
Abstract
Background
SHOX alterations have been reported in 67% of patients affected by Léri-Weill dyschondrosteosis (LWD), with a larger prevalence of gene deletions than point mutations. It has been recently demonstrated that these deletions can involve the SHOX enhancer region, rather that the coding region, with variable phenotype of the affected patients.
Here, we report a SHOX gene analysis carried out by MLPA in 14 LWD patients from 4 families with variable phenotype.
Case presentation
All patients presented a SHOX enhancer deletion. In particular, a patient with a severe bilateral Madelung deformity without short stature showed a homozygous alteration identical to the recently described 47.5 kb PAR1 deletion. Moreover, we identified, for the first time, in three related patients with a severe bilateral Madelung deformity, a smaller deletion than the 47.5 kb PAR1 deletion encompassing the same enhancer region (ECR1/CNE7).
Conclusions
Data reported in this study provide new information about the spectrum of phenotypic alterations showed by LWD patients with different deletions of the SHOX enhancer region.
Similar content being viewed by others


Background
SHOX deficiency represents a frequent cause of short stature, being associated with different pathological phenotypes such as Turner syndrome (TS), Idiopathic Short Stature (ISS; MIM ID 300582), Léri-Weill dyschondrosteosis (LWD; MIM ID 127300) and Langer mesomelic dysplasia (LS; MIM ID 249700) [1–10].
LWD is characterized by the presence of short stature associated with specific bone alterations, such as the Madelung deformity of the forearm. However, the full-blown LWD phenotype is frequently not determined in pre-schooler children because the specific features of this condition (i.e. mesomelic disproportion of the limbs and Madelung deformity), appears during the second decade of life [11–13]. As a consequence, in many cases short stature represents the only clinical sign at diagnosis. All together, mutations affecting the SHOX function in the different pathological conditions display an estimate frequency of less than 1:1000, thus representing the most common mendelian disease in the Caucasian population [14].
Due to this high frequency of alterations of the SHOX gene and to the recently demonstrated good response to the treatment with growth hormone (GH) in patients with SHOX deficiency, the early identification of SHOX alterations has become crucial for the diagnosis of the disease and the therapeutic strategy [15]. In this view, a phenotype scoring system assisting the identification of the most appropriate subjects for SHOX testing has been developed by Rappold et al. [16], recommending SHOX analysis in presence of a score greater than four out of a total possible score of 24. Moreover, Binder described an interesting algorithm approach to SHOX mutation screening in short children, promoting the clinical diagnosis supported by an auxological analysis of the body proportions (mesomelia), the presence of minor abnormalities, and the search for subtle radiographic signs and the molecular studies for confirming clinical data [17].
A large number of literature reports have demonstrated the presence of SHOX alterations in about 67% of LWD cases [18, 19]. On the other hand, SHOX alterations are not detected in the vast majority of cases with idiopathic short stature (85–98%) [9, 17, 20].
The use of the MLPA assay [18–21] has disclosed that deletions can involve not only the SHOX coding region, but also the upstream and downstream SHOX enhancer sequences [22–25].
Recently, Benito-Sanz et al. [26] provided a deep characterization of a relatively small deletion of PAR1, previously reported by Chen et al. [10] and Caliebe et al. [27], uncovering a novel downstream enhancer. This deletion is correlated with a remarkably variable phenotype of patients [28], confirming the evidence that deletion size is not related with the severity of the clinical phenotype [29], which, despite the high penetrance of SHOX deficiency, is very variable becoming more pronounced with age and being more severe in females [17].
The identification of PAR1 deletions not involving the SHOX coding regions have raised novel interest to the knowledge of the mechanisms leading to short stature in cases with SHOX deficiency.
In order to provide a contribute to this field of studies, we report on SHOX gene analysis in LWD patients with selected dysmorphic signs derived from four families, all evidencing deletions of the enhancer region, which was present in homozygous form in a patient with Madelung deformity but normal stature.
Case presentation
A written informed consent was obtained from each patient. The different techniques were performed according to standard procedures of the participating centers, with the purpose to reach a genetic diagnosis in the studied patient. It was not designed as an experimental study.
Family 1
The index case (Figure 1-n.1) was a 12-years and 10 months girl admitted to the outpatient Endocrine Clinic of the Department of Pediatrics, University of Chieti, Italy, for short stature. The girl was the first offspring of unrelated healthy parents. She has one 10-year-old sister, who had a normal linear and ponderal growth. The girl was born after 39 weeks of gestation after an unremarkable pregnancy. Birth anthropometric measurements were the following: weight 2.500 kg (3th-10th percentile), length 48 cm (10th-25th percentile). From the first months of life she had slow linear and ponderal growth, with normal psychomotor development. At our first clinical evaluation she showed short stature (140.6 cm, −2.18 SDS). Sitting height was 75 cm (−2.0 SDS), sitting height/height ratio was 0.53 (+1.0 SDS) and arm span/height ratio was 96.7%. BMI was between 25th-50th percentile and head circumference was normal. She had mesomelia with muscular hypertrophy. No facial dysmorphism was detected except for mild webbed neck. Radiological examination of the forearms showed bilaterally slight triangular deformation of distal radial epiphysis and mild bowing of radial diaphysis; legs were normal. Bone age according to Greulich and Pyle was 13.8 yr. Based on the detected clinical and radiological signs, the total Rappold’s score was 6 (Table 1).

Pedigree of the four families. Black icon: clinically affected; white symbol: clinically unaffected; Δ: deletion; +: no mutation; ?: not tested.
The 49-year-old mother of the proband (Figure 1-n.2) also showed short stature (149 cm, −2.0 SDS). Sitting height was 72 cm (< −2.0 SDS), sitting height/height ratio was 0.48 cm (< −2.5 SDS) and arm span/height ratio was 93%. BMI was >50th percentile. She had mesomelia with muscular hypertrophy. No facial dysmorphisms were detected except for mild webbed neck. Radiological examination of the radius and ulna showed bilateral Madelung deformity; legs were normal. The total Rappold’s score was 12 (Table 1).
Family 2
The index case (Figure 1-n.3) was a 3-years and 11 months girl admitted to the outpatient Endocrine Clinic of the Department of Pediatrics, University of Chieti, Italy, for short stature. The girl was the second offspring of unrelated healthy parents. She has one 6-year-old healthy sister, showing normal linear and ponderal growth. The girl was born after 38 weeks of gestation after an unremarkable pregnancy. Birth anthropometric measurements were the following: weight 2.820 kg (10th-25th percentile), length 48.5 cm (25th-50th percentile). From the first months of life she had slow linear growth, whereas ponderal growth and psychomotor development were normal. At our first clinical evaluation she showed short stature (91.3 cm, −2.53 SDS). Sitting height was 72.6 cm (<−2.5 SDS), sitting height/height ratio was 0.53 (−1.2 SDS) and arm span/height ratio was 100%. BMI was >95th percentile. The habitus was muscular, and she had mesomelia and facial dysmorphisms, such as mild hypertelorism, left epicanthus, high-arched palate, mild webbed neck, cubitus and genu valgus and lordosis. At radiological examination left radius showed a mild bowing of diaphysis, triangularization of distal epiphysis and bowing of diaphysis of radius, radiolucency of distal radio-ulnar articulation with normality of left radius, ulnas and wrists (Figure 2E); legs were normal. Bone age according to Greulich and Pyle was 3 yr. The total Rappold’s score was 12 (Table 1).

Female 39-year-old: Bilateral forearm radiographs show triangularization of distal epiphysis and bowing of diaphysis of radius bilaterally; distal radio-ulnar articulations appeared radiographically lucent. Palmar subluxation of the left carpus (A-D). Female 6-year-old: Left forearm radiography shows triangularization of distal epiphysis and bowing of diaphysis of radius, radiolucency of distal radio-ulnar articulation (E). Madelung deformity of family 3 index case (n.8) (F). The lower panel showes MLPA assay results. (G) Control: Normal peaks were classified as showing a ratio of 0.65-1.35 (H) Heterozygous deletion of the 13296-L15336, 05645-L05099 and 05646-L15507. MLPA probes have a ratio < 0.65. (I) Homozygous deletion of the 13296-L15336, 05645-L05099 and 05646-L15507 MLPA probes showing a ratio = 0. (L) Heterozygous deletion of the 05645-L05099 MLPA probes.
Her mother (Figure 1-n.4) (39-year-old) also showed short stature (146.4 cm, −2.43SDS). Sitting height was 61.4 cm (<−2.5 SDS), sitting height/height ratio was 41.9%and arm span/height ratio was 98%. BMI was between 50th-75thpercentile. Muscular hypertrophy was detected. She had mesomelia with facial dysmorphisms including mild high-arched palate and webbed neck. Mild scoliosis with cubitus and genu valgus were detected. The patient suffered from adolescence of diffuse muscular pain, which at adult age was imputed to fibromyalgia. Radiological examination showed triangularization of distal epiphysis and bowing of diaphysis of radius bilaterally;distal radio-ulnar articulations appeared radiographically lucent and legs did not show skeletal anomalies (Figure 2A-D). The total Rappold’s score was 12 (Table 1).
A 10-year-old cousin (Figure 1-n.5) of the index case was also admitted for short stature. The girl was the second offspring of unrelated healthy parents. She has one 18-year-old healthy brother and one 7-year-old healthy sister, both of them with normal linear and ponderal growth. The girl was born after 32 weeks of gestation after an unremarkable pregnancy. Birth anthropometric measurements were the following: weight 2.850 kg (>97th percentile), length 45.2 cm (90th-97th percentile). From the first months of life she had slow linear growth with normal ponderal growth. Psychomotor development was normal. At our first clinical evaluation she showed short stature (125.3 cm, −2.07 SDS). Sitting height was 60 cm (<−2.5 SDS), sitting height/height ratio was 47.8%, arm span /height ratio was 100%. BMI was between 85th-90th percentile. The habitus was slightly muscular, and she had mesomelia and facial dysmorphisms, including mild high-arched palate and mild webbed neck. She also had cubitus valgus and valgus knee. Bone age according to Greulich and Pyle was 8.6 yr. Radiological examination of the radius, ulnas and legs did not show significant bone alterations.The total Rappold’s score was 12 (Table 1).
Her mother (Figure 1-n.6) (46-year-old), the sister of index case’s mother, also showed mild linear impairment (149.8 cm, −1.86 SDS). Sitting height was 70.8 cm (<−2.5 SDS), sitting height/height ratio was 47.2 (<−2.5 SDS) and arm span /height ratio was 98%. BMI was between 25th-50th percentile. The habitus was muscular, and she had mesomelia, cubitus valgus without facial dysmorphisms. The patient suffered from adolescence of diffuse muscular pain, which at adult age was imputed to fibromyalgia. Radiological examination showed bilaterally triangularization of distal epiphysis and bowing of diaphysis of radius, radiolucency of distal radio-ulnar articulation; legs were normal.The total Rappold’s score was 8 (Table 1).
Another sister of index case’s mother (Figure 1-n.7) (48-year-age old) also showed mild linear impairment (150.4 cm, −1.76 SDS). Sitting height was 75 cm (<−2.5 SDS), sitting height/height ratio was 49 (<−2.25 SDS) and arm span /height ratio was 96%. BMI was between 85th-95th percentile. The habitus was muscular, and she had mesomelia. The patient suffered from adolescence of diffuse muscular pain, which at adult age was imputed to fibromyalgia. Radiological examination of the radius and ulna showed bilateral Madelung deformity. The total Rappold’s score was 12 (Table 1).
Family 3
The index case (Figure 1-n.8) was a 30-year-old women admitted to the outpatient Genetic Clinic, University of Chieti, Italy, for Madelung deformity surgically corrected at the age of 18 (Figure 2F). The women was the second offspring of unrelated healthy parents. She has one 26-year-old healthy brother, who had a normal linear and ponderal growth. The women was born at term after an unremarkable pregnancy. Birth anthropometric measurements were in the normal rage. From the first months of life she had slow linear growth with normal ponderal growth and psychomotor development. At our first clinical evaluation she did not show short stature (155.6 cm, −0.89 SDS). Sitting height was 84 cm (< −2.5 SDS), sitting height/height ratio was 54% (< −2.5 SDS) and arm span/height ratio was 98%. BMI was between 25th-50th percentile. The habitus was muscular, and she had mesomelic, facial dysmorphisms (hypertelorism) and cubitus and genu valgus with scoliosis. The patient suffered from adolescence of diffuse muscular pain. Radiological examination of the radius and ulna showed bilateral Madelung deformity (Figure 2F). The total Rappold’s score was 16 (Table 1).
This patient had two daughters. The first girl (Figure 1-n.9) was born after 37 weeks of gestation after an unremarkable pregnancy. Birth anthropometric measurements were the following: weight 2.560 kg (10th-25thpercentile), length 49.5 cm (75th-90th percentile). She showed decrease of linear growth from the age of 21 months, while poor ponderal growth was detected from the age of 1 year. Psychomotor development was normal. At our first clinical evaluation she was 2-years and 10 months old. She showed short stature (87 cm, −2.09 SDS). Sitting height was 49 cm (<−2.5 SDS), sitting height/height ratio was 56.9% (<−2.0 SDS) and arm span/height ratio was 94%. BMI was at 10th percentile. Head circumference was normal. The habitus was not muscular, and she had mesomelia and facial dysmorphisms, including hypertelorism, epicanthus and mild webbed neck. Bone age according to Greulich and Pyle was 2.5 yr. Radiological examination of the forearms was normal.The total Rappold’s score was 7 (Table 1).
The second girl (Figure 1-n.10) was born after 37 weeks of gestation after an unremarkable pregnancy. Birth anthropometric measurements were the following: weight 2.430 kg (3th-10th percentile), length 46 cm (10th-25th percentile). She showed poor linear and ponderal growth from the age of six months. Psychomotor development was normal.At our first clinical evaluation she was 11-month-old. She showed short stature (72 cm, −4.11 SDS). Sitting height was 42 cm (< −2.5 SDS), sitting height/height ratio was 58.3% (<−2.5 SDS) and arm span/height ratio was 94%. BMI was at 10th percentile. Head circumference was normal.The habitus was not muscular. She did not show mesomelia but facial dysmorphisms were detected, including epicanthus, hypertelorism and mild webbed neck. Bone age according to Greulich and Pyle was 1 yr. Radiological examination of the radius and ulna was normal.The total Rappold’s score was 7 (Table 1).
The index case’s mother (Figure 1-n.11) (54 year-old) did not show short stature (159 cm, −0.32 SDS). Sitting height was 75 cm (<−2.5 cm), sitting height/height ratio was 52% (<−2.5 SDS) and arm span/height ratio was 100%. BMI was between 25th-50th percentile. The habitus was muscular, and she had mesomelia with facial dysmorphisms (hypertelorism) and mild scoliosis. Radiological examination of the radius and ulna showed bilateral Madelung deformity, with left ulnar dorsal dislocation. The total Rappold’s score was 8 (Table 1).
Family 4
The index case (Figure 1-n.12) was a 14-years and 7 months girl admitted to the outpatient Endocrine Clinic of the Department of Pediatrics, University of Chieti, Italy, for short stature. The girl was the first offspring of unrelated healthy parents. She was born after 40 weeks of gestation after an unremarkable pregnancy. Birth anthropometric measurements were the following: weight 3.180 kg (25th-50th percentile), length 48 cm (10th-25th percentile). From the first months of life she had slow linear and ponderal growth, with normal psychomotor development. At our first clinical evaluation she showed short stature (147.6 cm, −1.79 SDS). Sitting height was 75 cm (−2.5 SDS), sitting height/height ratio was 0.50 (−2.0 SDS) and arm span/height ratio was 97%. BMI was between 50th-75th percentile and head circumference was normal. She had mild webbed neck, muscular hypertrophy, pectus carenatum, and cubitus and genu valgus. Radiological examination of the radius and ulna showed bilateral Madelung deformity. Bone age according to Greulich and Pyle was 16 yr. Based on the detected clinical and radiological signs, the total Rappold’s score was 9 (Table 1).
The sister of the proband (Figure 1-n.13) (11-year-old) showed a normal linear growth (138 cm, −1.0 SDS), sitting height was 67 cm (< −2.5 SDS), sitting height/height ratio was 0.48 cm (< −2.5 SDS) and arm span/height ratio was 97%. BMI was >85th percentile. She had muscular hypertrophy, pectus carenatum, and cubitus and genu valgus. Radiological examination showed bilaterally triangularization of distal epiphysis and bowing of diaphysis of radius, correlated to Madelung deformity. Bone age according to Greulich and Pyle was 12 yr. The total Rappold’s score was 9 (Table 1).
The mother of the proband (Figure 1-n.14) (42-year-old) also showed normal stature (160.2 cm, −0.12 SDS). Sitting height was 80 cm (< −2.5 SDS), sitting height/height ratio was 0.50 cm (< −2.0 SDS) and arm span/height ratio was 93%. BMI was >95th percentile. She had mild mesomelia with muscular hypertrophy. No facial dysmorphisms were detected except for webbed neck. The patient suffered from adolescence of diffuse muscular pain.Radiological examination of the radius and ulna showed bilateral Madelung deformity. The total Rappold’s score was 12 (Table 1).
Laboratory investigations allowed us to exclude thyroid dysfunction, abnormal IGF-1 levels and celiac disease in all the investigated patients, which all had normal diploid karyotype.
Molecular analysis
Genomic DNA was extracted from peripheral blood or buccal swab by QIAamp DNA Blood Midi Kit (Qiagen, Hilden, Germany). SHOX deletions were detected using the MRC-Holland MLPA kit (Salsa P018-E and F1; MRC-Holland, Netherlands – Resnova, Italy) according to the manufacturer’s instructions. The P018-D1 SHOX probemix contains 44 MLPA probes with amplification products between 130 and 463 nt. Seven of these probes are specific for each exon of the human SHOX gene and one is mapped just before the SHOX promoter region (4 kb before SHOX-PAR1). In addition, 14 probes are present detecting sequences in a region downstream of SHOX which has been implicated in regulation of SHOX transcription. Furthermore, nine probes on the X chromosome, out of PAR regions, are included in this probemix. Finally, ten autosomal reference probes are included. All the patients were also investigated with the last MLPA SHOX probemix (P018-F1) that as compared to oldest D1version three new probes near the PAR1 boundary have been included. Two probes (GPR143 and 13296-L15336) has been removed. The 88 and 96 nt control fragments have been replaced (QDX2) (http://www.mrc-holland.com). Data Analysis was performed using the Coffalyser software v. 9.4 (http://coffalyser.wordpress.com/).
In order to rule out false positive cases due to the presence of polymorphisms hampering the MLPA probe, we further investigated cases with deletion using PCR amplification with primers mapped within the probes region (SHOX_318 F: ACACCCAGTCATGAATGCAA; SHOX_318 R: CTTGGCTGGACAGACTCAGG; SHOX_432 F: ACATCGGCCTTTCCAAATAA; SHOX_432R: CTCGGGAGGCAGAGAGATTT), followed by direct sequencing on ABI 3130XL (ABI, Warrington, UK).
Results
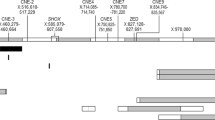
MLPA analysis, carried out in the four index patients and their familiars (N = 10), evidenced a heterozygous deletion of probes 13296-L15336, 05645-L05099 and 05646-L15507 (47543 bp deletion) in nine patients (patients 1–7, 9, 10) and a homozygous deletion of the same probes in the patient n. 8 (Figure 2H, I). A heterozygous deletion encompassing only the probe 05645-L05099 was revealed in three related patients (patients 12–14) (Figure 2L). All detected deletions involved a distinct SHOX enhancer region, not affecting the SHOX coding region.
All the data were confirmed using the latest MLPA SHOX probemix (P018-F1). Since in this last probe mix version the probe 13296-L15336 was removed because not reliable, the 47543pb deletion was characterized by the absence of the remaining two MLPA probes 05645-L05099 and 05646-L15507.
MLPA results were confirmed by PCR and DNA sequencing, showing absence of amplification in the patient with a homozygote deletion and absence of polymorphisms in all the observed heterozygous deletions.
Discussion
The SHOX gene belongs to a family of transcriptional regulators that are mainly expressed in the middle portion of the limbs where assure the correct balance between proliferation and apoptosis during bone development. The absence of wild type SHOX would promote atypical proliferation of the chondrocytes combined with defective differentiation, leading to retarded longitudinal bone growth [30, 31]. This specific pattern of expression can explain the wide phenotypic variability of SHOX deficiency patients with cases of normal stature but mesomelia and Madelung deformity.
In the present study we investigated SHOX region molecular defect in four LWD families, three of which showed the recurrent ~47.5 kb PAR1 deletion, previously described by Benito-Sanz et. al [26]. This deletion is mapped downstream of the SHOX gene and contains an enhancer sequence (ECR1/CNE7). The deletion was present in heterozygous state in all the analyzed members except one case of homozygous deletion, which surprisingly presented severe bilateral Madelung deformity but a normal stature within her target height range.
The fourth analyzed family is of particular interest, since we found, for the first time, a heterozygous deletion encompassing only one of the three classical MLPA probes characterizing the recurrent – 47.5 kb PAR1 deletions. The rearrangement cosegregated with the LWD phenotype in all the members of the family, with a clinical phenotype similar to the one showed by cases with larger deletion, confirming a pathogenic effect also of this shorter enhancer deletion. Looking in detail at this group of 14 patients it appears evident that the phenotype of patients with deletions in the 3’-PAR1 region is remarkably variable and not related to the extension or the homozygous and heterozygous form. Up to date only a few studies have been performed concerning the phenotype showed by patients carrier of enhancer deletions, all suggesting a great variability [7, 28]. Kant et al. described a case in which the enhancer deletion was associated with normal stature, although below the target height range [28].
In order to better assess the clinical expression in our cohort, we calculated the Rappold score of the investigated patients [16]. Interestingly, all patients had a score greater than 4 with a median value of 11.5 (range 6–13), but a great variability was found among the different clinical signs. In fact, short forearm and muscular hypertrophy were the two most observed dysmorphic abnormalities (100% and 80%, respectively), while cubitus valgus and BMI greater than 50th percentile were less frequent (60% and 40%, respectively). Unexpectedly, arm span/height ratio and sitting height/height ratio were uncommon (30% and 20%, respectively), whereas bowing of forearm and tibia as well as dislocation of ulna at the elbow were present only in one subject (Table 1).
The results obtained from the study of these four families suggest that, even if all the patients were eligible for the SHOX molecular analysis, some of the Rappold criteria are not very distinctive for SHOX deficiency. In fact, we had to take into account other characteristics, such as Madelung deformity, found in 60% of our sample. Therefore, it could be speculated that mutations in the SHOX enhancer region seem to be responsible mainly for bone deformities. In fact, both the homozygous case and her mother, who presented a heterozygous mutations in the SHOX enhancer region, showed bilateral Madelung deformity but not short stature.
The explanation of these findings probably is correlated with the etiology of the Madelung deformity, which originates with disorganized growth of part of the radial epiphysis, leading to radial bowing, premature fusion of that epiphysis, dorsal dislocation of the ulna, and wedged carpal bones. The premature fusion of the physis leads to cessation of longitudinal growth, and is always located in the ulnar zone of the distal radius but varies in the antero-posterior plane [32].
According to literature data, in our cohort, we did not find a correlation between bone defect, gender and the deletion size. These could be related to estrogens influence on the growth plate, which worse dyschondrosteosis leading to severe bone pain [33]. In this respect, it is interesting to underline that in this report many patients suffered from chronic widespread pain and received diagnosis of fibromyalgia. Since the pathogenesis of this musculoskeletal disorder is still unknown, a potential role of SHOX gene could be hypothesized, and further studies are required to clarify the relationship between SHOX deficit and fibromyalgia. In addition there isn’t an age-dependent phenotype in these subjects.
Conclusions
In conclusion, the present report confirms the usefulness to perform SHOX analysis including the enhancer sequence in patients with elevated Rappold’s score. In addition, it could be useful to follow-up these patients in order to verify the onset of fibromyalgia or other chronic idiopathic musculoskeletal disorders. Finally, the detection of a case of homozygous deletion of this region in a patient with severe bilateral Madelung deformity but a normal stature suggests the role of SHOX gene enhancer in contributing to different anomalies, which constitutes a wide spectrum of the disease. Therefore, the SHOX deficiency represents a complex and heterogeneous group of conditions ranging from the more severe phenotype (Langer syndrome) to milder forms (isolated short stature/isolated Madelung deformity).
Consent
Written informed consent was obtained from the patient for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Abbreviations
- SHOX :
-
Short stature homeobox
- PAR1:
-
Pseudoautosomal Region 1
- TS:
-
Turner syndrome
- ISS:
-
Idiopathic short stature
- LWD:
-
Léri-Weill dyschondrosteosis
- LS:
-
Langer mesomelic dysplasia
- SDS:
-
Standard deviation score
- GH:
-
Growth hormone
- FISH:
-
Fluorescence in situ hybridization
- MLPA:
-
Multiplex ligation-dependent probe amplification
- BMI:
-
Body mass index
- DNA:
-
Deoxyribonucleic acid.
References
Ellison JW, Wardak Z, Young MF, Robey PG, Laig-Webster M, Chiong W: PHOG, a candidate gene for involvement in the short stature of Turner syndrome. Hum Mol Genet. 1997, 6: 1341-1347.
Belin V, Cusin V, Viot G, Girlich D, Toutain A, Moncla A, Vekemans M, Le Merrer M, Munnich A, Cormier-Daire V: SHOX mutations in dyschondrosteosis (Léri-Weill syndrome). Nat Genet. 1998, 19: 67-69.
Stuppia L, Calabrese G, Borrelli P, Gatta V, Morizio E, Mingarelli R, Di Gilio MC, Crinò A, Giannotti A, Rappold GA, Palka G: Loss of the SHOX gene associated with Léri-Weill dyschondrosteosis in a 45, X male. J Med Genet. 1999, 36: 711-713.
Calabrese G, Fischetto R, Stuppia L, Capodiferro F, Mingarelli R, Causio F, Rocchi M, Rappold GA, Palka G: X/Y translocation in a family with Léri-Weill dyschondrosteosis. Hum Genet. 1999, 105: 367-368.
Palka G, Stuppia L, Guanciali Franchi P, Chiarelli F, Fischetto R, Borrelli P, Giannotti A, Fioretti G, Rinaldi MM, Mingarelli R, Rappold GA, Calabrese G: Short arm rearrangements of sex chromosomes with haploinsufficiency of the SHOX gene are associated with Léri-Weill dyschondrosteosis. Clin Genet. 2000, 57: 449-453.
Rappold GA, Fukami M, Niesler B, Schiller S, Zumkeller W, Bettendorf M, Heinrich U, Vlachopapadoupoulou E, Reinehr T, Onigata K, Ogata T: Deletions of the homeobox gene SHOX (short stature homeobox) are an important cause of growth failure in children with short stature. J Clin Endocrinol Metab. 2002, 87: 1402-1406.
Chen J, Wildhardt G, Zhong Z, Röth R, Weiss B, Steinberger D, Decker J, Blum WF, Rappold G: Enhancer deletions of the SHOX gene as a frequent cause of short stature: the essential role of a 250 kb downstream regulatory region. J Med Genet. 2009, 46: 834-839.
Sabherwal N, Bangs F, Röth R, Weiss B, Jantz K, Tiecke E, Hinkel GK, Spaich C, Hauffa BP, van der Kamp H, Kapeller J, Tickle C, Rappold G: Longrange conserved non-coding SHOX sequences regulate expression in developing chicken limb and are associated with short stature phenotypes in human patients. Hum Mol Genet. 2007, 16: 210-222.
Morizio E, Stuppia L, Gatta V, Fantasia D, Guanciali Franchi P, Rinaldi MM, Scarano G, Concolino D, Giannotti A, Verrotti A, Chiarelli F, Calabrese G, Palka G: Deletion of the SHOX gene in patients with short stature of unknown cause. Am J Med Genet A. 2003, 119A: 293-296.
Stuppia L, Calabrese G, Gatta V, Pintor S, Morizio E, Fantasia D, Guanciali Franchi P, Rinaldi MM, Scarano G, Concolino D, Giannotti A, Petreschi F, Anzellotti MT, Pomilio M, Chiarelli F, Tumini S, Palka G: SHOX mutations detected by FISH and direct sequencing in patients with short stature. J Med Genet. 2003, 40: E11-
Ross JL, Scott C, Marttila P, Kowal K, Nass A, Papenhausen P, Abboudi J, Osterman L, Kushner H, Carter P, Ezaki M, Elder F, Wei F, Chen H, Zinn AR: Phenotypes associated with SHOX deficiency. J Clin Endocrinol Metab. 2001, 86: 5674-5680.
Jorge AA, Souza SC, Nishi MY, Billerbeck AE, Libório DC, Kim CA, Arnhold IJ, Mendonca BB: SHOX mutations in idiopathic short stature and Léri-Weill dyschondrosteosis: frequency and phenotypic variability. Clin Endocrinol (Oxf). 2007, 66: 130-135.
Binder G, Ranke MB, Martin DD: Auxology is a valuable instrument for the clinical diagnosis of SHOX haploinsufficiency in schoolage children with unexplained short stature. J Clin Endocrinol Metab. 2003, 88: 4891-4896.
Niesler B, Röth R, Wilke S, Fujimura F, Fischer C, Rappold G: The novel human SHOX allelic variant database. Hum Mutat. 2007, 28: 933-938.
Binder G, Weidenkeller M, Blumenstock G, Langkamp M, Weber K, Franz AR: Rational approach to the diagnosis of severe growth hormone deficiency in the newborn. J Clin Endocrinol Metab. 2010, 95: 2219-2226.
Rappold G, Blum WF, Shavrikova EP, Crowe BJ, Roeth R, Quigley CA, Ross JL, Niesler B: Genotypes and phenotypes in children with short stature: clinical indicators of SHOX haploinsufficiency. J Med Genet. 2007, 44: 306-313.
Binder G: Short stature due to SHOX deficiency: genotype, phenotype, and therapy. Horm Res Paediatr. 2011, 75: 81-89.
Gatta V, Antonucci I, Morizio E, Palka C, Fischetto R, Mokini V, Tumini S, Calabrese G, Stuppia L: Identification and characterization of different SHOX gene deletions in patients with Léri-Weill dyschondrosteosys by MLPA assay. J Hum Genet. 2007, 52: 21-27.
Stuppia L, Gatta V, Antonucci I, Giuliani R, Palka G: Different approaches in the molecular analysis of the SHOX gene dysfunctions. J Endocrinol Invest. 2010, 33: 30-33.
Huber C, Rosilio M, Munnich A, Cormier- Daire V: French SHOX GeNeSIS Module: High incidence of SHOX anomalies in individuals with short stature. J Med Genet. 2006, 43: 735-739.
Stuppia L, Antonucci I, Palka G, Gatta V: Use of the MLPA Assay in the molecular diagnosis of gene copy number alterations in human genetic diseases. Int J Mol Sci. 2012, 13: 3245-3276.
Fukami M, Kato F, Tajima T, Yokoya S, Ogata T: Transactivation function of an approximately 800-bp evolutionarily conserved sequence at the SHOX 3' region: implication for the downstream enhancer. Am J Hum Genet. 2006, 78: 167-170.
Fukami M, Dateki S, Kato F, Hasegawa Y, Mochizuki H, Horikawa R, Ogata T: Identification and characterization of cryptic SHOX intragenic deletions in three Japanese patients with Léri-Weill dyschondrosteosis. J Hum Genet. 2008, 53: 454-459.
Durand C, Bangs F, Signolet J, Decker E, Tickle C, Rappold G: Enhancer elements upstream of the SHOX gene are active in the developing limb. Eur J Hum Genet. 2010, 18: 527-532.
Kenyon EJ, McEwen G, Callaway H, Elgar G: Functional analysis of conserved noncoding regions around the short Stature hox Gene (shox) in whole zebrafish embryos. PLoS One. 2011, 6: e21498-
Benito-Sanz S, Royo JL, Barroso E, Paumard-Hernández B, Barreda-Bonis AC, Liu P, Gracía R, Lupski JR, Campos-Barros Á, Gómez-Skarmeta JL, Heath KE: Identification of the first recurrent PAR1 deletion in Léri-Weill dyschondrosteosis and idiopathic short stature reveals the presence of a novel SHOX enhancer. J Med Genet. 2012, 49: 442-450.
Caliebe J, Broekman S, Boogaard M, Bosch CA, Ruivenkamp CA, Oostdijk W, Kant SG, Binder G, Ranke MB, Wit JM, Losekoot M: IGF1, IGF1R and SHOX mutation analysis in short children born small for gestational age and short children with normal birth size (idiopathic short stature). Horm Res Paediatr. 2012, 77: 250-260.
Kant SG, Broekman SJ, de Wit CC, Bos M, Bos M, Scheltinga SA, Bakker E, Oostdijk W, van der Kamp HJ, van Zwet EW, van der Hout AH, Wit JM, Losekoot M: Phenotypic characterization of patients with deletions in the 3'-flanking SHOX region. Peer J. 2013, 19: 1:e-
Schiller S, Spranger S, Schechinger B, Fukami M, Merker S, Drop SL, Tröger J, Knoblauch H, Kunze J, Seidel J, Rappold GA: Phenotypic variation and genetic heterogeneity in Léri–Weill syndrome. Eur J Hum Genet. 2000, 8: 54-62.
Marchini A, Marttila T, Winter A, Caldeira S, Malanchi I, Blaschke RJ, Häcker B, Rao E, Karperien M, Wit JM, Richter W, Tommasino M, Rappold GA: The short stature homeodomain protein SHOX induces cellular growth arrest and apoptosis and is expressed in human growth plate chondrocytes. J Biol Chem. 2004, 279: 37103-37114.
Munns CJ, Haase HR, Crowther LM, Hayes MT, Blaschke R, Rappold G, Glass IA, Batch JA: Expression of SHOX in human fetal and childhood growth plate. J Clin Endocrinol Metab. 2004, 89: 4130-4135.
Munns CF, Glass IA, LaBrom R, Hayes M, Flanagan S, Berry M, Hyland VJ, Batch JA, Philips GE, Vickers D: Histopathological analysis of Léri-Weill dyschondrosteosis: disordered growth plate. Hand Surg. 2001, 6: 13-23.
Ogata T: SHOX: pseudoautosomal homeobox containing gene for short stature and dyschondrosteosis. Growth Horm IGF Res. 1999, 9 (Suppl B): 53-57.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/15/87/prepub
Acknowledgements
The authors did not count with any funding source. The authors did not have funding sources for the preparation of the manuscript nor for its publication.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing and non-financial interests and also reveal any non-financial competing interests.
Authors’ contributions
VG: contributed to conception and design, MLPA data analysis and drafted the manuscript. CP: responsible of clinical, participated in its design and coordination and drafted the manuscript. VC: responsible of clinical data and participated in its design and coordination. SF: performed the SHOX gene study through MLPA. GC, MS and ARC: responsible of radiographic studies. AM: responsible of clinical data and participated in its design and coordination. FC: helped to draft the manuscript. LS: have given final approval of the version to be published. All authors read and approved the final manuscript.
Valentina Gatta, Chiara Palka contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Gatta, V., Palka, C., Chiavaroli, V. et al. Spectrum of phenotypic anomalies in four families with deletion of the SHOX enhancer region. BMC Med Genet 15, 87 (2014). https://doi.org/10.1186/1471-2350-15-87
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-15-87