
Abstract
Background
Congenital cataract is a Mendelian disorder that frequently causes blindness in infants. To date, various cataract-associated loci have been mapped; more than 30 genes have been identified by linkage analysis. However, the pathogenic loci in some affected families are still unknown, and new research strategies are needed. In this study, we used linkage-exome combinational analysis to further investigate the pedigree of a four-generation Chinese family with autosomal dominant coralliform cataract.
Methods
We combined whole exome sequencing and linkage analysis to identify the causative mutation. The exome capture and next-generation sequencing were used to sequence the protein-coding regions in the genome of the proband to identify rare mutations, which were further screened for candidate mutations in linkage regions. Candidate mutations were independently verified for co-segregation in the whole pedigree using Sanger sequencing.
Results
We identified a C to A transversion at nucleotide position c.70 in exon 2 of CRYGD, a cataract-associated gene. This mutation resulted in a threonine substitution for proline at amino acid residue 24.
Conclusions
We identified a missense P24T mutation in CRYGD that was responsible for coralliform cataract in our studied family. Our findings suggest that the combination of exome sequencing and linkage analysis is a powerful tool for identifying Mendelian disease mutations that might be missed by the classic linkage analysis strategy.
Similar content being viewed by others
Background
Congenital cataract is a Mendelian disorder resulting in blindness during infancy or early childhood. Non-syndromic congenital cataracts have an estimated frequency of 1 to 15 per 10,000 live births throughout the world [1–3]. Congenital cataract is primarily autosomal dominant, although autosomal recessive and X-linked inheritances have also been reported [4]. To date, various cataract-associated loci have been mapped, in which more than 30 genes were identified by linkage analysis. Most cataract-associated genes are crystallin genes, such as alpha crystallins (CRYAA and CRYAB), beta crystallins (CRYBB1, CRYBB2, CRYBB3, CRYBA1, CRYBA3, and CRYBA4), and gamma crystallins (CRYGA, CRYGC, CRYGD, and CRYGS). Approximately 25% of affected families have defects in membrane transport genes, including major intrinsic protein of lens fiber (MIP), gap junction proteins (GJA3 and GJA8), transmembrane protein 114 (TMEM114), and lens intrinsic membrane protein 2 (LIM2). The remaining known mutations are found in genes encoding cytoskeletal proteins, growth and transcription factors, v-maf musculoaponeurotic fibrosarcoma oncogene homolog, heat shock transcription factor, and others as outline in Additional file 1: Table S1.
Linkage analysis is a classic strategy for mapping disease-associated loci in Mendelian inheritance pedigrees. This method requires large families, commonly a multi-generation pedigree with at least 6 to 12 affected individuals, to obtain high reliability and statistical significance. However, significant linkage remains hard to establish despite large sample sizes, particularly due to a low density of microsatellite markers, misclassification of patients, low heterogeneity, low disease penetrance, or clinically identical phenocopies [5]. Furthermore, although significant linkage may be obtained, it is still difficult to further identify causal mutations in a large genomic interval including dozens of genes [6, 7]. Next-generation sequencing (NGS) technology provides new avenues for uncovering genetic causes of human diseases. Although whole genome sequencing is becoming more practical due to its falling cost and increased throughput, it still remains expensive for most applications. Whole exome sequencing is an economical method compared to whole genome sequencing. Recent studies showed that the human genome contains about 180,000 exons, accounting for about 1% of the total genome [8]. Thus, whole exome sequencing is especially promising for research on monogenic disorders [9, 10] since most of these disorders are caused by exonic mutations or splice-site mutations.
In a previous study, we presented evidence to suggest some candidate linkage regions in a four-generation Chinese family with autosomal dominant coralliform cataract, but the causal mutation was not identified [11]. In the current study, we have further investigated the same pedigree using whole exome sequencing and linkage combinational analysis to identify the causal mutation.
Methods
Clinical ascertainment and DNA sampling
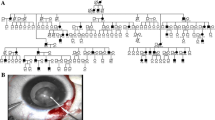
A total of 19 family members, including 9 affected and 10 unaffected individuals, were recruited for a previous study by Gao et al. in 2005 [11]. Three additional individuals (affected III:11, unaffected III:5 and III:8) were newly recruited for this study (Figure 1). All members of this family underwent an examination that included photography and slit-lamp microscopy of the lens. The research project was approved by the Ethics Committee of Harbin Medical University. All samples were collected with informed consent from the participants. Written informed consent was obtained from the parents of each child. All experiments carried out with human subjects were in compliance with the Helsinki Declaration.

Pedigree of a four-generation Chinese family with autosomal dominant coralliform cataract and the linked haplotypes. The black arrow indicates the proband. Black symbols and bars denote affected status.
DNA samples were extracted from peripheral blood leukocytes using the QIAamp Blood Mini DNA kit (Qiagen, Santa Clara, CA, USA). Before analysis of the samples, DNA aliquots were re-precipitated to remove proteins and fragments.
Exome capture and next-generation sequencing
A whole exome–enriched library was prepared from 3 μg of genomic DNA from the proband (II:6) using Agilent’s SureSelect Human All Exon 50 Mb solution-based capture reagent. Exome capture was performed according to the manufacturer’s protocol (Agilent, USA). The captured DNA was then sequenced using the Illumina HiSeq2000 platform. Raw image files were processed by Illumina Basecaller Software 1.7 (San Diego, CA, USA) for base-calling with default parameters.
Short-read alignment, mapping statistics, and variant annotation
The obtained sequence reads were aligned to the human genome (hg19) using the SOAP2 [12] and BWA [13] tools for single nucleotide polymorphism (SNP) and insertion/deletion (indel), respectively. The percentages of read alignment to both the reference genome and the targeted exome were calculated using Perl scripts. Similarly, Perl scripts were used for the detection of mismatch frequencies and error positions. SNP calling was done with SOAPsnp [14], and indels were identified through the alignment result with GATK [12]. Detailed annotation information was obtained from dbSNP, CCDS, UCSC Genome Browser, Ensembl, and Encode databases. Using these annotations, we screened the novel and likely deleterious variants for further study.
PCR and Sanger sequencing
Specific primers were designed for the target region, and the PCR products were sequenced on an ABI 3730 DNA analyzer following standard procedures (Life Technologies, USA). The sequence reads were analyzed using the Sequencher software package (GeneCodes Inc, USA). The sequencing traces were visually inspected in Finch TV v1.4 (Geospiza Inc, USA).
Linkage and haplotype analysis
Microsatellite markers were selected based on ABI PRISM Linkage Mapping Set (version 2.5, Applied Biosystems, USA) and the UCSC database. PCR products were electrophoresed on a 96-capillary automated DNA sequencer (MegaBACE 1000, Amersham, Germany) and were analyzed with Genetic Profiler software (version 1.5, Amersham, Germany). Two-point LOD scores were calculated using MLINK from the LINKAGE package (version 5.1). Autosomal dominant inheritance, disease-gene frequency of 0.0001, and 95% penetrance were assumed. Haplotyping was constructed using Cyrillic (version 2.1).
Results
Evaluation of exome sequencing data
A strategy of whole exome sequencing by hybrid capture and NGS was employed. The raw sequencing data obtained from the proband (II:6) was 9.4 Gb. The average read length was 90 bp. The efficiency of the hybrid capture was 81.6%; 71,968,280 out of 88,234,362 reads were uniquely mapped to targeted exome regions, and 99.39% of the whole exome was covered by reads. The distribution of per-base sequencing depth in target regions approximated a Poisson distribution, which showed that the captured exome region was evenly sampled (Additional file 1: Figure S1). Mean depth per base within the target regions was 111.85-fold, and 97.7% of these regions were covered by four or more reads (94.5% by 10 or more reads) by paired-end sequencing.
Combinational analysis of exome and linkage identified the causative gene
A total of 1868 genetic variations, including non-synonymous mutations, splice site variations, and indels, were identified from the proband (Table 1). Because numerous mutations were detected, we combined whole exome sequencing and linkage analysis to sift through the potential causative mutations. As described previously by Gao et al. [11], five loci with positive but non-significant LOD scores (>1) were identified by linkage analysis (Additional file 1: Table S2).
In these five positive loci, we identified 11 mutations in nine genes by sequencing, including 10 SNPs and one short indel (Table 2). A mutation of CRYGD, a known gene causing congenital cataract, was included in these mutations. This mutation at nucleotide position c.70 in exon 2 of CRYGD (Figure 2) results in a threonine substitution for proline at amino acid residue 24 (P24T). The mutation was present in 10 patients but absent in 12 unaffected members of the studied family and in 100 control chromosomes from unaffected individuals of matched geographical ancestry. The remaining 10 mutations were present in both patients and healthy relatives and showed no co-segregation with the disease. We also screened 35 known cataract genes (Additional file 1: Table S1) and identified five mutations in CRYGD, TMEM144, VIM, JAM3, and BFSP1 (Additional file 1: Table S3). These mutations also did not co-segregate with the disease, except for CRYGD.

Sequence and pedigree analysis of the C to A transversion in exon 2 of CRYGD . (A) Sequence of the wild-type CRYGD alleles in the unaffected family members. (B) Heterozygous C to A mutation of CRYGD exon 2, resulting in a substitution from proline (P) to threonine (T), was detected in affected patients. A single transversion was observed as a C/A double peak.
Linkage analysis with newly sampled family members and haplotyping
Considering the new findings by exome sequencing, three additional individuals were newly recruited from the same pedigree, and the linkage analysis was performed once again. The maximum LOD score was obtained at D2S2237 (Z=3.53, θ=0.0; Table 3). Recombination events in several individuals defined the proximal and distal borders of a significant cataract-associated locus within the region between D2S309 and D2S2178 on chromosome 2q33-34 (Figure 1). These results also suggest that CRYGD is the causative gene for the coralliform cataract observed in this family.
Discussion
We identified a C to A transversion at nucleotide position c.70 in exon 2 of CRYGD as the mutation responsible for congenital cataract in this family. CRYGD is an important structural protein essential for human lens transparency [15]. Based on the crystal structure of human CRYGD, the P24T mutation affects the N-terminal domain within the first Greek-key motif, causing the protein to have a slightly increased beta-sheet content, which may be attributed to the extension of an edge beta-strand due to the substitution of Pro24 with a residue capable of forming hydrogen bonds. The small increase in the fraction of beta-sheet content in the P24T mutant protein may contribute to the physical basis for precipitation of the protein [16, 17]. The P24T mutation in the CRYGD gene has been found in several pedigrees with various cataract phenotypes, including cerulean, coralliform, and fasciculiform [18–20], lending support to the conclusion that the P24T mutation in CRYGD identified in this study is the cause of congenital cataract in the affected family.
In order to contribute to the technological progress of mapping genetic causes of human disease, whole exome sequencing should be fast, comprehensive, and economical to identify protein-coding mutations, including missense, non-sense, splice site, and small deletion or insertion mutations. However, an individual typically varies from the reference genome at over 10,000 potential mutations [21]. In this study, a total of 1868 genetic variations were identified by whole exome sequencing. Due to this high number, sifting through the hundreds of gene variations to identify the causal mutation would be a difficult task. Therefore, we combined whole exome sequencing with linkage analysis to identify the causative mutation in five loci with positive but non-significant LOD scores. The majority (99.4%) of mutations were excluded, and only 11 candidate mutations in these linkage loci were identified. Finally, we identified CRYGD as the gene responsible for the cataract phenotype in these 11 mutations. Our observations show that the linkage-exome combinational analysis is an efficient strategy for identification of pathogenic mutations by remarkably reducing the pool of candidate genes.
For further confirmation, we recruited another three members from the same family and selected an additional seven microsatellite markers for fine linkage analysis. Significant linkage was easy to establish with these three newly recruited members. The maximum LOD score (Z=3.53, θ=0.0) was obtained for marker D2S2237 near the CRYGD gene (Table 3). These results further suggested that CRYGD was responsible for the coralliform cataract in this family. However, recruiting new family members is not always possible. Therefore, using linkage analysis in a small nuclear family followed by exome sequencing of a single patient to identify the causative gene is a feasible method. According to the present observations, whole exome sequencing is promising for the analysis of monogenic diseases and allows identification of pathogenic mutations when the linkage score is not significant or the candidate regions are too large to be investigated.
Conclusions
In conclusion, we identified the missense P24T mutation in CRYGD, which was responsible for the coralliform cataract afflicting a four-generation Chinese family. Notably, our study indicated that the linkage analysis-whole exome sequencing approach is a powerful tool for finding pathogenic genes of Mendelian inheritance and provides important guidance for developing an analytical framework in the near future.
References
Francis PJ, Berry V, Bhattacharya SS, Moore AT: The genetics of childhood cataract. J Med Genet. 2000, 37 (7): 481-488. 10.1136/jmg.37.7.481.
Apple DJ, Ram J, Foster A, Peng Q: Elimination of cataract blindness: A global perspective entering the new millennium?. Surv Ophthalmol. 2000, 45: S5-S196.
Gilbert C, Foster A: Childhood blindness in the context of VISION 2020–the right to sight. Bull World Health Organ. 2001, 79 (3): 227-232.
Hejtmancik JF: Congenital cataracts and their molecular genetics. Semin Cell Dev Biol. 2008, 19 (2): 134-149. 10.1016/j.semcdb.2007.10.003.
Kuhlenbaumer G, Hullmann J, Appenzeller S: Novel Genomic Techniques Open New Avenues in the Analysis of Monogenic Disorders. Hum Mutat. 2011, 32 (2): 144-151. 10.1002/humu.21400.
Zhao R, Yang Y, He X, Liu Z, Wang P, Zhou L, Tang J, Xu W, Li L, Zhu Y: An autosomal dominant cataract locus mapped to 19q13-qter in a Chinese family. Mol Vis. 2011, 17: 265-269.
Bateman JB, Richter L, Flodman P, Burch D, Brown S, Penrose P, Paul O, Geyer DD, Brooks DG, Spence MA: A new locus for autosomal dominant cataract on chromosome 19: linkage analyses and screening of candidate genes. Invest Ophthalmol Vis Sci. 2006, 47 (8): 3441-3449. 10.1167/iovs.05-1035.
Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, Shaffer T, Wong M, Bhattacharjee A, Eichler EE, et al: Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009, 461 (7261): 272-276. 10.1038/nature08250.
Lalonde E, Albrecht S, Ha KCH, Jacob K, Bolduc N, Polychronakos C, Dechelotte P, Majewski J, Jabado N: Unexpected Allelic Heterogeneity and Spectrum of Mutations in Fowler Syndrome Revealed by Next-Generation Exome Sequencing. Hum Mutat. 2010, 31 (8): 918-923. 10.1002/humu.21293.
Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, et al: Exome sequencing identifies the cause of a mendelian disorder. Nature Genetics. 2010, 42 (1): 30-U41. 10.1038/ng.499.
Gao L, Qin W, Cui H, Feng G, Liu P, Gao W, Ma L, Li P, He L, Fu S: A novel locus of coralliform cataract mapped to chromosome 2p24-pter. J Hum Genet. 2005, 50 (6): 305-310. 10.1007/s10038-005-0251-y.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al: The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20 (9): 1297-1303. 10.1101/gr.107524.110.
Li H, Durbin R: Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009, 25 (14): 1754-1760. 10.1093/bioinformatics/btp324.
Li R, Li Y, Fang X, Yang H, Wang J, Kristiansen K: SNP detection for massively parallel whole-genome resequencing. Genome Res. 2009, 19 (6): 1124-1132. 10.1101/gr.088013.108.
Bloemendal H, de Jong W, Jaenicke R, Lubsen NH, Slingsby C, Tardieu A: Ageing and vision: structure, stability and function of lens crystallins. Progress in biophysics and molecular biology. 2004, 86 (3): 407-485. 10.1016/j.pbiomolbio.2003.11.012.
Evans P, Wyatt K, Wistow GJ, Bateman OA, Wallace BA, Slingsby C: The P23T cataract mutation causes loss of solubility of folded gammaD-crystallin. J Mol Biol. 2004, 343 (2): 435-444. 10.1016/j.jmb.2004.08.050.
Basak A, Bateman O, Slingsby C, Pande A, Asherie N, Ogun O, Benedek GB, Pande J: High-resolution X-ray crystal structures of human gamma D crystallin (1.25 angstrom) and the R58H mutant (1.15 angstrom) associated with aculeiform cataract. Journal of Molecular Biology. 2003, 328 (5): 1137-1147. 10.1016/S0022-2836(03)00375-9.
Shentu X, Yao K, Xu W, Zheng S, Hu S, Gong X: Special fasciculiform cataract caused by a mutation in the gammaD-crystallin gene. Mol Vis. 2004, 10: 233-239.
Khan AO, Aldahmesh MA, Ghadhfan FE, Al-Mesfer S, Alkuraya FS: Founder heterozygous P23T CRYGD mutation associated with cerulean (and coralliform) cataract in 2 Saudi families. Mol Vis. 2009, 15 (149–50): 1407-1411.
Yang GX, Xiong CL, Li SL, Wang YY, Zhao JL: A recurrent mutation in CRYGD is associated with autosomal dominant congenital coralliform cataract in two unrelated Chinese families. Mol Vis. 2011, 17 (123): 1085-1089.
Altshuler DL, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, Collins FS, De la Vega FM, Donnelly P, Egholm M, et al: A map of human genome variation from population-scale sequencing. Nature. 2010, 467 (7319): 1061-1073. 10.1038/nature09534.
Kaul H, Riazuddin SA, Shahid M, Kousar S, Butt NH, Zafar AU, Khan SN, Husnain T, Akram J, Hejtmancik JF, et al: Autosomal recessive congenital cataract linked to EPHA2 in a consanguineous Pakistani family. Mol Vis. 2010, 16 (58–59): 511-517.
Bremond-Gignac D, Bitoun P, Reis LM, Copin H, Murray JC, Semina EV: Identification of dominant FOXE3 and PAX6 mutations in patients with congenital cataract and aniridia. Mol Vis. 2010, 16 (183–84): 1705-1711.
Zheng J-q, Liu P, Wang J-w, Liu J-j: Expression of mutation type GJA8 gene and wild type GJA8 gene of a congenital inherited nuclear cataract family in eukaryotic cells. Zhonghua yi xue za zhi. 2010, 90 (15): 1062-1066.
Khan K, Rudkin A, Parry DA, Burdon KP, McKibbin M, Logan CV, Abdelhamed ZI, Muecke JS, Fernandez-Fuentes N, Laurie KJ, et al: Homozygous mutations in PXDN cause congenital cataract, corneal opacity, and developmental glaucoma. Am J Hum Genet. 2011, 89 (3): 464-473. 10.1016/j.ajhg.2011.08.005.
Klopp N, Heon E, Billingsley G, Illig T, Wjst M, Rudolph G, Graw J: Further genetic heterogeneity for autosomal dominant human sutural cataracts. Ophthalmic Res. 2003, 35 (2): 71-77. 10.1159/000069134.
Santana A, Waiswol M, Arcieri ES, de Vasconcellos JPC, de Melo MB: Mutation analysis of CRYAA, CRYGC, and CRYGD associated with autosomal dominant congenital cataract in Brazilian families. Mol Vis. 2009, 15 (79–81): 793-800.
de Figueiredo ES, Giordano GG, Tavares A, da Silva MJ, de Vasconcellos JPC, Arieta CEL, de Melo MB: Novel human CRYGD rare variant in a Brazilian family with congenital cataract. Mol Vis. 2011, 17 (238–39): 2207-2211.
Chen JJ, Ma ZW, Jiao XD, Fariss R, Kantorow WL, Kantorow M, Pras E, Frydman M, Pras E, Riazuddin S, et al: Mutations in FYCO1 Cause Autosomal-Recessive Congenital Cataracts. Am J Hum Genet. 2011, 88 (6): 827-838. 10.1016/j.ajhg.2011.05.008.
Ma X, Li FF, Wang SZ, Gao C, Zhang M, Zhu SQ: A new mutation in BFSP2 (G1091A) causes autosomal dominant congenital lamellar cataracts. Mol Vis. 2008, 14: 1906-1911.
Vanita V, Singh JR, Singh D, Varon R, Sperling K: Novel mutation in the gamma-S crystallin gene causing autosomal dominant cataract. Mol Vis. 2009, 15: 476-481.
Riazuddin SA, Amiri-Kordestani L, Kaul H, Butt T, Jiao X, Riazuddin S, Hejtmancik JF: Novel SIL1 mutations in consanguineous Pakistani families mapping to chromosomes 5q31. Mol Vis. 2009, 15: 1050-1056.
Pras E, Raz J, Yahalom V, Frydman M, Garzozi HJ, Hejtmancik JF: A nonsense mutation in the glucosaminyl (N-acetyl) transferase 2 gene (GCNT2): association with autosomal recessive congenital cataracts. Invest Ophthalmol Vis Sci. 2004, 45 (6): 1940-1945. 10.1167/iovs.03-1117.
Azuma N, Hirakiyama A, Inoue T, Asaka A, Yamada M: Mutations of a human homologue of the Drosophila eyes absent gene (EYA1) detected in patients with congenital cataracts and ocular anterior segment anomalies. Hum Mol Genet. 2000, 9 (3): 363-366. 10.1093/hmg/9.3.363.
Muller M, Bhattacharya SS, Moore T, Prescott Q, Wedig T, Herrmann H, Magin TM: Dominant cataract formation in association with a vimentin assembly disrupting mutation. Hum Mol Genet. 2009, 18 (6): 1052-1057. 10.1093/hmg/ddn440.
Kloeckener-Gruissem B, Vandekerckhove K, Nurnberg G, Neidhardt J, Zeitz C, Nurnberg P, Schipper I, Berger W: Mutation of solute carrier SLC16A12 associates with a syndrome combining juvenile cataract with microcornea and renal glucosuria. Am J Hum Genet. 2008, 82 (3): 772-779. 10.1016/j.ajhg.2007.12.013.
Berry V, Francis PJ, Prescott Q, Waseem NH, Moore AT, Bhattacharya SS: A novel 1-bp deletion in PITX3 causing congenital posterior polar cataract. Mol Vis. 2011, 17 (140): 1249-1253.
Chen Q, Ma J, Yan M, Mothobi ME, Liu Y, Zheng F: A novel mutation in CRYAB associated with autosomal dominant congenital nuclear cataract in a Chinese family. Mol Vis. 2009, 15: 1359-1365.
Mochida GH, Ganesh VS, Felie JM, Gleason D, Hill RS, Clapham KR, Rakiec D, Tan WH, Akawi N, Al-Saffar M, et al: A Homozygous Mutation in the Tight-Junction Protein JAM3 Causes Hemorrhagic Destruction of the Brain, Subependymal Calcification, and Congenital Cataracts. Am J Hum Genet. 2010, 87 (6): 882-889. 10.1016/j.ajhg.2010.10.026.
Cai FC, Zhu JF, Chen W, Ke T, Wang F, Tu X, Zhang Y, Jin RM, Wu XY: A novel PAX6 mutation in a large Chinese family with aniridia and congenital cataract. Mol Vis. 2010, 16 (126–27): 1141-1145.
Yang GX, Zhang GS, Wu Q, Zhao JL: A novel mutation in the MIP gene is associated with autosomal dominant congenital nuclear cataract in a Chinese family. Mol Vis. 2011, 17 (148): 1320-1323.
Yang GX, Xing BG, Liu GC, Lu XQ, Jia XG, Lu XQ, Wang XL, Yu HY, Fu YJ, Zhao JL: A novel mutation in the GJA3 (connexin46) gene is associated with autosomal dominant congenital nuclear cataract in a Chinese family. Mol Vis. 2011, 17 (120): 1070-1073.
Jamieson RV, Farrar N, Stewart K, Perveen R, Mihelec M, Carette M, Grigg JR, McAvoy JW, Lovicu FJ, Tam PP, et al: Characterization of a familial t(16;22) balanced translocation associated with congenital cataract leads to identification of a novel gene, TMEM114, expressed in the lens and disrupted by the translocation. Hum Mutat. 2007, 28 (10): 968-977. 10.1002/humu.20545.
Hansen L, Eiberg H, Rosenberg T: Novel MAF mutation in a family with congenital cataract-microcornea syndrome. Mol Vis. 2007, 13: 2019-2022.
Sajjad N, Goebel I, Kakar N, Cheema AM, Kubisch C, Ahmad J: A novel HSF4 gene mutation (p.R405X) causing autosomal recessive congenital cataracts in a large consanguineous family from Pakistan. BMC Med Genet. 2008, 9: 99-10.1186/1471-2350-9-99.
Yang GX, Zhai XL, Zhao JL: A recurrent mutation in CRYBA1 is associated with an autosomal dominant congenital nuclear cataract disease in a Chinese family. Mol Vis. 2011, 17 (172–74): 1559-1563.
Ferrini W, Schorderet DF, Othenin-Girard P, Uffer S, Heon E, Munier FL: CRYBA3/A1 gene mutation associated with suture-sparing autosomal dominant congenital nuclear cataract: a novel phenotype. Invest Ophthalmol Vis Sci. 2004, 45 (5): 1436-1441. 10.1167/iovs.03-0760.
Ponnam SP, Ramesha K, Tejwani S, Matalia J, Kannabiran C: A missense mutation in LIM2 causes autosomal recessive congenital cataract. Mol Vis. 2008, 14: 1204-1208.
Vanita V, Hejtmancik JF, Hennies HC, Guleria K, Nurnberg P, Singh D, Sperling K, Singh JR: Sutural cataract associated with a mutation in the ferritin light chain gene (FTL) in a family of Indian origin. Mol Vis. 2006, 12: 93-99.
Conley YP, Erturk D, Keverline A, Mah TS, Keravala A, Barnes LR, Bruchis A, Hess JF, FitzGerald PG, Weeks DE, et al: A juvenile-onset, progressive cataract locus on chromosome 3q21-q22 is associated with a missense mutation in the beaded filament structural protein-2. Am J Hum Genet. 2000, 66 (4): 1426-1431. 10.1086/302871.
Shiels A, Bennett TM, Knopf HL, Yamada K, Yoshiura K, Niikawa N, Shim S, Hanson PI: CHMP4B, a novel gene for autosomal dominant cataracts linked to chromosome 20q. Am J Hum Genet. 2007, 81 (3): 596-606. 10.1086/519980.
Li F-F, Yang M, Ma X, Zhang Q, Zhang M, Wang S-Z, Zhu S-Q: Autosomal Dominant Congenital Nuclear Cataracts Caused by a CRYAA Gene Mutation. Current Eye Research. 2010, 35 (6): 492-498. 10.3109/02713681003624901.
Billingsley G, Santhiya ST, Paterson AD, Ogata K, Wodak S, Hosseini SM, Manisastry SM, Vijayalakshmi P, Gopinath PM, Graw GJ, et al: CRYBA4, a novel human cataract gene, is also involved in microphthalmia. Am J Hum Genet. 2006, 79 (4): 702-709. 10.1086/507712.
Wang KJ, Wang S, Cao NQ, Yan YB, Zhu SQ: A Novel Mutation in CRYBB1 Associated with Congenital Cataract-Microcornea Syndrome: The p. Ser129Arg Mutation Destabilizes the beta B1/beta A3-crystallin Heteromer But Not the beta B1-crystallin Homomer. Hum Mutat. 2011, 32 (3): E2050-E2060. 10.1002/humu.21436.
Yao K, Li JY, Jin CF, Wang W, Zhu YA, Shentu XC, Wang QW: Characterization of a novel mutation in the CRYBB2 gene associated with autosomal dominant congenital posterior subcapsular cataract in a Chinese family. Mol Vis. 2011, 17 (17–20): 144-152.
Riazuddin SA, Yasmeen A, Yao W, Sergeev YV, Zhang Q, Zulfiqar F, Riaz A, Riazuddin S, Hejtmancik JF: Mutations in betaB3-crystallin associated with autosomal recessive cataract in two Pakistani families. Invest Ophthalmol Vis Sci. 2005, 46 (6): 2100-2106. 10.1167/iovs.04-1481.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/14/107/prepub
Acknowledgements
We thank the family members for their participation. We are grateful to Professor Renke Li (Univ. Toronto, Canada) for helping us with the English language in the manuscript. This study was supported by the International Science & Technology Cooperation Program of China (No. 2013DFA31610), the Program for Changjiang Scholars and Innovative Research Team in University (No. IRT1230), the New Century Support Program for the Excellent Scholar, Ministry of Education of China (No. NCET-09-0322, NCET-10-0149, NCET-11-0954), and the National S&T Major Special Project (No.2011ZX09102-010-01).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
SF obtained the funding and designed the research. XJ carried out the laboratory work, analyzed most of the data, and drafted the manuscript. FZ participated in the design of the research and revision of the manuscript. JB and YY carried out sample collection and helped prepare the laboratory work. LG revised the manuscript and did the linkage analysis. WS supervised the research personnel. XZ and HS participated in the sequence alignment and revised the manuscript. RG and LX analyzed and interpreted the data. ZY and DS carried out the molecular biology experiments. All authors have read and approved the final manuscript.
Electronic supplementary material
12881_2013_1137_MOESM1_ESM.doc
Additional file 1: Table S1: Known genes involved in congenital cataract. Table S2. Two-point LOD score (> 1) for the microsatellite markers in our previous study on 19 family members. Table S3. Variations identified in 35 known cataract genes. Figure S1. The read-depth distribution of exome sequencing assay. X-axis shows sequencing depth, while Y-axis indicates the percentage of total target regions under a given sequencing depth [22–56]. (DOC 172 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Jia, X., Zhang, F., Bai, J. et al. Combinational analysis of linkage and exome sequencing identifies the causative mutation in a Chinese family with congenital cataract. BMC Med Genet 14, 107 (2013). https://doi.org/10.1186/1471-2350-14-107
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-14-107