
Abstract
Background
Asthma is a phenotypically diverse disease with genetic susceptibility. A single nucleotide polymorphism (SNP) in the CD14 gene at position -260 (also known as -159) C>T has been inconsistently associated with asthma. The aim of this study was to estimate the combined likelihood of developing asthma given the CD14 -260C>T genotype.
Methods
Following the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines, a systematic search and meta-analysis of the literature was conducted to estimate the association between this SNP and asthma. Planned subgroup analyses were performed to detect potential sources of heterogeneity from selected study characteristics. Post-hoc sensitivity analysis was performed to identify studies exerting excessive influence on among-study heterogeneity and combined effects.
Results
Meta-analysis of 23 studies yielded a non-significant overall association with high heterogeneity across studies. After restricting analysis to studies using atopic asthma and non-atopic non-asthma case-control phenotypes and excluding studies influencing heterogeneity, the genotype-specific odds ratios (ORs) suggested a codominant model. Carriers of the TT and CT genotypes were about 33% less likely (OR = 0.67, 95% CI: 0.54-0.84) and about 20% less likely (OR = 0.80, 95% CI: 0.66-0.95), respectively, to have atopic asthma compared to carriers of the CC genotype. Among-study heterogeneity may be explained by overly broad asthma phenotype definitions, gene-environment interactions, and gene-gene interactions.
Conclusions
A protective dose-response relationship between the CD14 -260T allele and atopic asthma susceptibility was observed. These results demonstrate the importance of precisely specified case-control groups as well as the need to assess interactions in the investigation of complex diseases such as asthma.
Similar content being viewed by others

Background
Asthma is a common, complex, chronic medical condition characterized by lung inflammation, reversible airflow obstruction, and enhanced airway responsiveness to a variety of environmental stimuli. Epidemiological evidence suggests increased asthma prevalence in recent decades with reduced international differences in asthma prevalence [1]. The most common asthma phenotype is atopic asthma, accounting for 56% of asthma cases in the United States [2]. Atopic asthma is an immunoglobulin E mediated hypersensitivity reaction triggered by environmental allergens, such as endotoxin and aero-allergens [3]. Although environmental factors are important determinants of asthma, numerous studies have revealed that asthma has a strong genetic component. Susceptibility genes have been identified from linkage, candidate gene association, and genome-wide association studies. As of 2010, over 250 different genes have been associated with asthma, including cluster of differentiation 14 (CD14) [4, 5].
A well studied common single nucleotide polymorphism (SNP) in the promoter region of CD14, -260C>T (rs2569190; also reported as CD14 -159), is the focus of this review. CD14 encodes a receptor protein that binds to lipopolysaccharide (LPS), its primary ligand, and interacts with co-receptors toll-like receptor 4 (TLR4) and lymphocyte antigen 96 (LY96). CD14 is expressed on the surface of monocytes, macrophages, and neutrophils as membrane CD14 and in the serum as soluble CD14 and its expression may be partially regulated at the genetic level [6]. LPS, a principle component of endotoxin, induces lung inflammation and originates from the outer membrane of Gram-negative bacteria. Ligand binding activates innate immune system pathways that may trigger atopic asthma [7]. Atopic asthmatic subjects are more sensitive to respirable endotoxin than non-asthmatic subjects [8] and also show increased expression of CD14 after acute allergen provocation [9] and LPS inhalation [10].
Two earlier meta-analyses found an overall null association between the CD14 -260C>T polymorphism and asthma, where no association was reported in some studies and the risk variant identified as either the T or C allele in others [11, 12]. Unfortunately, these meta-analyses lacked adequate reporting of methodology and included studies examining non-asthma phenotypes. A more recent meta-analysis found a significant decreased atopic asthma risk for the TT and CT genotypes compared with the CC genotype when analysis was restricted to studies of Asian populations and children [13]. However, that review had several significant errors regarding study inclusion, data abstraction, and analyses.
Due to the inconsistency of past meta-analyses, an updated review was conducted to estimate the meta-odds of developing asthma given the -260C>T genotype in CD14. Subgroup analyses were planned in order to explore potential sources of among-study heterogeneity by examining the effect of selected study characteristics on the combined effect estimate. Methodological issues in the literature studying this association are discussed.
Methods
Identification of eligible studies
Complete details of study methods are in Additional file 1. The review process followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines [14]. A PubMed, EMBASE, and Scopus search was conducted on April 29, 2011 using a sensitive strategy to identify relevant articles. The HuGE Literature Finder database was consulted for its listing of articles under the asthma phenotype and CD14. An article in press at time of search was added to the review [4]. Reference lists of articles retained for review and past meta-analyses were inspected for relevant publications. No publication date or language restrictions were imposed.
Article titles and abstracts of studies identified from the searches were screened and excluded from further analysis for the following reasons: ineligible phenotype, ineligible SNP, review article, basic science research, or animal research. The full-text of studies passing initial screening was reviewed and excluded based on the aforementioned and following criteria: not case-control or nested case-control study design, unreported genotype frequencies, or subjects included in another study. Studies must have an asthma outcome definition that followed accepted diagnostic guidelines, was physician diagnosed, or used a combination of questionnaire and clinical ascertainment.
For multiple publications based on related data sets, the study with the greatest number of subjects was included. Reviewers extracted study information independently and disagreements were resolved by discussion and consensus.
Statistical analysis
The general approach to meta-analysis has been described previously [15, 16]. The pooled frequency of the putative risk allele (-260T) was estimated in various ethnic groups using the inverse variance method. Heterogeneity of studies was assessed using the I 2 statistic [17] separately for the genotype-specific odds ratios (ORs) across studies: TT versus CC (OR1), CT versus CC (OR2), and TT versus CT (OR3). If no or low heterogeneity existed (I 2 < 25%), the inverse variance method was used to estimate the pooled OR and 95% confidence interval (CI), assuming a fixed effects model. Otherwise, a random effects model was used. Comparisons of OR1, OR2, and OR3 indicated the most appropriate genetic model for the -260T allele [16].
Subgroup analyses were planned when sufficient information was reported in at least four studies in each subgroup. The effect of having more homogeneous case and control phenotype definitions (atopic asthma versus non-atopic non-asthma), ethnicity, age, publication year, or study size on the association was examined to identify potential sources of heterogeneity. Post-hoc sensitivity analysis using the sequential algorithm [18] with an I 2 threshold of 25% was conducted in the presence of significant among-study heterogeneity to evaluate studies responsible for the heterogeneity. Influence analysis was conducted to allow identification of studies excessively perturbing the summary estimate. Publication bias was assessed visually using a funnel plot of the standard error of the logarithm of the effect estimate against the effect estimate of each study.
Review Manager Version 5.1.1 (Nordic Cochrane Centre, Cochrane Collaboration, Copenhagen, Sweden) was used to conduct the meta-analysis, sequential analysis, and publication bias assessment. MetaAnalyst Version Beta 3.13 (Tufts Medical Center, Boston, MA) was used to estimate the pooled -260T allele frequency and conduct the influence analysis.
Results
Study inclusion and characteristics
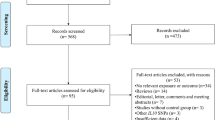
The literature search identified 204 potentially relevant articles. Initial screening of titles and abstracts excluded 159 studies which did not meet the eligibility criteria. The full-text of the remaining 45 studies was retrieved for review: 22 additional studies were excluded. Unpublished CD14 -260C>T SNP data was provided by the corresponding author for one study [4]. Multiple publications were discovered for two data sets [19–23]. The studies with the largest number of subjects were retained [20, 23]. Since Chan et al. [20] did not include genotype frequency data on atopic asthma cases and corresponding controls, this information was abstracted from the related paper with shared subjects by Leung et al. [21]. In total, this review yielded 23 studies [4, 11, 20, 23–42] for meta-analysis. Two studies were published in Chinese [25, 26] and one in Polish [32]. The search results revealed that it was necessary to search more than one database in order to capture all relevant studies. Figure 1 provides a summary of the search results.

Flow diagram of the systematic review and meta-analysis literature search results. HuGE is the Human Genome Epidemiology Network.
All studies retained for review used either a case-control or nested case-control design. Of the 23 studies, 15 included mixed asthma cases [11, 20, 23, 25, 29, 30, 33–40, 42], of which five separated asthma cases by atopic status [11, 20, 36, 37, 42], and eight included only atopic asthma cases [4, 24, 26–28, 31, 32, 41]. Thirteen studies investigated European populations [4, 11, 24, 27–36], eight investigated East Asian populations [20, 23, 25, 26, 37–40], and two investigated other populations [41, 42]. Appropriate diagnostic criteria and proper genotyping methods were used in all studies. Eight studies applied some form of genotyping quality control and only two reported that genotyping was blinded to case-control status. Deviation from Hardy-Weinberg equilibrium (HWE) was detected in the controls of three studies [27, 33, 42]. Genotype frequencies for the studies by Bjornvold et al. [24] and Hakonarson et al. [28] could not be ascertained and were estimated based on reported allele frequencies, assuming HWE. All studies used unique samples: a total of 4780 genotyped asthma cases and 5650 genotyped non-asthmatic controls were included in the meta-analysis. Study characteristics and genotype frequencies are summarized in Table 1 (see Table S1, Additional file 2, for a complete summary of abstracted study characteristics).
Pooled CD14 -260Tallele frequency in controls
Pooled CD14 -260T allele frequencies, using the inverse variance fixed effects model, were 0.457 (95% CI: 0.445-0.469) for overall European populations and 0.462 (95% CI: 0.449-0.475) for European populations excluding those not in HWE [27, 33]. The pooled frequency was 0.577 (95% CI: 0.562-0.592) for East Asian populations. The -260T allele frequency was 0.621 (95% CI: 0.576-0.665) in an Indian population.
Association between CD14 -260C>Tand asthma risk
The pooled ORs for each pair-wise genotype comparison and corresponding I 2 statistics are summarized in Table 2. For all studies, heterogeneity ranged from moderate to high for the non-significant genotype-specific ORs, suggesting no association between the polymorphism and asthma risk. Subgroup analyses (data not shown) did not show significant gene effects when studies were subset by ethnicity (European or East Asian), age range of cases and controls (adults or children), year of study publication (2006-2010 or 2001-2005), and genotyped study sample size (≥ 100 cases and ≥ 100 controls or < 100 cases or < 100 controls). Low to moderate among-study heterogeneity was present in all subgroups for OR2 and moderate to high heterogeneity for OR1 and OR3. Sensitivity analysis excluding studies that appeared to account for appreciable heterogeneity and influence did not meaningfully change the results for overall and subgroup meta-analyses (data not shown). Relatively symmetrical funnel plots indicated the absence of publication bias for the genotype-specific ORs (see Figures S1-S3, Additional files 3, 4 and 5).
Subgroup analysis by case-control phenotype definitions
Initial subgroup analysis of studies that had defined case-control phenotypes as atopic asthma and non-atopic non-asthma showed non-significant gene effects plus high among-study heterogeneity (Table 2). Further subgrouping of studies comparing atopic asthmatics and non-atopic non-asthmatics by ethnicity (European only), age (children only), year of study publication, and genotyped study sample size did not meaningfully change the results. Post-hoc sensitivity analysis identified three studies that may be responsible for the significant among-study heterogeneity: Kedda et al. [11], de Faria et al. [27], and Lachheb et al. [42]. While reported study characteristics for these three studies were not atypical compared to other studies, the controls for the studies by de Faria et al. and Lachheb et al. deviated significantly from HWE (p < 0.01). Influence analysis found moderate influence on the combined effects exerted by these three studies.
The genotype-specific ORs for the subgroup of studies with atopic asthma versus non-atopic non-asthma case-control groups, excluding the three studies identified by the post-hoc sequential analysis, implied a codominant model (Table 2). Compared to subjects with the CC genotype, the pooled ORs suggested that subjects with the TT genotype were some 33% less likely to have atopic asthma (OR1 = 0.67, 95% CI: 0.54-0.84, I 2 = 10%) (Figure 2) and subjects with the CT genotype were about 20% less likely to have atopic asthma (OR2 = 0.80, 95% CI: 0.66-0.95, I 2 = 0%) (Figure 3), showing a dose-response relationship for the T allele. No substantial heterogeneity was detected and publication bias was not evident in the funnel plots (see Figures S4 and S5, Additional files 6 and 7). Exclusion of any one particular study in the influence analysis did not meaningfully change the results (data not shown).

Forest plot of CD14 -260 ( -159 ) TT versus CC genotypes for studies with precisely defined phenotypes. The forest plot displays the meta-analysis results of studies included in the review that used atopic asthma versus non-atopic non-asthma case-control phenotypes, excluding heterogeneous studies identified by sequential analysis [11, 27, 42]. Meta-analysis was conducted using an inverse variance (IV), fixed effects model. For each study in the forest plot, the area of the black square is proportional to study weight and the horizontal bar represents the 95% confidence interval (CI). Atopic asthma and non-atopic non-asthma are abbreviated as AA and NANA, respectively.

Forest plot of CD14 -260 ( -159 ) CT versus CC genotypes for studies with precisely defined phenotypes. The forest plot displays the meta-analysis results of studies included in the review that used atopic asthma versus non-atopic non-asthma case-control phenotypes, excluding heterogeneous studies identified by sequential analysis [11, 27, 42]. Meta-analysis was conducted using an inverse variance (IV), fixed effects model. For each study in the forest plot, the area of the black square is proportional to study weight and the horizontal bar represents the 95% confidence interval (CI). Atopic asthma and non-atopic non-asthma are abbreviated as AA and NANA, respectively.
Discussion
The present meta-analysis found a non-significant association between the CD14 -260C>T polymorphism and overall asthma. There was also high among-study heterogeneity in the meta-analysis, possibly accounting for the inconsistently reported findings between this SNP and asthma [43]. Subgroup analysis of selected study characteristics did not reveal any significant associations or substantial decreases in the I 2 estimate of heterogeneity. When restricting analysis to studies that used atopic asthma versus non-atopic non-asthma case-control phenotypes and excluding studies influencing heterogeneity, the genotype-specific ORs suggested a codominant model.
A sequential analysis revealed three studies that appeared to account for the high among-study heterogeneity (see Additional file 1 for methodology). Two had controls that departed from HWE, which may represent possible sources of bias. The exploratory nature of post-hoc sequential analysis may present a weakness, but advantages include its objective approach and the fact that specific study characteristics that may contribute to heterogeneity are not always known or recorded. The latter is important: if various methodological nuances are not reported, subsequent meta-analysis would not account for these factors and the ability to assess sources of heterogeneity would be hampered. For example, reported study characteristics in the article by Kedda et al. [11], one of the studies identified to incur a large amount of heterogeneity, did not reveal any particular characteristic that deviated from other studies.
Stronger associations and significant relationships were found when analysis was restricted to studies with more homogeneously defined case-control phenotypes and with heterogeneous studies excluded. These results indicated that the -260T allele was significantly protective under the codominant model when comparing atopic asthmatics to non-atopic non-asthmatics. Observed among-study heterogeneity may be partially explained by the employment of overly broad case-control phenotype definitions. It has been suggested in genome-wide association studies that use of homogeneous case phenotypes and precisely specified control groups—those who unambiguously do not have the case phenotype—may improve study efficiency [44]. This principle, borrowed from extreme discordant sib-pair analysis [45], naturally extends to case-control selection in candidate gene association studies.
There is possible gene-environment interaction, in which the SNP acts as a modifier of asthma risk in individuals with different degrees of environmental endotoxin exposure. Carriers of the TT genotype have been found to have higher serum levels of CD14 than carriers of the CT or CC genotypes [43]. This epidemiologic evidence is supported by functional genomic studies that showed increased transcriptional activity of the -260T allele in a monocytic cell line [46]. An antagonistic interaction has been demonstrated between CD14 and endotoxin exposure: homozygotes for the T allele appear to be protective for asthma at low levels of endotoxin exposure, but may increase asthma risk at high levels of endotoxin exposure [43]. Based on these findings, Martinez [43] hypothesized that higher CD14 expression in TT homozygotes increased sensitivity to the protective effects of low level endotoxin exposure compared to carriers of other genotypes. However, at higher levels of endotoxin exposure, induced CD14 expression could be increased in carriers of the C allele, showing a reversed protective effect. The findings of the present meta-analysis, restricted to studies using the atopic asthma versus non-atopic non-asthma case-control phenotypes, are consistent with this hypothesis at low endotoxin exposure levels. The codominant model for the -260T allele implied a dose-response relationship in CD14 expression and reduction of atopic asthma risk. This gene-environment interaction may be a source of heterogeneity among studies in the present and earlier meta-analyses [11–13].
In addition to the promoter, many additional regulatory elements are necessary to influence gene expression, particularly for genes like CD14, which exhibit highly complex expression patterns. Regulatory elements, such as enhancers and repressors, may reside in intronic regions or up- and down-stream of the transcriptional unit [47]. A risk variant with no obvious and no known function may regulate a gene at a considerable genomic distance from the location of the SNP. Therefore, it is important to study the influence of gene-gene interaction as well as other polymorphisms in CD14 on the effects of this locus on asthma susceptibility.
Quality and methodology of studies
Assessing study quality was difficult due to inadequate reporting from all studies included in the meta-analysis. Many studies reported insufficient information about recruitment methodology and study participant characteristics, particularly for controls. Genotype distributions of controls departed from HWE in three studies [27, 33, 42]. Deviation from HWE in controls, or healthy populations, may indicate selection bias, population stratification, or genotyping errors [48]. Even in the absence of deviation from HWE, these biases could not be assessed given the inadequately reported information. Eight studies reported implementing some form of genotyping quality control [4, 20, 28, 30, 37, 38, 40, 41]. Only two published studies mentioned blinding of phenotype when genotyping [29, 38]. Furthermore, there is a potential for publication bias, where positive rather than negative findings tend to be published [49]. The completeness of evidence is also impeded by language bias. Studies conducted in non-English speaking countries tend to publish significant results in international journals and non-significant results in local journals, many of which are not indexed [50]. Selective publication of polymorphism and disease associations may obscure their true relationships.
Results from the pooled CD14 -260T allele frequency in controls revealed differences among the broad ethnic categories: 0.457 for European populations, 0.577 for East Asian populations, and 0.621 for an Indian population. In comparison, the International HapMap Project (Phase 3) reported the -260T allele frequency among Utah residents with Northern and Western European ancestry, Han Chinese in Beijing, China, Japanese in Tokyo, Japan, and Yoruba in Ibadan, Nigeria to be 0.474, 0.500, 0.488, and 0.293, respectively. The average heterozygosity reported in Build 132 of dbSNP is 0.488 ± 0.078 [51]. Interethnic differences in the allele frequencies of the CD14 -260C>T polymorphism is of concern as some studies included in this meta-analysis have different ethnic compositions between the cases and controls. Reported associations in studies of varying ethnic composition may have been influenced by population stratification. Even among apparently homogeneous ethnic groups, population stratification may be a problem [52]. The effect of this type of stratification has been reported to be small in most situations, but a small bias may be important in studies of genetic association, which typically consider small or moderate effects [53]. Only four studies included in this review reported an assessment of population stratification.
A commonly cited solution to addressing population stratification is the use of family-based designs to study genetic associations [44, 54]. However, the family-based design has its own inherent limitation to susceptibility variant discovery. It has been argued that neither common nor rare genetic variants are heritable, as they do not give rise to a substantial familial concentration of cases due to low penetrance [55]. Three family-based studies have explored the association of CD14 -260C>T and asthma with conflicting results [12, 41, 42]. Therefore, efforts should be made to accrue controls from the same source population as cases to avoid population stratification, particularly when ethnicity is not matched or controlled [44].
Conclusions
This meta-analysis provides a comprehensive examination of the available evidence concerning the association between the CD14 -260C>T polymorphism and asthma susceptibility. The significant association between this polymorphism and atopic asthma may be of clinical and public health importance. The genetics of asthma follow the "common disease, common variants" hypothesis, which posits that multiple genetic variants of interest are common to many individuals with the disease. These common variants typically have weak individual effects and low penetrance, but their high frequency confers a relatively large attributable risk in the population. Therefore, this common polymorphism, along with endotoxin exposure level information, has potential to be a useful and efficient predictor of atopic asthma risk. This review also emphasizes the importance of having precisely defined case-control groups to study complex diseases and demonstrates the need to incorporate gene-environment and gene-gene interaction analyses in future epidemiological investigations of asthma genetics.
References
Pearce N, Ait-Khaled N, Beasley R, Mallol J, Keil U, Mitchell E, Robertson C, the ISAAC Phase Three Study Group: Worldwide trends in the prevalence of asthma symptoms: phase III of the International Study of Asthma and Allergies in Childhood (ISAAC). Thorax. 2007, 62 (9): 758-766. 10.1136/thx.2006.070169.
Arbes SJ, Gergen PJ, Vaughn B, Zeldin DC: Asthma cases attributable to atopy: results from the Third National Health and Nutrition Examination Survey. J Allergy Clin Immunol. 2007, 120 (5): 1139-1145. 10.1016/j.jaci.2007.07.056.
Husain AN: The lung. Robbins and Cotran pathologic basis of disease. Edited by: Kumar V, Abbas AK, Fausto N, Aster JC. 2010, Philadelphia, PA: Saunders, 677-737. 8
Murk W, Walsh K, Hsu L-I, Zhao L, Bracken MB, DeWan AT: Attempted replication of 50 reported asthma risk genes identifies SNPs in RAD50 and PTPRE as associated with childhood atopic asthma. Hum Hered.
Ober C, Hoffjan S: Asthma genetics 2006: the long and winding road to gene discovery. Genes Immun. 2006, 7 (2): 95-100. 10.1038/sj.gene.6364284.
Ulevitch RJ, Tobias PS: Receptor-Dependent Mechanisms of Cell Stimulation by Bacterial Endotoxin. Annu Rev Immunol. 1995, 13: 437-457. 10.1146/annurev.iy.13.040195.002253.
Dobrovolskaia MA, Vogel SN: Toll receptors, CD14, and macrophage activation and deactivation by LPS. Microbes Infect. 2002, 4 (9): 903-914. 10.1016/S1286-4579(02)01613-1.
Peden DB, Tucker K, Murphy P, Newlin-Clapp L, Boehlecke B, Hazucha M, Bromberg P, Reed W: Eosinophil influx to the nasal airway after local, low-level LPS challenge in humans. J Allergy Clin Immunol. 1999, 104 (2 Pt 1): 388-394.
Virchow JC, Julius P, Matthys H, Kroegel C, Luttmann W: CD14 expression and soluble CD14 after segmental allergen provocation in atopic asthma. Eur Respir J. 1998, 11 (2): 317-323. 10.1183/09031936.98.11020317.
Alexis N, Eldridge M, Reed W, Bromberg P, Peden DB: CD14-dependent airway neutrophil response to inhaled LPS: role of atopy. J Allergy Clin Immunol. 2001, 107 (1): 31-35. 10.1067/mai.2001.111594.
Kedda MA, Lose F, Duffy D, Bell E, Thompson PJ, Upham J: The CD14 C-159T polymorphism is not associated with asthma or asthma severity in an Australian adult population. Thorax. 2005, 60 (3): 211-214. 10.1136/thx.2004.028449.
Nishimura F, Shibasaki M, Ichikawa K, Arinami T, Noguchi E: Failure to find an association between CD14-159C/T polymorphism and asthma: a family-based association test and meta-analysis. Allergol Int. 2006, 55 (1): 55-58. 10.2332/allergolint.55.55.
Zhang Y, Tian C, Zhang J, Li X, Wan H, He C, Guo L, Meilang Q, Peng C, Duo L, Huang J, Fan H: The -159C/T polymorphism in the CD14 gene and the risk of asthma: a meta-analysis. Immunogenetics. 2011, 63 (1): 23-32. 10.1007/s00251-010-0489-1.
Moher D, Liberati A, Tetzlaff J, Altman DG, the PRISMA Group: Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009, 6 (7): e1000097-10.1371/journal.pmed.1000097.
Zhang HF, Zhong BL, Zhu WL, Xie SL, Qiu LX, Zhu LG, Wang Y, Lei L: CD14 C-260T gene polymorphism and ischemic heart disease susceptibility: a HuGE review and meta-analysis. Genet Med. 2009, 11 (6): 403-408. 10.1097/GIM.0b013e3181a16cb0.
Thakkinstian A, McElduff P, D'Este C, Duffy D, Attia J: A method for meta-analysis of molecular association studies. Stat Med. 2005, 24 (9): 1291-1306. 10.1002/sim.2010.
Higgins JP, Thompson SG, Deeks JJ, Altman DG: Measuring inconsistency in meta-analyses. BMJ. 2003, 327 (7414): 557-560. 10.1136/bmj.327.7414.557.
Patsopoulos NA, Evangelou E, Ioannidis JP: Sensitivity of between-study heterogeneity in meta-analysis: proposed metrics and empirical evaluation. Int J Epidemiol. 2008, 37 (5): 1148-1157. 10.1093/ije/dyn065.
Chan IH, Leung TF, Tang NL, Li CY, Sung YM, Wong GW, Wong CK, Lam CW: Gene-gene interactions for asthma and plasma total IgE concentration in Chinese children. J Allergy Clin Immunol. 2006, 117 (1): 127-133. 10.1016/j.jaci.2005.09.031.
Chan IH, Tang NL, Leung TF, Huang W, Lam YY, Li CY, Wong CK, Wong GW, Lam CW: Study of gene-gene interactions for endophenotypic quantitative traits in Chinese asthmatic children. Allergy. 2008, 63 (8): 1031-1039. 10.1111/j.1398-9995.2008.01639.x.
Leung TF, Tang NL, Sung YM, Li AM, Wong GW, Chan IH, Lam CW: The C-159T polymorphism in the CD14 promoter is associated with serum total IgE concentration in atopic Chinese children. Pediatr Allergy Immunol. 2003, 14 (4): 255-260. 10.1034/j.1399-3038.2003.00048.x.
Tan CY, Chen YL, Wu LS, Liu CF, Chang WT, Wang JY: Association of CD14 promoter polymorphisms and soluble CD14 levels in mite allergen sensitization of children in Taiwan. J Hum Genet. 2006, 51 (1): 59-67. 10.1007/s10038-005-0323-z.
Wang JY, Liou YH, Wu YJ, Hsiao YH, Wu LS: An association study of 13 SNPs from seven candidate genes with pediatric asthma and a preliminary study for genetic testing by multiple variants in Taiwanese population. J Clin Immunol. 2009, 29 (2): 205-209. 10.1007/s10875-008-9256-6.
Bjornvold M, Munthe-Kaas MC, Egeland T, Joner G, Dahl-Jorgensen K, Njolstad PR, Akselsen HE, Gervin K, Carlsen KC, Carlsen KH, Undlien DE: A TLR2 polymorphism is associated with type 1 diabetes and allergic asthma. Genes Immun. 2009, 10 (2): 181-187. 10.1038/gene.2008.100.
Chen M, Wu B, Li W: [Influence of CD14 gene -159C/T polymorphism on IL-5 level in patients with asthma]. Shang Dong Yi Yao. 2009, 49 (5): 13-15.
Cui T, Jiang W, Wu J: [Genetic polymorphism of CD14 and allergic asthma in children]. Central China Medical Journal. 2003, 27 (5): 235-236.
de Faria IC, de Faria EJ, Toro AA, Ribeiro JD, Bertuzzo CS: Association of TGF-beta1, CD14, IL-4, IL-4R and ADAM33 gene polymorphisms with asthma severity in children and adolescents. J Pediatr (Rio J). 2008, 84 (3): 203-210.
Hakonarson H, Bjornsdottir US, Ostermann E, Arnason T, Adalsteinsdottir AE, Halapi E, Shkolny D, Kristjansson K, Gudnadottir SA, Frigge ML, Gislason D, Gislason T, Kong A, Gulcher J, Stefansson K: Allelic frequencies and patterns of single-nucleotide polymorphisms in candidate genes for asthma and atopy in Iceland. Am J Respir Crit Care Med. 2001, 164 (11): 2036-2044.
Heinzmann A, Dietrich H, Jerkic SP, Kurz T, Deichmann KA: Promoter polymorphisms of the CD14 gene are not associated with bronchial asthma in Caucasian children. Eur J Immunogenet. 2003, 30 (5): 345-348. 10.1046/j.1365-2370.2003.00414.x.
Koppelman GH, Reijmerink NE, Colin Stine O, Howard TD, Whittaker PA, Meyers DA, Postma DS, Bleecker ER: Association of a promoter polymorphism of the CD14 gene and atopy. Am J Respir Crit Care Med. 2001, 163 (4): 965-969.
Kowal K, Bodzenta-Lukaszyk A, Pampuch A, Szmitkowski M, Zukowski S, Donati MB, Iacoviello L: Analysis of -675 4 g/5 G SERPINE1 and C-159T CD14 polymorphisms in house dust mite-allergic asthma patients. J Investig Allergol Clin Immunol. 2008, 18 (4): 284-292.
Lis G, Kostyk E, Sanak M, Pietrzyk JJ: [Molecular studies in a population of children with bronchial asthma. I. Polymorphism in the promoter region of gene CD14]. Pneumonol Alergol Pol. 2001, 69 (5-6): 265-272.
Sengler C, Haider A, Sommerfeld C, Lau S, Baldini M, Martinez F, Wahn U, Nickel R, German Multicenter Allergy Study Group: Evaluation of the CD14 C-159 T polymorphism in the German Multicenter Allergy Study cohort. Clin Exp Allergy. 2003, 33 (2): 166-169. 10.1046/j.1365-2222.2003.01549.x.
Smit LA, Bongers SI, Ruven HJ, Rijkers GT, Wouters IM, Heederik D, Omland O, Sigsgaard T: Atopy and new-onset asthma in young Danish farmers and CD14, TLR2, and TLR4 genetic polymorphisms: a nested case-control study. Clin Exp Allergy. 2007, 37 (11): 1602-1608. 10.1111/j.1365-2222.2007.02831.x.
Smit LA, Siroux V, Bouzigon E, Oryszczyn MP, Lathrop M, Demenais F, Kauffmann F, Epidemiological Study on the Genetics and Environment of Asthma, Bronchial Hyperresponsiveness, and Atopy (EGEA) Cooperative Group: CD14 and toll-like receptor gene polymorphisms, country living, and asthma in adults. Am J Respir Crit Care Med. 2009, 179 (5): 363-368. 10.1164/rccm.200810-1533OC.
Woo JG, Assa'ad A, Heizer AB, Bernstein JA, Hershey GK: The -159 C- > T polymorphism of CD14 is associated with nonatopic asthma and food allergy. J Allergy Clin Immunol. 2003, 112 (2): 438-444. 10.1067/mai.2003.1634.
Hong SJ, Kim HB, Kang MJ, Lee SY, Kim JH, Kim BS, Jang SO, Shin HD, Park CS: TNF-alpha (-308 G/A) and CD14 (-159T/C) polymorphisms in the bronchial responsiveness of Korean children with asthma. J Allergy Clin Immunol. 2007, 119 (2): 398-404. 10.1016/j.jaci.2006.10.031.
Kuo Chou TN, Li YS, Lue KH, Liao CF, Lin CY, Tzeng PR, Wong RH: Genetic polymorphism of manganese superoxide dismutase is associated with childhood asthma. J Asthma. 2010, 47 (5): 532-538. 10.3109/02770901003686472.
Park S-O, Jo M-Y, Jung S-S, Ahn G-S: No association of polymorphisms in RANTES and CD14 genes with asthma in the Korean population. Kor J Genet. 2006, 28 (1): 75-81.
Wu X, Li Y, Chen Q, Chen F, Cai P, Wang L, Hu L: Association and gene-gene interactions of eight common single-nucleotide polymorphisms with pediatric asthma in middle China. J Asthma. 2010, 47 (3): 238-244. 10.3109/02770900903509099.
Sharma M, Batra J, Mabalirajan U, Goswami S, Ganguly D, Lahkar B, Bhatia NK, Kumar A, Ghosh B: Suggestive evidence of association of C-159T functional polymorphism of the CD14 gene with atopic asthma in northern and northwestern Indian populations. Immunogenetics. 2004, 56 (7): 544-547. 10.1007/s00251-004-0721-y.
Lachheb J, Dhifallah IB, Chelbi H, Hamzaoui K, Hamzaoui A: Toll-like receptors and CD14 genes polymorphisms and susceptibility to asthma in Tunisian children. Tissue Antigens. 2008, 71 (5): 417-425. 10.1111/j.1399-0039.2008.01011.x.
Martinez FD: CD14, endotoxin, and asthma risk: actions and interactions. Proc Am Thorac Soc. 2007, 4 (3): 221-225. 10.1513/pats.200702-035AW.
Bracken MB, DeWan A, Hoh J: Genomewide association studies. Molecular epidemiology: applications in cancer and other human diseases. Edited by: Rebbeck TR, Ambrosone CB, Shields PG. 2008, New York, NY: Informa Healthcare, 225-238.
Risch N, Zhang H: Extreme discordant sib pairs for mapping quantitative trait loci in humans. Science. 1995, 268 (5217): 1584-1589. 10.1126/science.7777857.
LeVan TD, Bloom JW, Bailey TJ, Karp CL, Halonen M, Martinez FD, Vercelli D: A common single nucleotide polymorphism in the CD14 promoter decreases the affinity of Sp protein binding and enhances transcriptional activity. J Immunol. 2001, 167 (10): 5838-5844.
Kleinjan DA, van Heyningen V: Long-range control of gene expression: emerging mechanisms and disruption in disease. Am J Hum Genet. 2005, 76 (1): 8-32. 10.1086/426833.
Little J, Higgins JPT, Eds: The HuGENet HuGE Review Handbook, version 1.0. [http://www.medicine.uottawa.ca/public-health-genomics/web/eng/index.html]
Rosenthal R: The "file drawer problem" and tolerance for null results. Psychol Bull. 1979, 86 (3): 638-641.
Juni P, Holenstein F, Sterne J, Bartlett C, Egger M: Direction and impact of language bias in meta-analyses of controlled trials: empirical study. Int J Epidemiol. 2002, 31 (1): 115-123. 10.1093/ije/31.1.115.
Database of Single Nucleotide Polymorphisms (dbSNP): Reference SNP (refSNP) Cluster Report: rs2569190 [build 132]. [http://www.ncbi.nlm.nih.gov/SNP/]
Thomas DC, Witte JS: Point: population stratification: a problem for case-control studies of candidate-gene associations?. Cancer Epidemiol Biomarkers Prev. 2002, 11 (6): 505-512.
Little J, Higgins JP, Ioannidis JP, Moher D, Gagnon F, von Elm E, Khoury MJ, Cohen B, Davey-Smith G, Grimshaw J, Scheet P, Gwinn M, Williamson RE, Zou GY, Hutchings K, Johnson CY, Tait V, Wiens M, Golding J, van Duijn C, McLaughlin J, Paterson A, Wells G, Fortier I, Freedman M, Zecevic M, King R, Infante-Rivard C, Stewart A, Birkett N: Strengthening the reporting of genetic association studies (STREGA): an extension of the STROBE Statement. Hum Genet. 2009, 125 (2): 131-151. 10.1007/s00439-008-0592-7.
Ziegler A, Konig IR, Thompson JR: Biostatistical aspects of genome-wide association studies. Biom J. 2008, 50 (1): 8-28. 10.1002/bimj.200710398.
Bodmer W, Bonilla C: Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet. 2008, 40 (6): 695-701. 10.1038/ng.f.136.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/12/93/prepub
Acknowledgements
The authors would like to thank Andrew T. DeWan for providing unpublished SNP data from the Perinatal Risk of Asthma in Infants of Asthmatic Mothers (PRAM) study. This work was supported by grants AI41040 and DA05484 from the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
Both authors conceived and designed the study, performed the statistical analysis and interpretation, drafted the manuscript, revised for important intellectual content, and read and approved the final manuscript.
Electronic supplementary material
12881_2011_825_MOESM2_ESM.DOC
Additional file 2: Table S1. Summary of abstracted characteristics of reviewed studies on CD14 -260(-159) C>Tand asthma. Complete summary of abstracted characteristics from studies included in the systematic review and meta-analysis. (DOC 80 KB)
12881_2011_825_MOESM3_ESM.PDF
Additional file 3: Figure S1. Funnel plot of CD14 -260(-159) TT versus CC genotypes for all reviewed studies. Standard error of the logarithm of the odds ratio (SE(log[OR])) was plotted against the OR of each study. (PDF 11 KB)
12881_2011_825_MOESM4_ESM.PDF
Additional file 4: Figure S2. Funnel plot of CD14 -260(-159) CT versus CC genotypes for all reviewed studies. Standard error of the logarithm of the odds ratio (SE(log[OR])) was plotted against the OR of each study. (PDF 10 KB)
12881_2011_825_MOESM5_ESM.PDF
Additional file 5: Figure S3. Funnel plot of CD14 -260(-159) TT versus CT genotypes for all reviewed studies. Standard error of the logarithm of the odds ratio (SE(log[OR])) was plotted against the OR of each study. (PDF 10 KB)
12881_2011_825_MOESM6_ESM.PDF
Additional file 6: Figure S4. Funnel plot of CD14 -260 ( -159 ) TT versus CC genotypes for studies with precisely defined phenotypes. The funnel plot displays studies included in the review that used atopic asthma cases and non-atopic non-asthmatic controls, excluding heterogeneous studies identified by sequential analysis [11, 27, 42]. Standard error of the logarithm of the odds ratio (SE(log[OR])) was plotted against the OR of each study. (PDF 11 KB)
12881_2011_825_MOESM7_ESM.PDF
Additional file 7: Figure S5. Funnel plot of CD14 -260 ( -159 ) CT versus CC genotypes for studies with precisely defined phenotypes. The funnel plot displays studies included in the review that used atopic asthma cases and non-atopic non-asthmatic controls, excluding heterogeneous studies identified by sequential analysis [11, 27, 42]. Standard error of the logarithm of the odds ratio (SE(log[OR])) was plotted against the OR of each study. (PDF 10 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zhao, L., Bracken, M.B. Association of CD14 -260 (-159) C>Tand asthma: a systematic review and meta-analysis. BMC Med Genet 12, 93 (2011). https://doi.org/10.1186/1471-2350-12-93
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-12-93