-R) [56]. Persons were categorized as having mild cognitive impairment if their DRS score fell below an age-adjusted threshold. Each case was discussed and a consensus "final" diagnosis was determined using all available information. For analytical purposes, the cases were classified as affected (demented), unclear (includes MCI), and unaffected (cognitively normal).
Five Amish pedigrees were included in this study. Three families were from Elkhart and LaGrange counties in Indiana, one extended family from Adams county Indiana, and one extended family from Holmes county Ohio. The extended pedigree from Adams county has been the subject of other previous and ongoing studies of dementia in the Amish [16, 57]. Among the 115 individuals who were genotyped, 40 were classified as having dementia, 9 were classified as unclear, and 66 individuals were unaffected. To minimize chance IBS inheritance, individuals selected for the CMS analysis were unrelated through the grandparental generation [49]. For this current study we identified 13 dementia cases and 14 cognitively normal individuals who met this requirement. This study was undertaken after Institutional Review Board review and approval.
Molecular analysis
Following informed consent, blood samples were collected from each individual and genomic DNA was extracted from blood using standard procedures. Cell lines have been initiated on most sampled individuals. All DNA samples were coded and stored at 4°C until used.
Markers were genotyped at both the Vanderbilt and Duke laboratories for all DNA samples. Laboratory personnel were blinded to pedigree structure, affection status, and location of quality control samples. Duplicate quality control samples were placed both within and across plates and equivalent genotypes were required for all quality control samples to ensure accurate genotyping. At the Vanderbilt laboratory, marker primer sequences were obtained from the Genome Database [58] or designed with Primer3 software [59] and synthesized by Invitrogen Life Technologies (Carlsbad, CA). Amplification was performed in a PCR Express machine (ThermoHybaid, Needham Heights, MA) with the following conditions: 94°C-4 min.; 94°C-15 sec., AT-30 sec., 72°C-45 sec. (35 cycles); 72°C-4 min. PCR products were denatured for 3 min. at 95°C and run on a 6% polyacrylamide gel (Sequagel-6®from National Diagnostics, Atlanta, GA) for ~1 hr. at 75 W. Gels were stained with a SybrGold®rinse (Molecular Probes, Eugene, OR) and scanned with the Hitachi Biosystems FMBIOII laser scanner (Brisbane, CA). Marker genotyping at the Duke laboratory was performed using fluorescence imaging (Molecular Dynamics SI Fluorimager) and a semi-automated allele calling system [60].
Hardy-Weinberg equilibrium calculations were performed for each marker and Mendelian inconsistencies were identified using PedCheck in the overall dataset [61]. Suspect genotypes were re-read by a different technician or re-run as necessary to reduce errors. All microsatellite markers were required to have >90% of possible genotypes to be included in the analysis.
Statistical analysis
Comparisons between case and control genotype data for the 407 microsatellite markers were conducted using the R software package to perform Fisher's exact tests in r × c Contingency Tables [62–64]. Each marker was examined for both allele and genotype differences between individuals affected with dementia and those without dementia. The first test was for IBS allele frequency inequality between the two classes, and the second test was for IBS genotype frequency inequality between the two classes. Fisher's exact test computes the probability p that the pattern of alleles observed in the sample would be obtained if there were truly no difference between the allele frequencies among affected and unaffected individuals. While our current sample size is adequate to detect moderate to major effects (odds ratio of >6 with 80% power), it does not preclude our ability to detect smaller effects given that these power calculations are based on the assumptions of complete independence of samples and random sampling of the population, neither of which is true.
To empirically evaluate the statistical significance of the p-values computed in the CMS study, we permuted our dataset. We randomly re-assigned affection status for each of the 27 individuals maintaining the original total of 13 cases and 14 controls. We then executed the Fisher's exact test using the same allele and genotype data in the original dataset for each of the 407 markers. The distribution of p-values obtained from Fisher's exact testing on 1000 randomized sets of data was then created for both the allele and genotype comparisons to assess the empiric thresholds. We would expect the Fisher's exact p-value to match the p-value within the large distribution. These permutations were needed to correct for any residual bias from unrecognized kinship correlation present.
Results
We tested 407 microsatellite markers for differences in both allele frequency and genotype frequency between Amish dementia cases and controls. We considered all pointwise p-values and have chosen to report only markers demonstrating Fisher's exact p-values < 0.05 for either allele or genotype frequency differences. This arbitrary threshold was chosen to limit the results to be displayed and to provide a reference point for discussion of markers demonstrating nominally significant (albeit within the null expectation given the number of markers examined) evidence of association to dementia within our population.
As an example, Table 1a shows the allele count data for marker D5S211. There are eight D5S211 alleles in the sample with 27 subjects being typed for 54 alleles. In the example, the probability of this data being obtained if there were no underlying difference between the allele distributions for the two classes is < 0.005 (Table 2). Table 1b shows the genotype data for marker D5S211. Note that out of the thirteen genotypes observed in the data only one genotype is present in both affected and unaffected classes. The probability of the data being obtained if there were no underlying difference between the genotype distributions for the case and controls is < 0.005 (Table 2). Markers demonstrating nominally significant (p ≤ 0.05) differences between cases and controls are listed in Table 2. There were 24 out of 407 markers (6%) demonstrating significant differences in allele frequency. The most significant markers were D3S1262, D5S211, and D19S1165. When examining the markers for genotype frequency differences, 21 out of 407 markers (5%) were significantly different between our dementia cases and controls. The most significant markers were D5S1480 and D5S211. While there were seven markers (D3S1262, D4S1625, D5S211, D6S1031, D8S1477, D8S272, D17S921, and D18S481) with p-values ≤ 0.05 for both the allelic and genotypic tests, only one marker (D5S211) was significant at the empiric p ≤ 0.005 level for both tests. Although all findings are uncorrected and there are no findings with genome-wide significance, markers in close proximity to those regions previously identified are of particular interest for future study.
Discussion
We have detected a few microsatellite markers of particular interest, which demonstrate significant differences between dementia cases and controls within our Amish founder population using the combinatorial mismatch scanning approach. The CMS concept is based on excess IBS allele/genotype sharing between individuals sharing a distant set of common founders [49]. The most noteworthy finding is on chromosome 5q35.2 at approximately 183 cM where we find evidence for both allele and genotype differences between our dementia cases and controls for marker D5S211. In their large genome-wide linkage study of Alzheimer's disease, Blacker et al. detected a multipoint lod score of 1.3 at this same marker [32]. In a recent study of consanguineous Israeli-Arab communities, Farrer and colleagues found significant evidence for allele frequency differences between AD cases and controls at the closest marker (D5S400 at 175 cM) on chromosome 5 run in their study [33]. Positive findings across three distinct study populations suggest that a gene or genes within this region of chromosome 5 may be involved in risk for dementia of the Alzheimer's type (DAT). Thus future examination of this region on chromosome 5 within our Amish families is warranted. While another marker on chromosome 5q31.3 (D5S1480 at 147 cM) demonstrated genotype differences between dementia cases and controls, this location is novel with respect to other previous studies.
We also found evidence of allele frequency differences on chromosome 3q27.3 at D3S1262 (201 cM). One study tested for association with AD in a geographically distinct Finnish population descended from a small group of original founders [65]. This group found significant association (empiric p = 0.007) at marker D3S1602 (also located at 201 cM) within their AD sample. An interesting candidate gene at this location is SST, the gene encoding somatostatin, which functions as a neurotransmitter in the central nervous system. Somatostatin inhibits the release of glucagon, growth hormone, gastrin, insulin, and secretin. Additional evidence for this region stems from our genome-wide linkage study within the Amish population, for which we observed a suggestive two-point lod score of 2.42 at the nearby marker D3S2398 (209 cM) [43].
An additional marker demonstrating suggestive allele frequency differences is located on chromosome 19p13.2 (D19S1165 at 36 cM). Hiltunen et al. had detected evidence for association at two nearby markers (D19S1034 and D19S433) spanning the region containing our significant results [65]. ICAM-1 (Intercellular Adhesion Molecule 1), a previously-reported AD candidate gene, also lies within this region of interest. Pola et al. showed that the ICAM-1 K469E gene polymorphism was associated with AD in an Italian population [66]. This association was not, however, supported in studies of the gene in Finnish and Spanish populations [67, 68]. Additional strong evidence from previous work indicates the presence of a late-onset AD locus within this region. A study by Wijsman et al. provides substantial evidence for a locus at approximately 35 cM affecting AD age at onset [69]. While our study does not address age at onset, it further suggests the involvement of this region in AD.
Another region of relative interest is on chromosome 4q31.2 at marker D4S1625. This marker located on chromosome 4q at approximately 146 cM lies between two markers (D4S2394 at 130 cM and D4S1548 at 154 cM) demonstrating highly suggestive evidence for linkage within our Amish population [43]. Further evidence for this region stems from work by Pericak-Vance et al. where they detect modest evidence for linkage to a marker only 4 cM away (D4S1629, lod = 1.32) from D4S1625 (Table 2) [26].
Given that our data may violate assumptions (i.e. normality and/or unrealized correlation) of the Fisher's exact test, we determined the empiric p-value for our results through permutation. We performed the Fisher's exact test on 1000 replicates containing the same original genotype data, but with randomized affection status. The resulting distribution of p-values was then used as an empiric measure of significance for our results (Table 2). On the whole, the empiric p-value thresholds for our study showed the Fisher's exact p-value to be somewhat more liberal than expected.
We have previously performed a genome-wide linkage screen for dementia within this population; however the complex nature of the Amish pedigrees provides a challenge for linkage analysis, given the size and number of consanguineous loops within these extended families. Accordingly the linkage analysis by itself does not allow taking full advantage of the data available to us. To examine our data more thoroughly, we performed the combinatorial mismatch scan. Both this approach and the linkage analysis utilize the high level of inter-relatedness, within the Amish population, to their advantage. The nature of the CMS analysis, allowed us to examine these data without being computationally burdened by the size or family structure of our population. These two methods complement each other by allowing the examination of the same data using both a family-based approach and a "pseudo" case-control approach to identify regions across the genome which are potentially involved in AD susceptibility. We are fully aware of the limited power of our current sample; however, these analyses should be viewed as an adjunct to our recent genomic screen.
Conclusion
We have reported several markers across the genome (chr3, 4, 5, and 19) to have significant allelic and/or genotypic frequency differences between dementia cases and controls within the combined Amish communities of Ohio and Indiana. While the evidence presented here is not overwhelming for any specific region, these results must be viewed in conjunction with not only our genomic screen but with findings across other studies within additional populations. In conclusion, our results provide the groundwork for future detailed study of these regions within our growing sample of Amish individuals.
References
Geldmacher DS, Whitehouse PJ: Differential diagnosis of Alzheimer's disease. Neurology. 1997, 48: S2-S9.
Brookmeyer R, Gray S, Kawas C: Projections of Alzheimer's disease in the United States and the public health impact of delaying disease onset. Am J Public Health. 1998, 88: 1337-1342.
Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA: Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol. 2003, 60: 1119-1122. 10.1001/archneur.60.8.1119.
St George-Hyslop PH, Tanzi RE, Polinsky RJ, Haines JL, Watkins PC, Myers RH, Feldman RG, Pollen D, Drachman D: The genetic defect causing familial Alzheimer's disease maps on chromosome 21. Science. 1987, 235: 885-890.
Goate AM, Chartier-Harlin MC, Mullan MC, Brown J, Crawford F, Fidani L, Guiffra L, Haynes A, Irving N, James L, Mant R, Newton P, Rooke K, Roques P, Talbot C, Pericak-Vance MA, Roses AD, Williamson R, Rossor M, Owen M, Hardy J: Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991, 33: 53-56.
St George-Hyslop P, Haines JL, Rogaev E, Mortilla M, Vaula G, Pericak-Vance MA, Foncin J-F, Montesi M, Bruni A, Sorbi S, Rainero I, Pinessi L, Pollen D, Polinsky R, Nee L, Kennedy J, Macciardi F, Rogaeva E, Liang Y, Alexandrova N, Lukiw W, Schlumpf K, Tanzi R, Tsuda T, Farrer L, Cantu J-M, Duara R, Amaducci L, Bergamini L, Gusella J, Roses A, McLachlan DC: Genetic evidence for a novel familial Alznheimer's disease locus on chromosome 14. Nature Genetics. 1992, 2: 330-334. 10.1038/ng1292-330.
Schellenberg GD, Bird TD, Wijsman EM, Orr HT, Anderson L, Nemens E, White JA, Bonnycastle L, Weber JL, Alonso ME: Genetic linkage evidence for a familial Alzheimer's disease locus on chromosome 14. Science. 1992, 258: 668-671.
Van Broeckhoven C, Backhovens H, Cruts M, De Winter G, Bruyland M, Cras P, Martin JJ: Mapping of a gene predisposing to early-onset Alzheimer's disease to chromosome 14q24.3. Nat Genet. 1992, 2: 335-339. 10.1038/ng1292-335.
Sherrington R, Rogaev E, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin J-F, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, DaSilva HAR, Haines JL, Pericak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St George-Hyslop PH: Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995, 375: 754-760. 10.1038/375754a0.
Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, Crowley AC, Fu Y-H, Guenette SY, Galas D, Nemens E, Wijsman EM, Bird TD, Schellenberg GD, Tanzi RE: Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995, 269: 973-977.
Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang G, Chi H, Lin C, Holman K, Tsuda T, Mar L, Sorbi S, Nacmias B, Placentini S, Amaducci L, Chumakov I, Cohen D, Lannfelt L, Fraser PE, Rommens JM, St George-Hyslop PH: Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature. 1995, 376: 775-778. 10.1038/376775a0.
Pericak-Vance MA, Bebout JL, Gaskell PC, Yamaoka LH, Hung W-Y, Alberts MJ, Walker AP, Bartlett RJ, Haynes CS, Welsh KA, Earl NL, Heyman A, Clark CM, Roses AD: Linkage studies in familial Alzheimer's disease: evidence for chromosome 19 linkage. Am J Hum Genet. 1991, 48: 1034-1050.
Menzel H-J, Kladetzky RG, Asman G: Apolipoprotein E polymorphism and coronary artery disease. Arteriosclerosis. 1983, 3: 310-315.
Saunders AM, Strittmatter WJ, Breitner JC, Schmechel D, St George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Growden J, Alberts MJ, Hulette C, Crain B, Goldgaber D, Roses AD: Association of apolipoprotein E allele 4 with late-onset familial and sporadic Alzheimer's disease. Neurology. 1993, 43: 1467-1472.
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA: Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993, 261: 921-923.
Pericak-Vance MA, Johnson CC, Rimmler JB, Saunders AM, Robinson LC, D'Hondt EG, Jackson CE, Haines JL: Alzheimer's disease and apolipoprotein E-4 allele in an Amish population. Annals of Neurology. 1996, 39: 700-704. 10.1002/ana.410390605.
Pericak-Vance MA, Yamaoka LH, Haynes CS, Speer MC, Haines JL, Gaskell PC, Hung W-Y, Clark CM, Heyman A, Trofatter J, Eisenmenger JP, Gilbert JR, Lee JE, Alberts MJ, Dawson DV, Bartlett RJ, Earl NL, Siddique T, Vance JM, Conneally PM, Roses AD: Genetic linkage studies in Alzheimer's disease families. Experimental Neurology. 1988, 102: 271-279. 10.1016/0014-4886(88)90220-8.
Pericak-Vance MA, Bass MP, Yamaoka LH, Gaskell PC, Scott WK, Terwedow HA, Menold MM, Conneally PM, Small GW, Vance JM, Saunders AM, Roses AD, Haines JL: Complete genomic screen in late-onset familial Alzheimer disease: evidence for a new locus on chromosome 12. JAMA. 1997, 278: 1237-1241. 10.1001/jama.278.15.1237.
Scott WK, Hauser ER, Schmechel DE, Welsh-Bohmer KA, Small GW, Roses AD, Saunders AM, Gilbert JR, Vance JM, Haines JL, Pericak-Vance MA: Ordered-subsets linkage analysis detects novel Alzheimer disease loci on chromosomes 2q34 and 15q22. Am J Hum Genet. 2003, 73: 1041-1051. 10.1086/379083.
Pericak-Vance MA, Bass ML, Yamaoka LH, Gaskell PC, Scott WK, Terwedow HA, Menold MM, Conneally PM, Small GW, Saunders AM, Roses AD, Haines JL: Complete genomic screen in late-onset familial Alzheimer's disease. Neurobiol Aging. 1998, 19: S39-S42. 10.1016/S0197-4580(98)00037-2.
Zubenko GS, Hughes HB, Stiffler JS, Hurtt MR, Kaplan BB: A genome survey for novel Alzheimer disease risk loci: results at 10-cM resolution. Genomics. 1998, 50: 121-128. 10.1006/geno.1998.5306.
Zubenko GS, Stiffler JS, Hughes HB, Hurtt MR, Kaplan BB: Initial results of a genome survey for novel Alzheimer's disease risk genes: association with a locus on the X chromosome. Am J Med Genet. 1998, 81: 196-205. 10.1002/(SICI)1096-8628(19980328)81:2<196::AID-AJMG14>3.0.CO;2-C.
Rogaeva E, Premkumar S, Song Y, Sorbi S, Brindle N, Paterson A, Duara R, Levesque G, Yu G, Nishimura M, Ikeda M, O'Toole C, Kawarai T, Jorge R, Vilarino D, Bruni AC, Farrer LA, St George-Hyslop PH: Evidence for an Alzheimer disease susceptibility locus on chromosome 12 and for further locus heterogeneity. JAMA. 1998, 280: 614-618. 10.1001/jama.280.7.614.
Kehoe P, Wavrant-De VF, Crook R, Wu WS, Holmans P, Fenton I, Spurlock G, Norton N, Williams H, Williams N, Lovestone S, Perez-Tur J, Hutton M, Chartier-Harlin MC, Shears S, Roehl K, Booth J, Van Voorst W, Ramic D, Williams J, Goate A, Hardy J, Owen MJ: A full genome scan for late onset Alzheimer's disease. Hum Mol Genet. 1999, 8: 237-245. 10.1093/hmg/8.2.237.
Scott WK, Grubber JM, Conneally PM, Small GW, Hulette CM, Rosenberg CK, Saunders AM, Roses AD, Haines JL, Pericak-Vance MA: Fine mapping of the chromosome 12 late-onset alzheimer disease locus: potential genetic and phenotypic heterogeneity. Am J Hum Genet. 2000, 66: 922-932. 10.1086/302828.
Pericak-Vance MA, Grubber J, Bailey LR, Hedges D, West S, Santoro L, Kemmerer B, Hall JL, Saunders AM, Roses AD, Small GW, Scott WK, Conneally PM, Vance JM, Haines JL: Identification of novel genes in late-onset Alzheimer's disease. Exp Gerontol. 2000, 35: 1343-1352. 10.1016/S0531-5565(00)00196-0.
Myers A, Holmans P, Marshall H, Kwon J, Meyer D, Ramic D, Shears S, Booth J, DeVrieze FW, Crook R, Hamshere M, Abraham R, Tunstall N, Rice F, Carty S, Lillystone S, Kehoe P, Rudrasingham V, Jones L, Lovestone S, Perez-Tur J, Williams J, Owen MJ, Hardy J, Goate AM: Susceptibility locus for Alzheimer's disease on chromosome 10. Science. 2000, 290: 2304-2305. 10.1126/science.290.5500.2304.
Bertram L, Blacker D, Mullin K, Keeney D, Jones J, Basu S, Yhu S, McInnis MG, Go RC, Vekrellis K, Selkoe DJ, Saunders AJ, Tanzi RE: Evidence for genetic linkage of Alzheimer's disease to chromosome 10q. Science. 2000, 290: 2302-2303. 10.1126/science.290.5500.2302.
Ertekin-Taner N, Graff-Radford N, Younkin LH, Eckman C, Baker M, Adamson J, Ronald J, Blangero J, Hutton M, Younkin SG: Linkage of plasma Abeta42 to a quantitative locus on chromosome 10 in late-onset Alzheimer's disease pedigrees. Science. 2000, 290: 2303-2304. 10.1126/science.290.5500.2303.
Myers A, Wavrant De-Vrieze F, Holmans P, Hamshere M, Crook R, Compton D, Marshall H, Meyer D, Shears S, Booth J, Ramic D, Knowles H, Morris JC, Williams N, Norton N, Abraham R, Kehoe P, Williams H, Rudrasingham V, Rice F, Giles P, Tunstall N, Jones L, Lovestone S, Williams J, Owen MJ, Hardy J, Goate A: Full genome screen for Alzheimer disease: stage II analysis. Am J Med Genet. 2002, 114: 235-244. 10.1002/ajmg.10183.
Mayeux R, Lee JH, Romas SN, Mayo D, Santana V, Williamson J, Ciappa A, Rondon HZ, Estevez P, Lantigua R, Medrano M, Torres M, Stern Y, Tycko B, Knowles JA: Chromosome-12 mapping of late-onset Alzheimer disease among Caribbean Hispanics. Am J Hum Genet. 2002, 70: 237-243. 10.1086/324773.
Blacker D, Bertram L, Saunders AJ, Moscarillo TJ, Albert MS, Wiener H, Perry RT, Collins JS, Harrell LE, Go RC, Mahoney A, Beaty T, Fallin MD, Avramopoulos D, Chase GA, Folstein MF, McInnis MG, Bassett SS, Doheny KJ, Pugh EW, Tanzi RE: Results of a high-resolution genome screen of 437 Alzheimer's disease families. Hum Mol Genet. 2003, 12: 23-32. 10.1093/hmg/ddg007.
Farrer LA, Bowirrat A, Friedland RP, Waraska K, Korczyn AD, Baldwin CT: Identification of multiple loci for Alzheimer disease in a consanguineous Israeli-Arab community. Hum Mol Genet. 2003, 12: 415-422. 10.1093/hmg/ddg037.
Holmans P, Hamshere M, Hollingworth P, Rice F, Tunstall N, Jones S, Moore P, DeVrieze FW, Myers A, Crook R, Compton D, Marshall H, Meyer D, Shears S, Booth J, Ramic D, Williams N, Norton N, Abraham R, Kehoe P, Williams H, Rudrasingham V, O'donovan M, Jones L, Hardy J, Goate A, Lovestone S, Owen M, Williams J: Genome screen for loci influencing age at onset and rate of decline in late onset Alzheimer's disease. Am J Med Genet B Neuropsychiatr Genet. 2005, 135: 24-32.
Avramopoulos D, Fallin MD, Bassett SS: Linkage to chromosome 14q in Alzheimer's disease (AD) patients without psychotic symptoms. Am J Med Genet B Neuropsychiatr Genet. 2005, 132: 9-13.
Schellenberg GD, D'Souza I, Poorkaj P: The genetics of Alzheimer's disease. Curr Psychiatry Rep. 2000, 2: 158-164.
Bertram L, Tanzi RE: Alzheimer's disease: one disorder, too many genes?. Hum Mol Genet. 2004, 13 (Spec No 1): R135-R141. 10.1093/hmg/ddh077.
Jackson CE, Symon WE, Pruden EL, Kaer IM, Mann JD: Consanguinity and blood group distribution in an Amish isolate. Am J Hum Genet. 1968, 20: 522-527.
Gingerich HF, Kreider RW: Amish and Amish Mennonite Genealogies. 1986, Gordonville, PA: Pequea Publishers
Hostetler J: Amish Society. 1993, Baltimore, MD: Johns hopkins University Press, 4
Kraybill DB: The Riddle of Amish Culture. 2001, Baltimore, MD: Johns Hopkins University Press
Van Der Walt JM, Scott WK, Slifer S, Gaskell PC, Martin ER, Welsh-Bohmer K, Creason M, Crunk A, Fuzzell D, McFarland L, Kroner CC, Jackson CE, Haines JL, Pericak-Vance MA: Maternal lineages and Alzheimer disease risk in the Old Order Amish. Hum Genet. 2005, 118: 115-122. 10.1007/s00439-005-0032-x.
Hahs DW, McCauley JL, Crunk AE, McFarland LL, Gaskell PC, Jiang L, Slifer SH, Vance JM, Scott WK, Welsh-Bohmer KA, Johnson SR, Jackson CE, Pericak-Vance MA, Haines JL: A genome-wide linkage analysis of dementia in the Amish. Am J Med Genet B Neuropsychiatr Genet. 2006, 141: 160-166.
Agarwala R, Biesecker LG, Schaffer AA: Anabaptist genealogy database. Am J Med Genet C Semin Med Genet. 2003, 121: 32-37. 10.1002/ajmg.c.20004.
Sobel E, Lange K: Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet. 1996, 58: 1323-1337.
Abney M, Ober C, McPeek MS: Quantitative-trait homozygosity and association mapping and empirical genomewide significance in large, complex pedigrees: fasting serum-insulin level in the Hutterites. Am J Hum Genet. 2002, 70: 920-934. 10.1086/339705.
Bourgain C, Hoffjan S, Nicolae R, Newman D, Steiner L, Walker K, Reynolds R, Ober C, McPeek MS: Novel case-control test in a founder population identifies P-selectin as an atopy-susceptibility locus. Am J Hum Genet. 2003, 73: 612-626. 10.1086/378208.
Bourgain C, Genin E: Complex trait mapping in isolated populations: Are specific statistical methods required?. Eur J Hum Genet. 2005, 13: 698-706. 10.1038/sj.ejhg.5201400.
Heath S, Robledo R, Beggs W, Feola G, Parodo C, Rinaldi A, Contu L, Dana D, Stambolian D, Siniscalco M: A novel approach to search for identity by descent in small samples of patients and controls from the same mendelian breeding unit: a pilot study on myopia. Hum Hered. 2001, 52: 183-190. 10.1159/000053375.
Moore JH: The ubiquitous nature of epistasis in determining susceptibility to common human diseases. Hum Hered. 2003, 56: 73-82. 10.1159/000073735.
Agarwala R, Schaffer AA, Tomlin JF: Towards a complete North American Anabaptist Genealogy II: analysis of inbreeding. Hum Biol. 2001, 73: 533-545.
Agarwala R, Biesecker LG, Hopkins KA, Francomano CA, Schaffer AA: Software for constructing and verifying pedigrees within large genealogies and an application to the Old Order Amish of Lancaster County. Genome Res. 1998, 8: 211-221.
Folstein MF, Folstein SE, McHugh PR: "Mini-Mental State": a practical method for grading the cognitive state of patients for the clinician. Journal of Psychiatric Research. 1975, 12: 189-198. 10.1016/0022-3956(75)90026-6.
Mattis S: Mental status examination for organic mental syndrome in the elderly patient. Geriatric Psychiatry. Edited by: Bellak L, Karasu TB. 1976, New Your, NY: Grune & Stratton
Kaplan E, Goodglass H, Weintraub S: The Boston Naming Test. 1976, Philadelphia, PA: Lea & Febiger
Jastak S, Wilkinson GS: Wide Range Achievement Test-Revised. 1984, Wilmington, Delaware: Jastak Associates
Ashley-Koch AE, Shao Y, Rimmler JB, Gaskell PC, Welsh-Bohmer KA, Jackson CE, Scott WK, Haines JL, Pericak-Vance MA: An autosomal genomic screen for dementia in an extended Amish family. Neurosci Lett. 2005, 379: 199-204. 10.1016/j.neulet.2004.12.065.
Genome Database. [http://www.gdb.org/]
Primer3. [http://www-genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi]
Vance JM, Othmane KB: Methods of genotyping: Design and implementation of mapping studies ofr complex human diseases. Current Protocols in Human Genetics. Edited by: Haines JL, Pericak-Vance MA. 1998, New York: John Wiley & Sons Inc, New York
O'Connell JR, Weeks DE: PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998, 63: 259-266. 10.1086/301904.
Ihaka R, Gentleman R: A Language for Data Analysis and Graphics. Journal of Computational and Graphical Statistics. 1996, 5: 299-314.
Mehta CR, Patel NR: Algorithm 643. FEXACT: A Fortran subroutine for Fisher's exact test on unordered r*c contingency tables. ACM Transactions on Mathematical Software. 1986, 12: 154-161. 10.1145/6497.214326.
Clarkson DB, Fan Y, Joe H: A Remark on Algorithm 643: FEXACT: An Algorithm for Performing Fisher's Exact Test in r × c Contingency Tables. ACM Transactions on Mathematical Software. 1993, 19: 484-488. 10.1145/168173.168412.
Hiltunen M, Mannermaa A, Thompson D, Easton D, Pirskanen M, Helisalmi S, Koivisto AM, Lehtovirta M, Ryynanen M, Soininen H: Genome-wide linkage disequilibrium mapping of late-onset Alzheimer's disease in Finland. Neurology. 2001, 57: 1663-1668.
Pola R, Flex A, Gaetani E, Santoliquido A, Serricchio M, Pola P, Bernabei R: Intercellular adhesion molecule-1 K469E gene polymorphism and Alzheimer's disease. Neurobiol Aging. 2003, 24: 385-387. 10.1016/S0197-4580(02)00087-8.
Mattila KM, Hiltunen M, Rinne JO, Mannermaa A, Roytta M, Alafuzoff I, Laippala P, Soininen H, Lehtimaki T: Absence of association between an intercellular adhesion molecule 1 gene E469K polymorphism and Alzheimer's disease in Finnish patients. Neurosci Lett. 2003, 337: 61-63. 10.1016/S0304-3940(02)01296-X.
Rodero L, Infante J, Palacio E, Llorca J, Berciano J, Combarros O: Polymorphism at codon 469 of the intercellular adhesion molecule-1 gene is not associated with sporadic Alzheimer's disease. Am J Med Genet B Neuropsychiatr Genet. 2004, 126: 66-68. 10.1002/ajmg.b.20140.
Wijsman EM, Daw EW, Yu CE, Payami H, Steinbart EJ, Nochlin D, Conlon EM, Bird TD, Schellenberg GD: Evidence for a novel late-onset Alzheimer disease locus on chromosome 19p13.2. Am J Hum Genet. 2004, 75: 398-409. 10.1086/423393.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/7/19/prepub
Acknowledgements
This work was funded through NIH/NIA grants AG19085, AG19757, AG19726, the Claude Pepper Center (AG11268), and a discovery grant from Vanderbilt University. Additional work was performed using the Vanderbilt Center for Human Genetics Research Core facilities, the Vanderbilt General Clinical Research Center (M01 RR-00095), and the Duke Center for Human Genetics Core facilities. We thank all of the family participants and the Amish community members for so kindly agreeing to participate in our studies. Without their involvement, none of this research would have been possible.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
JLM directed and performed some of the analyses, collated the results, and was responsible for preparing and editing the manuscript and tables therein. DWH was involved in the drafting of the manuscript and providing input on the analyses. LJ was responsible for data management and analysis. WKS provided input for the analysis, helped in editing the manuscript, and provided financial support through grant funding. KAW was key in all clinical evaluations and provided input for the manuscript. CEJ has been a longtime consultant for ascertainment and recruitment from the Amish community due to his extensive interaction within this isolated population. JMV coordinated the genotyping of the microsatellite markers used within this study. JLH and MPV are Principal Investigator (PI) and co-PI, respectively. Both PIs were instrumental in providing the infrastructure, aiding in the study design, providing input in the manuscript, and supporting this project as well as additional projects surrounding this manuscript through their grant funding.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
McCauley, J.L., Hahs, D.W., Jiang, L. et al. Combinatorial Mismatch Scan (CMS) for loci associated with dementia in the Amish. BMC Med Genet 7, 19 (2006). https://doi.org/10.1186/1471-2350-7-19
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-7-19