
Abstract
Background
Thirty-nine patients have been described with deletions involving chromosome 6p25. However, relatively few of these deletions have had molecular characterization. Common phenotypes of 6p25 deletion syndrome patients include hydrocephalus, hearing loss, and ocular, craniofacial, skeletal, cardiac, and renal malformations. Molecular characterization of deletions can identify genes that are responsible for these phenotypes.
Methods
We report the clinical phenotype of seven patients with terminal deletions of chromosome 6p25 and compare them to previously reported patients. Molecular characterization of the deletions was performed using polymorphic marker analysis to determine the extents of the deletions in these seven 6p25 deletion syndrome patients.
Results
Our results, and previous data, show that ocular dysgenesis and hearing impairment are the two most highly penetrant phenotypes of the 6p25 deletion syndrome. While deletion of the forkhead box C1 gene (FOXC1) probably underlies the ocular dysgenesis, no gene in this region is known to be involved in hearing impairment.
Conclusions
Ocular dysgenesis and hearing impairment are the two most common phenotypes of 6p25 deletion syndrome. We conclude that a locus for dominant hearing loss is present at 6p25 and that this locus is restricted to a region distal to D6S1617. Molecular characterization of more 6p25 deletion patients will aid in refinement of this locus and the identification of a gene involved in dominant hearing loss.
Similar content being viewed by others

Background
Delineation of deletion syndromes is important to human health and patient care. Careful description of deletion syndromes allows improved diagnosis and more accurate prognosis for patients suspected to have chromosomal deletions. In addition to improving patient care, we can also glean more about the biology of the genes involved in the deletions. Characteristic phenotypes of patients with 6p25 deletion syndrome are hydrocephalus, hearing impairment, and ocular, craniofacial, skeletal, cardiac, and renal malformations [1]. Excluding ring chromosomes or products of unbalanced translocations, thirty nine patients have been reported with terminal [1–19] or interstitial [4, 18, 20–22] 6p25 deletions. Unfortunately, relatively few of these patients have had the deletions characterized at a molecular level. We present detailed clinical reports and molecular characterizations of three patients with terminal deletions of chromosome 6p that were briefly reported previously (Z.K., C.C., and GMO6222 from [9]), and four patients with terminal deletions of chromosome 6p as a result of unbalanced translocations (E.S. from [23] and three unreported cases). By comparing our patients to patients with molecularly characterized terminal deletions in the literature, we determined that ocular malformations and hearing loss are the two most highly penetrant phenotypes. While ocular malformations are likely due to deletions of the FOXC1 gene, the cause of the hearing impairment is unknown. Based on our results, we are able to define the minimal critical interval containing a locus for dominant hearing impairment as a 4 megabase (Mb) region distal to D6S1617.
Methods
Microsatellite analysis
Patient 1 DNA was isolated from a cell line supplied from Coriell Institute For Medical Research Cell Repository (#GMO6222). For all other patients, DNA was extracted from peripheral blood leukocytes [24] collected in EDTA tubes after informed written consent was obtained. To determine the number of alleles for polymorphic loci, 35S-dATP was directly incorporated into PCR products. The PCR products were then separated on 6% polyacrylamide gels. Patients were considered to be heterozygous for a locus when two alleles were observed, and homozygous/hemizygous for a locus when only one allele was observed. To maximize the chances of detecting heterozygosity for a particular marker, markers with heterozygosity in excess of 75% were chosen with the exception of D6S942 and D6S1006, which have heterozygosity of approximately 50% and 61% respectively. Marker information can be obtained from http://www.ncbi.nlm.nih.gov:80/entrez/query.fcgi?db=unists.
Clinical reports
Clinical reports for each patient were obtained from the physician who referred the patient. To be thorough, in addition to reporting phenotypes observed, we also reported when a specific phenotype was looked for but not observed. The number of patients in the literature with the same clinical phenotypes are indicated in Table 1. Table 2 is a summary of the patients for whom molecular characterizations of the deletion have been determined.
Patient 1 – 46, XX, del(6)(p24)
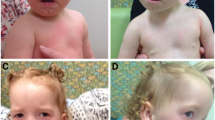
Patient 1 sample and clinical details were obtained from Coriell Institute For Medical Research Cell Repository (#GMO6222). The patient had Dandy-Walker cyst, hydrocephalus, Peters' anomaly, hypertelorism, ear tags, low set ears, and normal Hageman factor level in blood.
Patient 2 – 46, XY, del(6)(p24)
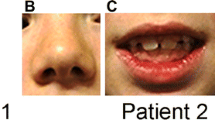
Patient 2 had bilateral microphthalmia, corneal opacities, interstitial keratitis and nystagmus. Craniofacial malformations included microcephaly, hypertelorism, low set, posteriorly angulated ears, and a high arched palate. The patient had severe, progressive, congenital scoliosis and profound bilateral clubfoot. An examination of the chest revealed widely spaced nipples. An x-ray was conducted but was unremarkable. Cardiac findings included a grade II to grade III murmur, a patent ductus arteriosus (PDA), a patent foramen ovale (PFO), right ventricular hypertrophy and left axis deviation. There was no ventricular septal defect (VSD) and the PFO closed with time. Hydrocephalus was noted at birth and a brain MRI revealed Dandy-Walker variant. The corpus callosum was thin and upwardly bowed and the middle and inferior portions of the cerebellar vermis were absent. The patient had severe hearing impairment. Genitourinary malformations included micropenis and hydronephrosis secondary to left ureteropelvic junction obstruction. The patient has a history of chronic oliguria, and chronic neutropenia. Results of liver and renal function tests were normal.
Patient 3 – 46, XY, del(6)(p24)
Patient 3 had slanted palpebral fissures, shallow orbits, bilateral hyperopia, Rieger malformation, hypoplastic irides, mild megalocornea, sclerocornea, and bilateral crescentic hyperpigmentation of the retina. The lenses were clear and the intraocular pressures (IOP) were normal. There was no nystagmus or strabismus and the optic nerve head, macula and vessels appeared normal. Craniofacial malformations included hypertelorism, brachycephaly, flat mid-face, low-set ears, small nose, broad nasal bridge, frontal bossing, and mild micrognathia. The patient also had a short neck, scoliosis, proximately flexed thumbs, club feet, and mild clinodactyly of the 5th fingers. His nipples were widely spaced and there were five supernumerary nipples. At birth he had a PFO, a VSD, and tricuspid valve regurgitation; however, all three resolved by the age of one year without intervention. Dandy Walker variant, agenesis of the corpus callosum and brainstem, seizures and profound bilateral deafness were all present. His knees were normal and hips were stable in all ranges of motion but there was delayed ossification of the femoral and humeral heads. Bone age was consistent with a one-year-old child when the patient was two years of age. Fingernails and toenails were soft and the patient had eczema. Genitalia, liver and spleen were all normal.
Patient 4 – 46, XYder(6)t(4;6)(p14;p25)
Patient 4 had an unbalanced translocation resulting in monosomy for 6p25-ter and trisomy of 4p14-ter. The patient had congenital glaucoma, nystagmus, strabismus and Rieger malformation including posterior embryotoxon, iris adhesions, bilateral elevated IOP and a diffuse corneal opacity on the right eye. This patient had no craniofacial malformations, cardiac murmur, neurological abnormalities, or remarkable abnormalities of the chest, hands and feet or kidneys.
Patient 5 – 46, XY, der(6)t(6;8)(p25;q24.1)
Patient 5 had an unbalanced translocation resulting in monosomy for 6p25-ter and trisomy for 8q24.1-ter. At birth the infant was in the 10th centile for height and weight, was hypotonic and had little spontaneous movement. He had Reiger anomaly including ectropian uveae with adhesions around the entire iridocorneal angle and bilateral congenital glaucoma. The optic nerve head appeared normal. Facial malformations included turricephaly, hypertelorism and low set, anteriorly displaced ears. The patient had kyphoscoliosis, hemivertebrae (T5-T6), and did not have a full complement of ribs. Abdominal ultrasound, shoulders and pelvis were all unremarkable. Nail hypoplasia was observed and the patient had sparse eyebrows and lashes. Cardiac malformations were atrial septal defect (ASD), PDA and PFO. He had mild hydrocephalus and hearing loss, however, the cerebellum was normal and an EEG was unremarkable.
Patient 6 – 46, XX, der(6)t(2;6)(q32;p24)
Patient 6 had an unbalanced translocation resulting in monosomy for 6p24-ter and trisomy for 2q32-ter. She had bilateral aniridia, and a vascularized corneal opacification but IOP was normal in each eye. The left eye was smaller than the right and the right eye had extensive retinal pigment epithelial washout and thinning of the retina. Craniofacial malformations included microdolichocephaly, telecanthus, hypertelorism, low-set ears, a broad nasal bridge, cleft palate, micrognathia, long philtrum, prominent premaxilla and a short neck. She had adducted thumbs and camptodactyly of her hands and a valgus deformity of the left foot. The patient also had limited flexion and extension of elbows, knees, and hips and had asymmetrical hypoplasia of vertebrae T12. As an infant the patient had apneic spells with cyanosis and bradycardia while feeding although the cardiovascular system, chest, and abdomen were unremarkable upon examination. A computed tomography scan of the head was normal. The patient had progressive hearing loss and was fitted for a hearing aid at the age of 7 months. Genitalia were normal but right hydronephrosis and a bilateral cystouretero reflux were documented.
Patient 7
Patient 7 had an unbalanced translocation resulting in monosomy of 6p25-ter and a trisomy of the terminal portion of chromosome 10p. She had hypotonia, mild developmental delay and Rieger anomaly. Craniofacial malformations included hypertelorism, a flat nasal bridge, small, low set ears, micrognathia, prognathia and a small mouth with protruding tongue. The patient had scoliosis, a short neck, widespread nipples and malformation of both hands. There was no heart murmur, neurological malformations or hearing impairment.
Results
In cases where DNA was not available from a transmitting parent, we could only define the maximum size of the deletion. A terminal deletion can be no larger than the boundary defined by the most distal marker that is heterozygous. At markers distal to this, where the patient has a single allele, the patient may be homozygous or may be deleted and therefore is hemizygous. By using highly polymorphic markers we increased the odds that a single allele, at consecutive markers, indicates a deletion.
No parental DNA was available for Patients 1, 2, 4 or 6. Patients 1 and 2 were both homozygous for D6S1006 but heterozygous for D6S277. All markers tested distal to D6S277 had single alleles and therefore the maximum deletion sizes for Patients 1 and 2 are 8.4 Mb distal to D6277 (Figure 1). A DNA sample was provided from the mother of Patient 3. Microsatellite analysis indicated that the deletion in Patient 3 was not of maternal origin so only the maximum deletion size could be determined. Patient 3 had a single allele for all markers tested except D6S1006 which was heterozygous. Therefore the maximum deletion size for Patient 3 is 13.0 Mb distal to D6S1006 (Figure 1). Patient 4 had a single allele for D6S942 and was heterozygous for D6S967 thus the maximum deletion for this patient is 1.4 Mb distal to D6S967 (Figure 1). We determined Patient 5 to have a deletion of paternal origin. The patient is deleted for D6S942, D6S967, and D6S344 but not D6S1617 indicating the deletion breakpoint is between 1.5 Mb and 4.2 Mb (Figure 1). Patient 6 had single alleles for D6S942, D6S967, and D6S344 but was heterozygous for D6S1617. Therefore the maximum deletion for Patient 6 is 4.2 Mb distal to D6S1617 (Figure 1). We determined Patient 7 to have a deletion of maternal origin. The patient was deleted for D6S942, D6S967, D6S344, D6S1617, D6S1713, but not D6S477 indicating the deletion breakpoint is between 4.3 and 6.1 Mb (Figure 1).

Schematic diagram of the distal tip of human chromosome 6p. The black boxes indicate regions where the patients are not deleted. White boxes indicate the minimum extent of the deletion. Grey boxes indicate the maximum extent of the deletion. Markers used are listed to the left of the patients chromosomes under locus. Genes in this region are listed in bold print. The genetic distance of markers, in Mb (megabases) from the telomere, are also indicated.
Discussion and conclusions
We have presented clinical and molecular characterizations of seven patients with 6p25 terminal deletion syndrome. Of 39 described cases of terminal deletions of human chromosome 6p25, patients frequently present with ocular, craniofacial, skeletal, cardiac, and renal malformations, hearing loss, and hydrocephalus (Table 1). The phenotypic variation between these patients may be due to a variety of genetic or non-genetic factors (including additional chromosomal rearrangements as in patients 4, 5, 6, and 7). Molecular characterization of the deletions can allow us to begin to determine the genetic factors involved. However, only 19 patients (7 reported here and 12 from the literature) have had deletions characterized at a molecular level (Table 2). By molecular characterization of the deletions in our new patients and comparing them to previously described patients we have determined that ocular dysgenesis and hearing impairment are highly penetrant phenotypes associated with the 6p25 deletion syndrome.
Ocular dysgenesis
Ocular anterior segment dysgenesis, is present in 17 of the 19 patients with molecularly characterized deletions. The two patients without anterior segment dysgenesis were from the same report [25], however, both patients had fundus exams and therefore it is unlikely that anterior segment dysgenesis would have been overlooked unless it was extremely mild. Mutations in FOXC1, including deletions or duplications, are well established to cause anterior segment dysgenesis [9, 18, 26–32]. Patients 1, 2, 3, 5, 6, and 7 have maximum deletions that include FOXC1 and have ocular malformations consistent with those previously reported. Interestingly, the maximum deletion for Patient 4 does not include FOXC1, however, this patient has an ocular phenotype similar to that associated with FOXC1 mutations. It is possible that the deletion in this patient perturbs FOXC1 regulatory elements. Alternatively, since this patient has an unbalanced translocation, the phenotype may be caused by some other aspect of the chromosomal rearrangement, however, patients with 4p trisomy do not typically have these types of ocular malformations [33].
Hearing impairment
The second phenotype observed with a high penetrance is hearing impairment. Of 15 patients assessed, 14 of them had hearing impairment (Table 2). This proportion is very similar to that of patients with ocular dysgenesis and suggests that there may be a dominant locus for hearing impairment. Since there is phenotypic variability, and since absence of a reported phenotype may reflect the failure to assay the phenotype, it is prudent to only use affected individuals to refine this genetic locus. Two recent reports suggest that this locus is distal to a marker D6S1574 (which is between D6S477 and D6S1617) [34] and distal to GMDS (at 1.6 Mb) [19] respectively. Data from our study confirms this observation. Patients 5 and 6 both have hearing loss and both exclude marker D6S1617 from the critical interval. Therefore, we conclude that a dominant locus for hearing loss exists distal to D6S1617. Since FOXC1 is in this critical interval, it is possible that deletion of FOXC1 underlies the hearing impairment. Similarly, some patients mapped to 13q14 have anterior segment dysgenesis and hearing defects that may be caused by a single gene[35].
Variably associating phenotypes
Patients 1, 2, 3, and 5, had hydrocephalus. This is consistent with these patients being deleted for FOXC1. Mice that are homozygous for a null allele of Foxc1 also have congenital hydrocephalus [9, 32] and deletion of one copy of FOXC1 is suggested to cause hydrocephaly in humans [9]. However, not all patients deleted for FOXC1 (here or in the literature) have hydrocephalus (for example, Patient 8 is deleted for FOXC1 but did not have hydrocephalus) and thus other factors must contribute to the development of this disorder.
FOXC1 is also involved in heart, renal, and skeletal development [9, 32, 36–38] and has been implicated in human congenital heart disease [28, 37, 38]. Deletion of FOXC1 may contribute to the cardiac malformations observed in Patients 1, 2, 3 and 5. Renal examinations were only reported for three patients in this study. Patients 2 and 6 had hydronephrosis and FOXC1 is within the deletion interval for these patients. All patients potentially deleted for FOXC1 in this study have malformations of the spine or vertebrae and often of the hands and feet.
Bone morphogenetic proteins (BMPs) are involved in a large number of developmental processes [39] and thus a deletion may contribute to many aspects of the developmental phenotypes. Patients 1, 2, and 3 have maximum deletions encompassing BMP6. BMP6 is involved in ossification of the sternum [40] and thus may be involved in delayed bone ossification observed in Patient 3.
Patient 3 has the largest maximal deletion in our study. The maximal deletion includes the genes TFAP2A (Transcription factor AP2 alpha), and ET-1 (Endothelin 1). Both genes are involved in otic, craniofacial, and cardiac development [41–44], while TFAP2A is further involved in renal and skeletal development [41–43]. Thus possible deletion of these genes in Patient 3 may influence many aspects of his phenotype including scoliosis, cardiac, renal, craniofacial, and ocular malformations and hearing impairment.
We have presented clinical and molecular characterization of 7 patients with 6p25 terminal deletion syndrome. Our data, and data from additional patients in the literature, define the 6p25 deletion syndrome as having highly penetrant ocular malformations and hearing impairment. Additionally, there is variable association with hydrocephaly, craniofacial, skeletal, cardiac, and renal malformations. Based on our data, we suggest that the minimal critical interval for a dominant locus for hearing impairment be reduced to the interval distal to marker D6S1617. Employment of other molecular characterization techniques (such as FISH) would provide more informativity in cases where a transmitting parent is not available and resolve ambiguities between homozygous and hemizygous in the absence of two alleles. Additionally, characterization of more deletion patients, family linkage analysis or animal models with targeted mutations of candidate genes will be useful approaches to refine this critical interval and possibly identify the gene involved.
Author's contributions
D.B. Gould prepared DNA from patient blood, compiled patient clinical descriptions, performed marker analysis, interpreted data and wrote manuscript. M.S. Jaafar, M.K. Addison, I.M. MacDonald, F. Munier, and R. Ritch identified and diagnosed the patients and provided blood samples and clinical reports. M.A. Walter was involved in experimental design and oversight, interpretation of the data and editing of the manuscript.

Schematic of extent of deletions for 12 patients in the literature with deletions characterized at a molecular level. The black boxes indicate regions where the patients are not deleted. White boxes indicate the minimum extent of the deletion. Grey boxes indicate markers that may or may not be deleted. Genes in this region are listed in bold print. The genetic distance of markers, in Mb (megabases) from the telomere, are also indicated. The source of the information is as follows: Patient A [19], Patient B [28], Patient C is case 2 from [25] and CA from 1, Patients D-G [18], Patient H [10] and SG from [1], Patient I is case 1 from [25] and BD from [1], Patient J is HH from [1], Patient K is JW from [1], Patient L is case 2 from [5].
References
Davies AF, Mirza G, Sekhon G, Turnpenny P, Leroy F, Speleman F, Law C, van Regemorter N, Vamos E, Flinter F, Ragoussis J: Delineation of two distinct 6p deletion syndromes. Hum Genet. 1999, 104: 64-72. 10.1007/s004390050911.
Alashari M, Chen E, Poskanzer L: Partial deletion of chromosome 6p: autopsy findings in a premature infant and review of the literature. Pediatr Pathol Lab Med. 1995, 15: 941-947.
D'Alessandro E, Santiemma V, Lo Re ML, Ligas C, Del Porto G: 6p23 deletion mosaicism in a woman with recurrent abortions and idiopathic hypoprolactinemia. Am J Med Genet. 1992, 44: 220-222.
Davies AF, Mirza G, Flinter F, Ragoussis J: An interstitial deletion of 6p24-p25 proximal to the FKHL7 locus and including AP-2alpha that affects anterior eye chamber development. J Med Genet. 1999, 36: 708-710.
Davies AF, Stephens RJ, Olavesen MG, Heather L, Dixon MJ, Magee A, Flinter F, Ragoussis J: Evidence of a locus for orofacial clefting on human chromosome 6p24 and STS content map of the region. Hum Mol Genet. 1995, 4: 121-128.
Guillen-Navarro E, Chan WC, Ragoussis J, Davies AF, Ostrer H, Perle MA: A rare de novo microdeletion of distal chromosome 6p: clinical phenotype and molecular cytogenetic characterisation. Am J Hum Genet. 1997, 61: A719-
Jalal SM, Macias VR, Roop H, Morgan F, King P: Two rare cases of 6p partial deletion. Clin Genet. 1989, 36: 196-199.
Kormann-Bortolotto MH, Farah LM, Soares D, Corbani M, Muller R, Adell AC: Terminal deletion 6p23: a case report. Am J Med Genet. 1990, 37: 475-477.
Kume T, Deng KY, Winfrey V, Gould DB, Walter MA, Hogan BL: The forkhead/winged helix gene Mf1 is disrupted in the pleiotropic mouse mutation congenital hydrocephalus. Cell. 1998, 93: 985-996. 10.1016/S0092-8674(00)81204-0.
Law CJ, Fisher AM, Temple IK: Distal 6p deletion syndrome: a report of a case with anterior chamber eye anomaly and review of published reports. J Med Genet. 1998, 35: 685-689.
Ouchi K, Kasai R: A case of partial monosomy 6p. Jpn J Hum Genet. 1982, 27: 214-
Palmer CG, Bader P, Slovak ML, Comings DE, Pettenati MJ: Partial deletion of chromosome 6p: delineation of the syndrome. Am J Med Genet. 1991, 39: 155-160.
Plaja A, Vidal R, Soriano D, Bou X, Vendrell T, Mediano C, Pueyo JM, Labrana X, Sarret E: Terminal deletion of 6p: report of a new case. Ann Genet. 1994, 37: 196-199.
Reid CS, Stamberg J, Phillips JA: Monosomy for distal segment 6p: clinical description and use in localising a region imortant for expression of Hageman factor. Pediatr Res. 1983, 17: A217-
Sachs ES, Hoogeboom AJ, Niermeijer MF, Schreuder GM: Clinical evidence for localisation of HLA proximal of chromosome 6p22. Lancet. 1983, 1: 659-10.1016/S0140-6736(83)91839-1.
Walsh LM, Lynch SA, Clarke MP: Ocular abnormalities in a patient with partial deletion of chromosome 6p. A case report. Ophthalmic Genet. 1997, 18: 151-156.
Zurcher VL, Golden WL, Zinn AB: Distal deletion of the short arm of chromosome 6. Am J Med Genet. 1990, 35: 261-265.
Lehmann OJ, Ebenezer ND, Ekong R, Ocaka L, Mungall AJ, Fraser S, McGill JI, Hitchings RA, Khaw PT, Sowden JC, Povey S, Walter MA, Bhattacharya SS, Jordan T: Ocular developmental abnormalities and glaucoma associated with interstitial 6p25 duplications and deletions. Invest Ophthalmol Vis Sci. 2002, 43: 1843-1849.
Anderlid BM, Schoumans J, Hallqvist A, Stahl Y, Wallin A, Blennow E, Nordenskjold M: Cryptic subtelomeric 6p deletion in a girl with congenital malformations and severe language impairment. Eur J Hum Genet. 2003, 11: 89-92. 10.1038/sj.ejhg.5200907.
Davies AF, Olavesen MG, Stephens RJ, Davidson R, Delneste D, Van Regemorter N, Vamos E, Flinter F, Abusaad I, Ragoussis J: A detailed investigation of two cases exhibiting characteristics of the 6p deletion syndrome. Hum Genet. 1996, 98: 454-459. 10.1007/s004390050239.
Moriarty AP, Kerr-Muir MG: Sclerocornea and interstitial deletion of the short arm of chromosome 6--(46XY del[6] [p22 p24]). J Pediatr Ophthalmol Strabismus. 1992, 29: 177-179.
van Swaay E, Beverstock GC, van de Kamp JJ: A patient with an interstitial deletion of the short arm of chromosome 6. Clin Genet. 1988, 33: 95-101.
MacDonald IM, Clarke WN, Clifford BG, Reid JC, Cox DM, Hunter AG: Corneal pathology and aniridia associated with partial trisomy 2q, due to a maternal (2;6) translocation. Ophthalmic Paediatr Genet. 1984, 4: 75-80.
Miller SA, Dykes DD, Polesky HF: A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16: 1215-
Pierquin G, Van Regemorter N, Hayez Delatte, Fourneau C, Bormans J, Foerster M, Damis E, Cremer-Perlmutter N, Lapiere CM, Vamos E: Two unrelated children with partial trisomy 1q and monosomy 6p, presenting with the phenotype of the Larsen syndrome. Hum Genet. 1991, 87: 587-591.
Mears AJ, Jordan T, Mirzayans F, Dubois S, Kume T, Parlee M, Ritch R, Koop B, Kuo WL, Collins C, Marshall J, Gould DB, Pearce W, Carlsson P, Enerback S, Morissette J, Bhattacharya S, Hogan B, Raymond V, Walter MA: Mutations of the forkhead/winged-helix gene, FKHL7, in patients with Axenfeld-Rieger anomaly. Am J Hum Genet. 1998, 63: 1316-1328. 10.1086/302109.
Nishimura DY, Searby CC, Alward WL, Walton D, Craig JE, Mackey DA, Kawase K, Kanis AB, Patil SR, Stone EM, Sheffield VC: A spectrum of FOXC1 mutations suggests gene dosage as a mechanism for developmental defects of the anterior chamber of the eye. Am J Hum Genet. 2001, 68: 364-372. 10.1086/318183.
Nishimura DY, Swiderski RE, Alward WL, Searby CC, Patil SR, Bennet SR, Kanis AB, Gastier JM, Stone EM, Sheffield VC: The forkhead transcription factor gene FKHL7 is responsible for glaucoma phenotypes which map to 6p25. Nat Genet. 1998, 19: 140-147. 10.1038/493.
Mirzayans F, Gould DB, Heon E, Billingsley GD, Cheung JC, Mears AJ, Walter MA: Axenfeld-Rieger syndrome resulting from mutation of the FKHL7 gene on chromosome 6p25. Eur J Hum Genet. 2000, 8: 71-74. 10.1038/sj.ejhg.5200354.
Smith RS, Zabaleta A, Kume T, Savinova OV, Kidson SH, Martin JE, Nishimura DY, Alward WL, Hogan BL, John SW: Haploinsufficiency of the transcription factors FOXC1 and FOXC2 results in aberrant ocular development. Hum Mol Genet. 2000, 9: 1021-1032. 10.1093/hmg/9.7.1021.
Kidson SH, Kume T, Deng K, Winfrey V, Hogan BL: The forkhead/winged-helix gene, Mf1, is necessary for the normal development of the cornea and formation of the anterior chamber in the mouse eye. Dev Biol. 1999, 211: 306-322. 10.1006/dbio.1999.9314.
Hong HK, Lass JH, Chakravarti A: Pleiotropic skeletal and ocular phenotypes of the mouse mutation congenital hydrocephalus (ch/Mf1) arise from a winged helix/forkhead transcriptionfactor gene. Hum Mol Genet. 1999, 8: 625-637. 10.1093/hmg/8.4.625.
Patel SV, Dagnew H, Parekh AJ, Koenig E, Conte RA, Macera MJ, Verma RS: Clinical manifestations of trisomy 4p syndrome. Eur J Pediatr. 1995, 154: 425-431. 10.1007/s004310050318.
Zhang HZ, Li P, Wang D, Huff S, Nimmakayalu M, Qumsiyeh M, Pober BR: FOXC1 gene deletion is associated with eye anomalies in ring chromosome 6. Am J Med Genet. 2004, 124A: 280-287. 10.1002/ajmg.a.20413.
Phillips JC, del Bono EA, Haines JL, Pralea AM, Cohen JS, Greff LJ, Wiggs JL: A second locus for Rieger syndrome maps to chromosome 13q14. Am J Hum Genet. 1996, 59: 613-619.
Kume T, Deng K, Hogan BL: Murine forkhead/winged helix genes Foxc1 (Mf1) and Foxc2 (Mfh1) are required for the early organogenesis of the kidney and urinary tract. Development. 2000, 127: 1387-1395.
Swiderski RE, Reiter RS, Nishimura DY, Alward WL, Kalenak JW, Searby CS, Stone EM, Sheffield VC, Lin JJ: Expression of the Mf1 gene in developing mouse hearts: implication in the development of human congenital heart defects. Dev Dyn. 1999, 216: 16-27. 10.1002/(SICI)1097-0177(199909)216:1<16::AID-DVDY4>3.3.CO;2-T.
Winnier GE, Kume T, Deng KY, Rogers R, Bundy J, Raines C, Walter MA, Hogan BLM, Conway SJ: Roles for the winged helix transcription factors MF1 and MFH1 in cardiovascular development revealed by nonallelic noncomplementation of null alleles. Dev Biol. 1999, 213: 418-431. 10.1006/dbio.1999.9382.
Hogan BL: Bone morphogenetic proteins: multifunctional regulators of vertebrate development. Genes Dev. 1996, 10: 1580-1594.
Solloway MJ, Dudley AT, Bikoff EK, Lyons KM, Hogan BL, Robertson EJ: Mice lacking Bmp6 function. Dev Genet. 1998, 22: 321-339. 10.1002/(SICI)1520-6408(1998)22:4<321::AID-DVG3>3.3.CO;2-7.
Nottoli T, Hagopian-Donaldson S, Zhang J, Perkins A, Williams T: AP-2-null cells disrupt morphogenesis of the eye, face, and limbs in chimeric mice. Proc Natl Acad Sci U S A. 1998, 95: 13714-13719. 10.1073/pnas.95.23.13714.
Mitchell PJ, Timmons PM, Hebert JM, Rigby PW, Tjian R: Transcription factor AP-2 is expressed in neural crest cell lineages during mouse embryogenesis. Genes Dev. 1991, 5: 105-119.
West-Mays JA, Zhang J, Nottoli T, Hagopian-Donaldson S, Libby D, Strissel KJ, Williams T: AP-2alpha transcription factor is required for early morphogenesis of the lens vesicle. Dev Biol. 1999, 206: 46-62. 10.1006/dbio.1998.9132.
Kurihara Y, Kurihara H, Suzuki H, Kodama T, Maemura K, Nagai R, Oda H, Kuwaki T, Cao WH, Kamada N, et al.: Elevated blood pressure and craniofacial abnormalities in mice deficient in endothelin-1. Nature. 1994, 368: 703-710. 10.1038/368703a0.
Kelly PC, Blake WW, Davis JR: Tandem Y/6 translocation with partial deletion 6 (p23----pter). Clin Genet. 1989, 36: 204-207.
Sivak LE, Esbenshade J, Brothman AR, Issa B, Lemons RS, Carey JC: Multiple congenital anomalies in a man with (X;6) translocation. Am J Med Genet. 1994, 51: 9-12.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/5/17/prepub
Acknowledgements
The authors extend gratitude to the patients and families who participated in this study. Thank you to Farideh Mirzayans for technical assistance. D.B.G. is supported by a Canadian Institute of Health Research (CIHR) doctoral fellowship. F.M. is supported by a grant from the Swiss National Science Foundation (#32-065250.01). M.A.W. is an Alberta Heritage Foundation for Medical Research (AHFMR) senior scholar and CIHR investigator.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
None declared.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Gould, D.B., Jaafar, M.S., Addison, M.K. et al. Phenotypic and molecular assessment of seven patients with 6p25 deletion syndrome: Relevance to ocular dysgenesis and hearing impairment. BMC Med Genet 5, 17 (2004). https://doi.org/10.1186/1471-2350-5-17
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-5-17