
Abstract
Background
To examine the effect of genetic variation in APOE, IDE and IL1B on the response to induced ketosis in the Alzheimer's Disease Assessment Scale-Cognitive subscale (ADAS-Cog) in subjects with mild to moderate Alzheimer's disease (AD).
Methods
Genotype effects on ADAS-Cog scores from a randomized, double-blind, placebo-controlled study in mild to moderate AD were examined by an overall two way analysis of variance. In addition, interactions with the carriage status of the epsilon 4 allele of the APOE gene (APOE4) were examined.
Results
Significant differences in response to induced ketosis were found among non-carriers of putative gain-of-function polymorphisms in rs1143627 and rs16944 in the IL1B gene and among variants of the polymorphism rs2251101 in the IDE gene. Significant differences were found among non-carriers of the APOE4 gene, with notable improvement among the E3/E3 genotype group.
Conclusions
Variants in APOE, IL1B and IDE may influence the cognitive response to induced ketosis in patients with mild to moderate AD.
Trial registration
This trial was registered with ClinicalTrials.gov, registry number NCT00142805.
Similar content being viewed by others


Background
Alzheimer's disease (AD) is a progressive neurodegenerative disease. The major risk factors for the most common form of AD, known as late onset or sporadic AD, are age and possession of one or more copies of the epsilon 4 variant of the apolipoprotein E gene (APOE4). APOE4 behaves in a dominant dose-dependent manner. One copy of APOE4 increases the risk of developing AD by about 3 fold, while two copies increases the risk approximately 10 fold [1, 2].
Alzheimer's disease is characterized by an early and progressive decrease in the cerebral metabolic rate of glucose (CMRglc) [3–5]. The primary regions affected in AD are the posterior cingulate and the parietal, temporal, and prefrontal cortices. These regions correlate with the highly metabolically active default network, suggesting a metabolic link between hypometabolism, amyloid deposition, and cell atrophy (for review see [6]). The declines in CMRglc in AD could be attributed to loss of cells or synaptic fields, however, decreased rates of glucose phosphorylation [7] and low expression of energy generating genes [8] have been observed in AD, suggesting an underlying metabolic defect in these brain regions [9].
Under normal conditions, the brain is dependent almost exclusively on glucose and few other substrates are metabolized [10]. Therefore, declines in glucose utilization can result in severe impairment. Under conditions of low glucose availability, such as during fasting or low carbohydrate intake, the body will mobilize ketone bodies, from energy-rich fat stores, which can provide an alternative substrate for glucose metabolism in the brain [11]. The endogenously produced compounds β-hydroxybutyrate, acetoacetate and acetone are normally referred to as ketone bodies. Due to their efficient metabolism and ability to substitute for glucose, ketone bodies offer a potential therapeutic benefit for AD [12], as well as other neurological disorders [13].
Previous studies have demonstrated that the induction of ketosis in mild to moderate AD patients improves scores in the Alzheimer's Disease Assessment Scale -Cognitive subscale (ADAS-Cog) relative to placebo among non-carriers of the APOE4 allele using both acute [14] and chronic [15] dosing regimens. Despite the replication of these findings, it is unclear why E4(-) subjects would respond to ketosis while E4(+) subjects would not. Some evidence suggests that E4(+) subjects may have greater mitochondrial dysfunction relative to E4(-) subjects (for review see [16]) and therefore may not metabolize ketone bodies as well. Alternatively, differential insulin signaling seen in AD patients based on E4 carriage status may affect transport and metabolism of ketone bodies.
To gain further insight into this phenomenon, and to provide direction for future research, other genetic markers were tested for their ability to modulate performance on the ADAS-Cog test during induced ketosis in mild to moderate AD patients. Here we report the effects of polymorphisms in insulin degrading enzyme (IDE) and interleukin 1-beta (IL1B) on ADAS-Cog scores after 45 and 90 days of a ketogenic therapy followed by a two week washout, day 104. A report detailing the present study population, the overall results, and the APOE4 effects on cognitive outcomes was previously published [15].
Ketosis was induced by the administration of AC-1202, a formulation of medium chain triglycerides (MCTs). MCTs are triglycerides with fatty acid chains of between 5 and 12 carbons The catabolism of MCTs differs substantially from the more common long chain triglycerides (LCTs). MCTs are immune to the regulation of LCT catabolism and are well known for their ability to induce ketosis after oral administration (for review see [17]).
Insulin degrading enzyme is a zinc binding metalloprotease encoded by the IDE gene located on chromosome 10. The Ide protein degrades a variety of short polypeptides including insulin and amyloid beta [18]. Polymorphisms in the region of the IDE gene have been implicated as risk factors in AD (for review see [2]). Neuroinflammation has also been considered a feature of AD and may influence risk and rate of progression of the disease [19]. Interleukin 1beta is a proinflammatory cytokine encoded by the IL1B gene located on chromosome 2. Interleukin levels are normally low in the CNS but are elevated after acute injury and in chronic neurodegenerative diseases such as AD [20]. In addition, for each marker, the interaction with E4 carriage status was examined.
Methods
The analysis presented here is an extension of previously reported results of a study examining the induction of mild ketosis in patients with mild to moderate AD. As reported in the earlier study [15], an oral ketogenic compound, AC-1202, was tested in patients with probable mild to moderate AD to examine if ketosis could improve cognitive performance. AC-1202 was administered daily for 90 days in 152 subjects in a US-based, randomized, double-blind, placebo-controlled, parallel-group study. Subjects were not asked to change their diets and continued taking approved AD medications. The results of the 90-day study found a significant difference between AC-1202 and Placebo in mean change from Baseline in ADAS-Cog score on Day 45. Based on previous acute dosing study [14], results of cognitive tests were stratified by APOE4 carriage status. Significant differences were reported between AC-1202 and Placebo in mean change from Baseline in ADAS-Cog score on both Day 45 and Day 90 among participants who were non-carriers of the APOE4 allele. Supporting the improvement relative to placebo by administration of AC-1202, a significant pharmacologic response was observed between serum β-hydroxybutyrate levels and change in ADAS-Cog scores in the non-carriers of APOE4. Detailed description of this study and its outcomes has been previously published [15].
Ethics
The trial was carried-out in accordance with the principles of the Declaration of Helsinki and with institutional review board approval (Essex Institutional Review Board, Lebanon, NJ). Subjects and their caregivers provided written informed consent, which included an optional written provision for genotyping. For genetic consent, participants could consent to be tested for APOE genotype only, for any additional DNA markers only, for both, or for neither. Genetic consent was not required for entry into the study. Genetic information was not shared with physicians, site personnel, or study participants.
Registration
This trial was registered with ClinicalTrials.gov, registry number NCT00142805, information available at http://clinicaltrials.gov/ct2/show/NCT00142805.
Study design
This was a randomized, double-blind, placebo-controlled, parallel, multi-center trial sponsored by Accera, Inc. of Broomfield, CO. It was conducted between October 5, 2004 and June 29, 2006 at 23 treatment centers located within the United States. The study recruited outpatients with a diagnosis of probable AD of mild to moderate severity according to National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association (NINCDS-ADRDA) and DSM IV criteria, with a MMSE score of between 14 and 24 (inclusive) at Screen. A CT or MRI within 24 months prior to Screen had to show no signs of tumor, structural abnormality, or degenerative disease. Subjects were required to have a Modified Hachinski Ischemia Scale score ≤4. Participants were randomized to receive either daily doses of AC-1202 or matching Placebo for 90 days. To acclimate participants to investigational product, subjects received a one-half dose daily during the first week of the study. After this one week titration, participants were instructed to take a full dose. A full dose of active contained 20 grams of MCTs. The effects of the intervention on cognitive performance were measured at 45 and 90 days post-Baseline and after a two week washout on Day 104. Primary cognitive outcome was change from Baseline in ADAS-Cog scores compared to Placebo. Participants in the study were allowed to remain on currently prescribed AD medications, provided they were on stable dosing for at least three months prior to enrollment and did not change dosing during the course of the study. Most participants (> 75%) were on one or more currently approved AD medications. Placebo and AC-1202 groups were well matched for demographic parameters. For detailed description of study and participants see Henderson et. al. [15].
Subject disposition
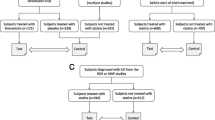
Two hundred fifty-three subjects were screened at 23 clinical sites located within the United States. One hundred one of these participants did not meet inclusion/exclusion criteria or refused to participate in the study. One hundred fifty-two participants were enrolled in the study. At their discretion, participants could consent to APOE genotyping only, additional genetic markers only, or both. One hundred thirty-five of the enrolled subjects consented to genotyping for the APOE locus. One hundred thirty-one subjects consented to both APOE genotyping and additional markers. Of these 131 subjects, 11 lacked a post-Baseline visit and no ADAS-Cog data was available for analysis (Figure 1).

Subject Disposition.
Study material
AC-1202 (caprylic triglyceride, NeoBee 895, Stepan Chemical Company) is a structured medium chain triglyceride (MCT) in which greater than 95% of the fatty acids are caprylic acid (C8:0). NeoBee 895 is an approved food additive. Consumption of MCTs may give rise to gastrointestinal distress such as cramping, nausea and diarrhea. To improve tolerability, caprylic triglyceride was formulated as an emulsified spray dried powder consisting of 33% AC-1202 (NeoBee 895), 64% gum Acacia (Instagum, CNI) and 2.6% syloid (244FP, Grace Davison). Placebo was formulated to be isocaloric to the active formulation and consisted of a mixture of 51% gum acacia, 37% dextrose, 10% safflower oil and 2% syloid (prepared by The Chemins Company). Investigational product was given as a powder packaged in 30 gram sachets containing either active (equivalent to 10 grams of AC-1202) or matching Placebo. A full dose consisted of two sachets (equivalent to 20 grams of AC-1202). Product was reconstituted in water or other liquids.
β-hydroxybutyrate testing
Blood samples were taken for serum BHB levels prior to dosing and 2 hr post-dosing. Levels of serum β-hydroxybutyrate were determined using the Stanbio Laboratory β-hydroxybutyrate test kit (StanBio Inc). Briefly, in the presence of NAD, ß-hydroxybutyrate is converted to acetoacetate and NADH by the enzyme ß-hydroxybutyrate dehydrogenase. The NADH produced reacts with p-iodonitrotetrazolium in the presence of diaphorase to produce a color that is read at 505 nm to determine the concentration.
Genotyping
High molecular weight DNA was isolated from whole blood using standard techniques. Apolipoprotein E (APOE) epsilon 2, 3 and 4 genotyping was performed using allele specific extension as previously described [21]. For single nucleotide polymorphisms, genotyping was performed by amplification of genomic DNA isolated from whole blood and sequencing samples using standard procedures. Primer pair sequences used for amplification and sequencing were: IDE rs2251101 CAGCACTTTAGGAGGCCAAG/CTGCCCTTACAGGGATGAAA; IL1B rs1143627 and rs16944 CACAAAGAGGCAGAGAGACAGA/GTCTTGCAGGGTTGTGTGAG. In some cases an unambiguous genotype could not be determined and no genotype was assigned.
Cognitive Testing
Subjects were administered the ADAS-Cog test at Baseline and Days 45, 90 and 104. The ADAS-Cog is one of the most widely used scales for anti-dementia drugs in the United States. The ADAS-Cog subscale consists of 11 tasks measuring cognitive abilities in memory, language, orientation, and praxis, with a total score ranging from 0 (no impairment) to 70 (severe impairment). The higher the ADAS-Cog score, the more impaired the subject. Therefore, lowering of the ADAS-Cog score is a measure of cognitive improvement.
Statistical Methods
Analysis was done comparing change from Baseline in ADAS-Cog scores using reported scores between Active and Placebo treated groups on Days 45, 90 and 104. Only actual scores for each time point were used in this analysis. No data was imputed. An overall two way analysis of variance was used to evaluate cognitive scores by genotype and treatment interactions at Days 45, 90 and 104.
Results and Discussion
Single Nucleotide Polymorphisms (SNPs) effects on ADAS-Cog
Single nucleotide polymorphisms (SNPs) rs1143627 and rs16944 in the promoter region of IL1B and rs2251101 in the 3' untranslated region of IDE were analyzed for their effects on ADAS-Cog scores after administration of AC-1202 or Placebo. The genotypic frequencies of the SNPs in the study population are listed in Table 1. Cognitive outcomes associated with common APOE genotypes were also examined. Mean change from Baseline in ADAS-Cog scores are shown in Table 2.
For SNP rs2251101 near the IDE gene, heterozygous C/T subjects administered AC-1202 demonstrated significant improvement relative to Placebo at Days 45 (Δ3.96; CI 1.27- 6.66; p = 0.004) and Day 90 (Δ5.71; CI 1.88-9.54; p = 0.004). When participants were categorized as either IDE rs2251101 C/C(-) or IDE rs2251101 C/C(+) based on their carriage status of the C/C genotype, significant improvement was found in IDE rs2251101 C/C(-) subjects administered AC-1202 relative to Placebo at Days 45 (Δ2.85; CI 1.10-4.61; p = 0.002) and Day 90 (Δ3.21;CI 0.766-5.652; p = 0.011) (Table 2).
For SNPs rs1143627 and rs16944 in the promoter region of IL1B gene, identical significant effects were seen for rs1143627 T/T and rs16944 C/C carriers. Both genotypes demonstrated significant improvement among those administered AC-1202 relative to Placebo at Days 45 (Δ2.79; CI 0.26-5.33; p = 0.031) and Day 90 (Δ3.70; CI 0.09-7.30; p = 0.044). Note, rs1143627 T and rs16944 C were in complete linkage disequilibrium in the sample population.
No significant genotype interactions for any SNPs were found between AC-1202 and Placebo at Day 104 (Table 2).
APOEgenotype effects on ADAS-Cog
Among APOE genotypes, homozygous carriers of the epsilon 3 allele administered AC-1202 demonstrated significant improvement relative to Placebo at Days 45 (Δ5.31; CI 2.15- 8.48; p = 0.0012) and Day 90 (Δ3.98; CI 0.41-7.55; p = 0.030). Carriers of the E4 allele did not demonstrate significant effects on ADAS-Cog scores. Subjects who carried a single E4 allele, for example E4/E3 genotype did not differ significantly from Placebo at either Day 45 or Day 90 (See Table 2). Consistent with earlier results, when participants were categorized as either E4(+) or E4(-) based on their carriage status of the epsilon 4 allele, significant improvement were found in E4(-) subjects administered AC-1202 relative to Placebo at Days 45 (Δ5.35; CI 2.37-8.321; p = 0.0006) and Day 90 (Δ3.9; CI 0.45-7.35; p = 0.027). Due to the small number of E4/E4 carriers, it was not possible to examine dosage effects of E4 on ADAS-Cog scores.
Note, the dataset used in the present study included only ADAS-Cog scores from a subset of participants who signed genetic consent for additional markers and hence the means and p-values differ slightly from the population who consented to APOE testing as previously reported [15]. However, the overall findings of significance did not change.
Interactions with APOE4
Previous studies have demonstrated a pharmacogenetic response in cognitive performance by induction of ketosis based on APOE4 carriage status. In the present study, each of the SNPs were examined for an interaction between the SNP genotype and E4 carriage status, as defined as either E4(-) or E4(+).
Evidence of possible interactions with APOE4 were defined as genetic combinations that produced enhanced cognitive improvement at Day 45, Day 90 and Day 104 when compared to the cognitive results for E4(-) subjects alone. As described above, among E4(-) subjects administered AC-1202, change from Baseline scores on ADAS-Cog improved an average of -2.16 points on Day 45, -2.27 points on Day 90, and -0.87 points on Day 104.
Specific genotype combinations of IDE and E4(-) as well as IL1B and E4(-) produced additive improvements in cognitive performance. Among E4(-) subjects administered AC-1202 who were also heterozygous C/T for the IDE rs2251101SNP, scores on ADAS-Cog improved from Baseline an average of -4.5 points on Day 45, -7.73 points on Day 90, and -4.5 points on Day 104, all of which were significantly different from Placebo means (p < 0.05) (Figure 2A, Table 3). Among E4(-) subjects administered AC-1202 who were also homozygous for T/T for the IL1B rs1143627 SNP (all of these subjects were also homozygous for the C/C allele of rs16944), scores on ADAS-Cog improved from Baseline an average of -3.6 points on Day 45, -5.7 points on Day 90 and -3.3 points on Day 104, all of which were significantly different from Placebo means (p < 0.05) (Figure 2B, Table 3). No significant differences between Active and Placebo were found among E4(+) subjects and any genotypes of IDE or IL1B (Table 4).

Change from Baseline in ADAS-Cog scores among responder genotypes over time. Red markers represent mean change from Baseline among subjects administered AC-1202. Blue markers represent mean change from Baseline among subjects administered Placebo. Error bars represent standard error of the mean. A Solid lines and solid markers represent subjects who are APOE4(-). Dotted lines and solid markers represent mean scores of subjects who are heterozygous for the IDE SNP rs2251101. Dashed lines and open markers represent mean scores of subjects who were E4(-) and heterozygous for the IDE SNP rs2251101. B Solid lines and solid markers represent subjects who are APOE4(-). Dotted lines and solid markers represent mean scores of subjects who are homozygous for the IL1B SNP rs1143627 T allele. Dashed lines and open markers represent mean scores of subjects who were E4(-) and homozygous for the IL1B SNP rs1143627 T allele. Asterisks represent significant difference between AC-1202 and Placebo means (p-value < 0.05).
Changes in serum β-hydroxybutyrate levels
To examine if differences in cognitive performance were due to differences in circulating ketone body levels, serum β-hydroxybutyrate (BHB) levels were compared between potential responder genotypes. Serum BHB levels among subjects receiving AC-1202 were compared between genotypes at Baseline, Day 45 and Day 90. At Baseline 1/2 dose (10 grams of AC-1202) was administered and on Days 45 and 90 full dose (20 grams of AC-1202). Significant differences were found at the Baseline visit between E4 carriers and non-carriers (p-value 0.03). However, this finding did not reproduce at later time points. Among E4(-) and IDE and IL1B genotypes administered AC-1202, there were no significant differences between BHB levels at any study visit (Table 5).
Induction of ketosis by the oral administration of the ketogenic compounds MCTs improved cognitive performance in mild to moderate AD patients relative to Placebo. Yet, the improvement was largely restricted to non-carriers of the AD risk factor APOE4 [14, 15]. In the present study, additional genetic markers were examined for their ability to influence cognitive performance at specific time points during a 90 day dosing of MCTs. In general, the low number of subjects in the study did not allow for definitive analysis, and the results should be considered exploratory.
Ketone bodies are produced mainly by the liver from fatty acids (FA) during periods of low carbohydrate availability. Ketogenesis and the utilization of ketone bodies is regulated at several key steps that, under normal feeding conditions, prevent substantial amounts of ketone bodies from being produced. Ketogenesis requires abundant circulating free fatty acid (FFA) levels for oxidation in the liver. Thus, conditions, such as high carbohydrate diet and elevated insulin signaling prevent ketone body production.
Ketone bodies have several properties that make them attractive for treating neurodegenerative disorders. During starvation conditions ketone bodies can substitute for the majority of the brain's energy requirements [11]. Studies with infused ketone bodies have demonstrated that even in the presence of normal glucose, the brain will metabolize ketone bodies [22]. Therefore, in conditions where glucose use is impaired, ketone bodies may offer a substitute fuel. This is the rationale for the successful use of ketogenic diets in GLUT1 deficiency syndrome, where the brain cannot transport sufficient glucose for normal function and ketones can substitute for the lack of glucose [23]. Similarly, the regional decreases in CMR(glc) seen in AD may benefit from the exogenous supplementation with ketone bodies (for review see [12]).
The SNP rs2251101 is located in the 3' un-translated region of the IDE gene. The C allele has been associated with a putative reduction of function haplotype of IDE. Homozygous carriers of the C allele were found to present high fasting and post-prandial insulin levels, as well as greater body mass index, suggestive of low Ide activity [24]. In the present study, heterozygous C/T carriers as well as grouped non-C/C carriers administered AC-1202 had significantly improved ADAS-Cog scores at Days 45 and 90 relative to Placebo subjects of the same genotype. Homozygous C/C carriers may have reduced Ide activity and hence higher insulin levels. High insulin levels may reduce levels of monocarboxylate transporters and inhibit the ability to respond to induced ketosis. It is well recognized that insulin signaling promotes fatty acid storage and reduces fatty acid oxidation, while conditions of low insulin signaling, such as fasting, promote ketogenesis and ketolysis. This mechanism is likely to operate in the brain [25].
Two SNPs (rs1143627 and rs16944) found in the promoter region of the IL1B gene have been associated with clinically observed differences in the levels of Il1β protein in vivo. The SNP rs1143627 C/T is located at position -31 in the putative TATA box of the IL1B gene. The SNP rs16944 C/T is located at position -511. A haplotype, composed of the T allele at -511 and the C allele at -31, is significantly associated with a two to threefold increase in lipopolysaccharide (LPS) -induced Il1β protein secretion [26]. Thus, carriers of this haplotype may produce an enhanced inflammatory response and inhibit their ability to respond to ketosis, while non-carriers of this haplotype, (subjects who were homozygous for rs1143627 T and rs16944 C) showed significant response at Days 45 and 90.
Similar to polymorphisms in IDE, enhanced inflammatory response may inhibit the ability of the cells to utilize ketone bodies by influencing insulin signaling. Inflammation is well recognized to diminish the rate of hepatic ketogenesis, possibly by increasing circulating insulin levels [27]. Notably, due to their unique metabolism, medium chain fatty acids, such as caprylic acid generated by AC-1202, are immune to the inflammatory inhibition of ketogenesis [28]. Of note, no significant differences in serum BHB levels were noted between IL1B rs1143627 and rs16944 genotypes. One mechanism by which elevated circulating insulin levels may inhibit the uptake of ketone bodies is by reduction of monocarboxylate transporter proteins, such as monocarboxyate transporter 1 (MCT1). Consistent with this view, inflammation of the intestine has been associated with decreased levels of MCT1 [29] and increased reliance on glucose [30], suggesting that inflammation could induce a shift away from ketone body metabolism towards glucose. In addition, inflammation may directly inhibit cells' ability to metabolize ketone bodies. For example, treating rats with LPS was found to cause nitration and reduced activity of the protein succinyl-CoA:3-oxoacid CoA transferase (SCOT; EC 2.8.3.5) [31]. SCOT catalyzes the formation of acetyl-CoA from acetoacetate and is the rate limiting step in the metabolism of ketone bodies. Therefore, inhibition of uptake or metabolism of ketone bodies by an enhanced inflammatory response due to polymorphisms in IL1B may reduce the ability of an AD patient to respond to induced ketosis (Figure 3).

A model of genotypic effects on ketone body metabolism in mild-to-moderate Alzheimer's disease. Non-carriers of the APOE4 allele may have decreased insulin signaling allowing for increased ketone body metabolism. Carriers of the T allele of rs2251101 have elevated Ide activity relative to C carriers and therefore reduced insulin signaling. Carriers of the rs1143627 T and rs16944 C alleles have a reduced inflammatory response relative to rs1143627 C and rs16944 T carriers. This reduced inflammatory response may allow for improved ketone body metabolism. Each of the polymorphisms that reduce insulin signaling may allow for better response to induced ketosis in AD.
Such a model may explain the additive effects seen among E4(-) subjects without the reduction of function IDE genotype rs2251101 C/C and without the proinflammatory genotype of IL1B rs1143627 C/C. Absence of these genotypes is predicted to lead to decreased insulin levels. The lower levels of insulin in an E4(-) background may be insufficient to overcome the mild insulin resistance and hence these subjects can respond to induced ketosis (Figure 3). In contrast, the relative insulin sensitivity of E4(+) subjects may prevent them from responding to ketosis in any genetic background [32, 33].
When IDE and IL1B responder genotypes were examined in E4(+) carriers, no significant effects were observed, yet an additive or synergistic effect appears to be present in E4(-) subjects, suggesting that the effects seen in the overall genotyped population may have been driven by the strong response among E4(-) subjects who also carried the IDE or IL1B responder genotypes. This is notable by improvement in performance relative to the Placebo after the two week washout, which suggests that ketone bodies may confer a durable effect on cognition, possibly by improving mitochondrial efficiency and reducing oxidative damage [34] or through improvement in cerebral lipid environment [35].
Conclusions
In conclusion, despite the relatively small size of this study, genetic influences on cognitive scores in response to induced ketosis were noted. The main modulator of induced ketosis appears to be the carriage status of APOE4. It may not be a coincidence that APOE4 is also the major genetic risk factor for late onset AD. The failure of APOE4 carriers to respond to ketosis may indicate a more insidious metabolic problem. APOE4 carriers may be overly reliant on glucose and hence, over a lifetime, cerebral neurons are deprived of the metabolic advantages conferred by ketone body metabolism and this may be crucial to etiology of AD [36]. Importantly, this type of pharmacogenomic profiling not only offers insights into the disease process, it also allows targeting of patients who are most likely to respond to therapy. In this way, better and more effective therapeutics can be developed.
Abbreviations
- ADAS-Cog:
-
Alzheimer's Disease Assessment Scale-Cognitive subscale
- AD:
-
Alzheimer's disease
- CMRglc:
-
Cerebral Metabolic Rate of Glucose
- MMSE:
-
Mini Mental State Exam
- NINCDS-ADRDA:
-
National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association
- CT:
-
computerized tomography
- MRI:
-
magnetic resonance imaging
- MCT:
-
medium chain triglyceride
- CI:
-
confidence interval
- SCOT:
-
succinyl-CoA:3-oxoacid CoA transferase.
References
Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM: Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. Jama. 1997, 278 (16): 1349-1356. 10.1001/jama.278.16.1349.
Bertram L, Tanzi RE: Thirty years of Alzheimer's disease genetics: the implications of systematic meta-analyses. Nature reviews. 2008, 9 (10): 768-778. 10.1038/nrn2494.
de Leon MJ, Ferris SH, George AE, Christman DR, Fowler JS, Gentes C, Reisberg B, Gee B, Emmerich M, Yonekura Y, et al: Positron emission tomographic studies of aging and Alzheimer disease. AJNR Am J Neuroradiol. 1983, 4 (3): 568-571.
Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J: Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer's dementia. Proceedings of the National Academy of Sciences of the United States of America. 2004, 101 (1): 284-289. 10.1073/pnas.2635903100.
Mosconi L, Brys M, Glodzik-Sobanska L, De Santi S, Rusinek H, de Leon MJ: Early detection of Alzheimer's disease using neuroimaging. Exp Gerontol. 2007, 42 (1-2): 129-138. 10.1016/j.exger.2006.05.016.
Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, Sheline YI, Klunk WE, Mathis CA, Morris JC, et al: Molecular, structural, and functional characterization of Alzheimer's disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005, 25 (34): 7709-7717. 10.1523/JNEUROSCI.2177-05.2005.
Piert M, Koeppe RA, Giordani B, Berent S, Kuhl DE: Diminished glucose transport and phosphorylation in Alzheimer's disease determined by dynamic FDG-PET. J Nucl Med. 1996, 37 (2): 201-208.
Liang WS, Reiman EM, Valla J, Dunckley T, Beach TG, Grover A, Niedzielko TL, Schneider LE, Mastroeni D, Caselli R, et al: Alzheimer's disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proceedings of the National Academy of Sciences of the United States of America. 2008, 105 (11): 4441-4446. 10.1073/pnas.0709259105.
Cunnane S, Nugent S, Roy M, Courchesne-Loyer A, Croteau E, Tremblay S, Castellano A, Pifferi F, Bocti C, Paquet N, et al: Brain fuel metabolism, aging, and Alzheimer's disease. Nutrition (Burbank, Los Angeles County, Calif. 2011, 27 (1): 3-20. 10.1016/j.nut.2010.07.021.
Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD, Sakurada O, Shinohara M: The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. Journal of neurochemistry. 1977, 28 (5): 897-916. 10.1111/j.1471-4159.1977.tb10649.x.
Owen OE, Morgan AP, Kemp HG, Sullivan JM, Herrera MG, Cahill GF: Brain metabolism during fasting. The Journal of clinical investigation. 1967, 46 (10): 1589-1595. 10.1172/JCI105650.
Henderson ST: Ketone bodies as a therapeutic for Alzheimer's disease. Neurotherapeutics. 2008, 5 (3): 470-480. 10.1016/j.nurt.2008.05.004.
Prins ML: Cerebral metabolic adaptation and ketone metabolism after brain injury. J Cereb Blood Flow Metab. 2008, 28 (1): 1-16. 10.1038/sj.jcbfm.9600543.
Reger MA, Henderson ST, Hale C, Cholerton B, Baker LD, Watson GS, Hyde K, Chapman D, Craft S: Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiology of aging. 2004, 25 (3): 311-314. 10.1016/S0197-4580(03)00087-3.
Henderson ST, Vogel JL, Barr LJ, Garvin F, Jones JJ, Costantini LC: Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer's disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutrition & metabolism. 2009, 6 (1): 31-
Roses AD, Sauders AM: Perspective on a pathogenesis and treatment of Alzheimer's disease. Alzheimer's and Dementia. 2006, 2: 59-70. 10.1016/j.jalz.2005.12.001.
Bach AC, Ingenbleek Y, Frey A: The usefulness of dietary medium-chain triglycerides in body weight control: fact or fancy?. Journal of lipid research. 1996, 37 (4): 708-726.
Miners JS, Baig S, Palmer J, Palmer LE, Kehoe PG, Love S: Abeta-degrading enzymes in Alzheimer's disease. Brain pathology (Zurich, Switzerland). 2008, 18 (2): 240-252. 10.1111/j.1750-3639.2008.00132.x.
McNaull BB, Todd S, McGuinness B, Passmore AP: Inflammation and anti-inflammatory strategies for Alzheimer's disease--a mini-review. Gerontology. 2010, 56 (1): 3-14. 10.1159/000237873.
Shaftel SS, Griffin WS, O'Banion MK: The role of interleukin-1 in neuroinflammation and Alzheimer disease: an evolving perspective. Journal of neuroinflammation. 2008, 5: 7-10.1186/1742-2094-5-7.
Nalbantoglu J, Gilfix BM, Bertrand P, Robitaille Y, Gauthier S, Rosenblatt DS, Poirier J: Predictive value of apolipoprotein E genotyping in Alzheimer's disease: results of an autopsy series and an analysis of several combined studies. Annals of neurology. 1994, 36 (6): 889-895. 10.1002/ana.410360614.
Hasselbalch SG, Madsen PL, Hageman LP, Olsen KS, Justesen N, Holm S, Paulson OB: Changes in cerebral blood flow and carbohydrate metabolism during acute hyperketonemia. The American journal of physiology. 1996, 270 (5 Pt 1): E746-751.
Wang D, Pascual JM, Yang H, Engelstad K, Jhung S, Sun RP, De Vivo DC: Glut-1 deficiency syndrome: clinical, genetic, and therapeutic aspects. Annals of neurology. 2005, 57 (1): 111-118. 10.1002/ana.20331.
Gu HF, Efendic S, Nordman S, Ostenson CG, Brismar K, Brookes AJ, Prince JA: Quantitative trait loci near the insulin-degrading enzyme (IDE) gene contribute to variation in plasma insulin levels. Diabetes. 2004, 53 (8): 2137-2142. 10.2337/diabetes.53.8.2137.
Pan JW, Rothman TL, Behar KL, Stein DT, Hetherington HP: Human brain beta-hydroxybutyrate and lactate increase in fasting-induced ketosis. J Cereb Blood Flow Metab. 2000, 20 (10): 1502-1507.
Hall SK, Perregaux DG, Gabel CA, Woodworth T, Durham LK, Huizinga TW, Breedveld FC, Seymour AB: Correlation of polymorphic variation in the promoter region of the interleukin-1 beta gene with secretion of interleukin-1 beta protein. Arthritis and rheumatism. 2004, 50 (6): 1976-1983. 10.1002/art.20310.
Neufeld HA, Pace JG, Kaminski MV, George DT, Jahrling PB, Wannemacher RW, Beisel WR: A probable endocrine basis for the depression of ketone bodies during infectious or inflammatory state in rats. Endocrinology. 1980, 107 (2): 596-601. 10.1210/endo-107-2-596.
Neufeld HA, Kaminski MV, Wannemacher RW: Effect of inflammatory and noninflammatory stress on ketone bodies and free fatty acids in rats. The American journal of clinical nutrition. 1977, 30 (8): 1357-1358.
Thibault R, De Coppet P, Daly K, Bourreille A, Cuff M, Bonnet C, Mosnier JF, Galmiche JP, Shirazi-Beechey S, Segain JP: Down-regulation of the monocarboxylate transporter 1 is involved in butyrate deficiency during intestinal inflammation. Gastroenterology. 2007, 133 (6): 1916-1927. 10.1053/j.gastro.2007.08.041.
Thibault R, Blachier F, Darcy-Vrillon B, de Coppet P, Bourreille A, Segain JP: Butyrate utilization by the colonic mucosa in inflammatory bowel diseases: a transport deficiency. Inflammatory bowel diseases. 2010, 16 (4): 684-695.
Marcondes S, Turko IV, Murad F: Nitration of succinyl-CoA:3-oxoacid CoA-transferase in rats after endotoxin administration. Proceedings of the National Academy of Sciences of the United States of America. 2001, 98 (13): 7146-7151. 10.1073/pnas.141222598.
Ariza MJ, Sanchez-Chaparro MA, Baron FJ, Hornos AM, Calvo-Bonacho E, Rioja J, Valdivielso P, Gelpi JA, Gonzalez-Santos P: Additive effects of LPL, APOA5 and APOE variant combinations on triglyceride levels and hypertriglyceridemia: results of the ICARIA genetic sub-study. BMC medical genetics. 2010, 11: 66-
Craft S, Asthana S, Schellenberg G, Cherrier M, Baker LD, Newcomer J, Plymate S, Latendresse S, Petrova A, Raskind M, et al: Insulin metabolism in Alzheimer's disease differs according to apolipoprotein E genotype and gender. Neuroendocrinology. 1999, 70 (2): 146-152. 10.1159/000054469.
Studzinski CM, Mackay WA, Beckett TL, Henderson ST, Murphy MP, Sullivan PG, Burnham WM: Induction of ketosis may improve mitochondrial function and decrease steady-state amyloid-beta precursor protein (APP) levels in the aged dog. Brain research. 2008, 1226: 209-217.
Taha AY, Henderson ST, Burnham WM: Dietary enrichment with medium chain triglycerides (AC-1203) elevates polyunsaturated fatty acids in the parietal cortex of aged dogs: implications for treating age-related cognitive decline. Neurochemical research. 2009, 34 (9): 1619-1625. 10.1007/s11064-009-9952-5.
Henderson ST: Ketone bodies as a therapeutic for Alzheimer's disease. Emerging Drugs and Targets for Alzheimer's Disease: Volume 1: Beta-Amyloid, Tau Protein and Glucose Metabolism. Edited by: Martinez A. 2010
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/12/137/prepub
Acknowledgements and Funding
Accera funded the study, designed the protocol, and either conducted or commissioned the data analysis and interpretation. The data are maintained on file at the offices of Accera, Inc Broomfield, CO.
Author information
Authors and Affiliations
Corresponding author
Additional information
Statement of Competing interests
Some authors may benefit from this publication. Author SH is an employee of the sponsor of this trial, Accera Inc. SH has very minor stock ownership in Accera.
SH is the sole inventor of one issued patent (US 6835750) entitled: Use of medium chain triglycerides for the treatment and prevention of Alzheimer's disease and other diseases resulting from reduced neuronal metabolism II. SH and Accera have other published pending patent applications in this area: US 2002/0006959 A1, US 2003/0059824 A1, US 2006/0122270 A1, US 2008/0009467 A1, US 2006/0252775 A1, US 2007/0135376 A1, US 2008/0287372 A1, US 2007/0179197 A1.
Authors' contributions
SH designed the research approach and wrote the paper. SH and JP analyzed data. All authors read and approved the final manuscript
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Henderson, S.T., Poirier, J. Pharmacogenetic analysis of the effects of polymorphisms in APOE, IDE and IL1B on a ketone body based therapeutic on cognition in mild to moderate Alzheimer's disease; a randomized, double-blind, placebo-controlled study. BMC Med Genet 12, 137 (2011). https://doi.org/10.1186/1471-2350-12-137
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-12-137