
Abstract
Background
Individuals born small for gestational age (SGA) are at increased risk of rapid postnatal weight gain, later obesity and diseases in adulthood such as type 2 diabetes, hypertension and cardiovascular diseases. Environmental risk factors for SGA are well established and include smoking, low pregnancy weight, maternal short stature, maternal diet, ethnic origin of mother and hypertension. However, in a large proportion of SGA, no underlying cause is evident, and these individuals may have a larger genetic contribution.
Methods
In this study we tested the association between SGA and polymorphisms in genes that have previously been associated with obesity and/or diabetes. We undertook analysis of 54 single nucleotide polymorphisms (SNPs) in 546 samples from the Auckland Birthweight Collaborative (ABC) study. 227 children were born small for gestational age (SGA) and 319 were appropriate for gestational age (AGA).
Results and Conclusion
The results demonstrated that genetic variation in KCNJ11, BDNF, PFKP, PTER and SEC16B were associated with SGA and support the concept that genetic factors associated with obesity and/or type 2 diabetes are more prevalent in those born SGA compared to those born AGA. We have previously determined that environmental factors are associated with differences in birthweight in the ABC study and now we have demonstrated a significant genetic contribution, suggesting that the interaction between genetics and the environment are important.
Similar content being viewed by others


Background
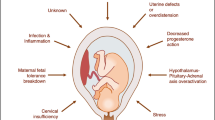
Small for gestational age (SGA) babies (typically defined as birthweight below the 10th centile according to gestational age [1, 2]) are not only at increased risk of neonatal morbidity and mortality, but are also at increased risk of rapid postnatal weight gain, later obesity and diseases in adulthood such as type 2 diabetes, hypertension and ischemic heart disease [3–7] which are major causes of adult morbidity and mortality worldwide. Although the cause of this association is unknown, several hypotheses have been proposed. The fetal insulin hypothesis [8] proposes that common genes inherited by the fetus affect both birth size and predisposition to adult diseases. In contrast, the Barker hypothesis [4, 5, 9–11] suggests the association to be the result of fetal programming - permanent changes in physiology and metabolism in response to adverse maternal uterine environment in pregnancy that result in increased metabolic disease risk in adulthood. The increased risk of adult metabolic diseases in those who are born small at birth is further amplified by an accelerated pattern of growth during childhood. The thrifty phenotype hypothesis explains this phenomenon by suggesting that the fetal nutrient conserving adaptations in response to intrauterine under nutrition is overwhelmed by nutrient abundance post-natally and manifests in adult metabolic diseases [9]. Singhal and Lucas [12] propose that it is not low birth weight per se, but this rapid postnatal growth that is responsible for the increased risk for disease in later life.
Environmental risk factors for SGA are well established and include smoking, low pregnancy weight, maternal short stature, maternal diet, ethnic origin of mother and hypertension [13]. These risk factors have been studied and confirmed in the Auckland Birthweight Collaborative (ABC) study [14, 15]. However, in a large proportion of SGA, no underlying cause is evident, and these individuals may have a larger genetic contribution. The existence of such genetic factors is supported by the observation that SGA births tend to cluster in families and to recur in successive generations [16, 17]. However, genetic association publications for SGA to date have been rare and inconsistent. Infante-Rivard et al., 2007 [18] provides a summary overview of genetic association studies for SGA. In brief, only the thrombophilia pathway including genetic variants in MTHFR, FV, and FII [19, 20], and xenobiotic-metabolizing pathways with variants in CYP1A1, GSTT and GSTM [21, 22] have been studied with any frequency. Other pathways that have been studied with less frequency include the vascular dysfunction or atherosclerosis pathway with variants in APOE, PON and ACE [23–25] and the insulin resistance pathway with variants in IGF-1 [26]. However, none of these pathways have shown robust associations with susceptibility to adult diseases with which SGA has been linked.
As children born SGA often become obese in later life and/or develop type 2 diabetes we tested the association between SGA and polymorphisms in genes that have previously been associated with obesity and/or type 2 diabetes under the hypothesis that a common genetic denominator might predispose to both SGA and obesity and/or type 2 diabetes. The hypothesis was tested in subjects from the Auckland Birthweight Collaborative (ABC) study. Since we began this study there have been several publications examining the relationship between type 2 diabetes susceptibility genes and birthweight (these studies are described in the discussion), however we have included both obesity and type 2 diabetes susceptibility genes and we have focused on SGA status.
Methods
Samples
The ABC study has been fully described previously [14].
In summary, between 16 October 1995 and 12 August 1996, babies born and resident in the Waitemata Health or Auckland Healthcare regions were eligible for inclusion and from 12 August 1996 to 30 November 1997, babies born and resident in the Auckland Healthcare region were eligible for inclusion. Preterm infants (< 37 completed weeks of gestation), multiple births and those with congenital abnormalities were excluded. The sample was selected disproportionately to include all infants born at term and SGA (≤10th percentile for sex and gestation for New Zealand) [27] and a random sample of appropriate for gestational age (AGA) infants (> 10th percentile for sex and gestation). Gestational age was estimated using the date of the last menstrual period where it was available and was within 2 weeks of the best clinical estimate of gestational age at birth; otherwise the best clinical estimate was used.
Data have been collected at birth, 1, 3.5, 7 and most recently 11 years of age. The original sample at birth resulted in a sample of 1714 subjects, of which 871 mothers were identified in the obstetric data to be of European ethnicity. At the age of 1 and 3.5 follow up of non-European ethnicities was poor resulting in a lack of ability to generalise the results from these children to their particular populations. As a result follow-up from the age of 7 has only been carried out on those children whose mothers were identified as European ethnicity at birth. At 11 years 546 participants consented to collection of peripheral blood (n = 397) or a buccal swab (n = 149) for DNA extraction and genotyping. 227 samples were from children born small for gestational age (SGA) and 319 were from children born appropriate for gestational age (AGA).
DNA was extracted from the blood/buccal samples using Qiagen's DNA extraction kit and following the manufacturer's instructions.
The study received ethical approval from the Northern Regional Ethics Committee. Signed consent for the study and extraction of DNA was given by the parents of the children and assent also given by the child.
SNP selection
The SNPs were selected from a systematic literature search to identify genetic variants demonstrating association with obesity and/or type 2 diabetes. We included 18 diabetes SNPs and 46 obesity SNPs identified from published candidate gene or genome-wide association studies, totalling 64 SNPs which were located in 42 genes. See table 1 for the list of genes/SNPs selected for investigation.
Genotyping
Genotyping was performed with the MassARRAY and iPlex systems of the Sequenom genotyping platform (Sequenom, San Diego, CA), which uses the MALDI-TOF primer extension assay [28, 29] according to manufacturers' recommendations.
Assays were optimized in 24 samples consisting of 20 reference Centre d'Etude du Polymorphisme Humain (CEPH) samples and 4 blanks.
All sample plates contained cases, controls, blanks, CEPH and duplicate samples. Quality control measures included independent double genotyping, blind to sample identity and blind to the other caller, and where available comparison of our CEPH genotypes to those in the HapMap http://www.hapmap.org.
Statistical analysis
SNPs were tested for deviation from Hardy Weinberg Equilibrium (HWE) in both cases (SGA) and controls (AGA), and for the weighted population using a chi-square goodness-of-fit test.
To determine if there were differences between children born SGA and those born AGA, genotype and allele frequencies for each SNP were analyzed by logistic regression using the major allele as the reference for allele analyses and the major homozygote group as the reference for the genotype analysis. Odds ratios show the increased risk (OR > 1) or decreased risk (OR < 1) for the minor allele, or genotype group, of being SGA in relation to the reference group. Univariable logistic regression was carried out to assess the relationship of each SNP with SGA and those found to be significant at the 10% level were carried through to multivariable analyses. The multivariable model controlled for the previously published model for SGA in this population [14] namely gestational age, gender, socio-economic status, age mother left school, marital status, attendance at antenatal classes, primiparity, maternal smoking during pregnancy, marijuana use during pregnancy, maternal height and weight, maternal age at index pregnancy and maternal hypertension.
Statistical analyses were carried out using R [30] and SAS (V9.1 SAS Institute., Cary, NC, USA).
Results
We do not have genotypic data for 10 SNPs: rs1387153 (MTNR1B), rs6020339 (CTNNBL1), rs2388399 (PFKP), rs477181 (MC4R), rs3820152 (ADIPOR1), rs7561317 (TMEM18), rs7647305 (ETV5 - DGKG), rs2844479 (NCR3 - AIF1), rs8050136 (FTO) and rs10487818 (NAMPT). These SNPs either could not be multiplexed into our sequenom assays, failed or did not pass our quality control measure for inclusion in the analysis. The remaining 54 SNPs produced genotypic data for analysis. Each of these SNPs had a genotyping call rate greater than 90% and the genotyping calls did not differ significantly from HWE criteria.
Nine SNPs were significant at the 10% level in the univariable analysis at either the genotypic or allelic level and each of these SNPs was then taken forward to multivariable analysis (see tables 2 and 3 for results).
Six SNPs demonstrated statistical significance at p < 0.05 in either the univariable or multivariable analysis: rs9939609 which is an intronic SNP in FTO, rs5219 which is a missense (Lys-Glu) SNP in KCNJ11, rs925946 which is located 9,240 bp from BDNF, rs6602024 which is an intronic SNP in PFKP, rs10508503 which is located 179,016 bp from PTER and rs10913469 which is an intronic SNP in SEC16B.
As we have two SNPs in BDNF and these two SNPs are in LD (D' = 1, r2 = 0.116) and exist within the same haplotype block (confirmed using haploview 4.2) we conducted a haplotype analysis to determine if the haplotype would give stronger results than the individual SNPs (data not shown). The haplotype analysis of rs6265 and rs925946 found an overall effect (Global Stat = 5.92, p = 0.05). In univariable analysis compared to the GG haplotype the AG haplotype did not show a statistically increased risk (OR = 1.22, 95%CI = 0.86-1.68), whilst those with the GT haplotype had a borderline decreased risk (OR = 0.78, 95%CI = 0.59-1.04). In multivariable analyses these odds ratios moved towards unity and were not statistically significant suggesting that the haplotype analysis does not add anything further to the analysis of each SNP individually.
We have examined anthropometric characteristics of the children at 11 years of age (table 4) and found that the children born SGA remain significantly lighter, shorter and still have lower BMI than the AGA children. Hence not all of the SGA children have shown catch up growth.
Discussion
We have found associations (with p values less than 0.05) for SGA with the diabetes related SNP in KCNJ11 and the obesity related SNPs in FTO, PFKP, PTER, SEC16B and BDNF. After controlling for potential confounders the association with the FTO SNP did not remain significant, whilst the other 5 SNPs were positively associated with SGA in the multivariable model.
The T allele of KCNJ11 SNP rs5219 is associated with type 2 diabetes in adults and has been shown to be associated with reduced insulin secretion (see table 1 for references) so our study result finding that this risk allele is associated with being SGA is compatible with the fetal insulin hypothesis where genetically mediated reduced insulin secretion beginning in-utero results in reduced birthweight, and later increases the risk of developing T2 D. Two previous studies evaluated the diabetes related KCNJ11 variant with birthweight and found no association [31, 32]. It is possible that this variant interacts with other factors, either genetic or environmental, that exist within the ABC cohort but are not present in the other two studies.
Our study found that the high risk allele for obesity in the PTER SNP (C allele at rs10508503) was associated with being SGA. This finding would fit with the fetal insulin hypothesis only if this allele had a direct effect on increasing insulin resistance prior to manifesting as increased BMI later in life, and thus manifests as low birth weight and later leads to obesity. Alternatively, the association of this obesity gene in SGA babies may be due to some survival advantage of being a "thin-fat" baby in terms of inappropriate fat mass for body size [33], not discerned by the simple measure of birth weight. It may be that most of these SGA babies grow into genetically predisposed obese children and adults; hence we are observing the association with post-natal obesity in our SGA cohort.
Conversely, our study found an association of SGA with the low risk alleles for obesity in the BDNF and SEC16B genes suggesting that these alleles may confer a propensity to small size beginning in-utero, since the same SNP in BDNF has been associated with thinness in women [34]. It would be interesting to examine whether this sub-group of SGA babies go on to have improved metabolic outcomes later in life by having a lower risk of obesity.
Since we began this study associations between common variants in type 2 diabetes susceptibility genes have been tested in several large birthweight cohorts. Freathy et al, 2009 [35] looked at five type 2 diabetes susceptibility genes and found that the CDKAL1 and HHEX-IDE loci were associated with reduced birth weight. They did not detect an association with CDKN2A/B, IGF2BP2, and SLC30A8. All 5 of these loci were included in our study and we did not detect an association for any of them with SGA. Zhao et al, 2009 [36] also observed an association between lower birth weight and the CDKAL1 locus. However, no association was found with 19 other diabetes genes examined, including KCNJ11 for which we found an association with SGA. Pulizzi et al, 2009 [37] investigated 9 diabetes genes, all of which are included in our study but were not found to be associated with SGA. Of the tested variants, the risk variant in HHEX showed a trend towards a low birthweight and the risk variant in the CDKN2A/2B locus was associated with high birthweight. The three studies described above investigated birthweight. Only TCF7L2 has been studied for association with SGA [38, 39]. The gene was not associated with SGA in these two cohorts or our own. However, an association has been described between TCF7L2 and birthweight, although the effect was strongest with maternal genotype and after adjustment for maternal genotype fetal TCF7L2 genotype was not associated with birth weight [40].
We examined the publically available British 1958 birth cohort database http://www.b58cgene.sgul.ac.uk/ for our significant genes. SNPs in BDNF and FTO were associated with birthweight but KCNJ11, PTER, PFKP and SEC16B did not show any associations.
The failure to replicate the associations reported by Freathy et al, Pulizzi et al and Zhao et al and our reporting of significant results for different genes may be due to the different phenotype used (birthweight vs. SGA) and/or due to different study populations with different environmental and genetic influences.
To summarise, we have identified five SNPs/genes which are associated with SGA. While noting that replication in independent samples is essential, our data provides evidence that genetic variation in type 2 diabetes and obesity susceptibility genes such as KCNJ11, BDNF, PFKP, PTER and SEC16B have a possible role in SGA as well as their established roles in obesity and/or diabetes.
We recognise that the association observed with these SNPs are unlikely to survive any adjustment for multiple testing and could thus be false positives. But it is possible that we may be seeing small genetic effects here and as our sample size is small compared to the majority of genetic association studies today we have low power to detect these associations with a high level of statistical significance. Calculations of statistical power using PS 2.1.31 [41] show that for the ABC study we have 31.61% power to detect an odds ratio of 1.2, 56.84% power to detect an odds ratio of 1.3 and 78.01% power to detect an odds ratio of 1.4 for a SNP with a minor allele frequency of 0.48 (such as rs864745 in JAZF11). For SNPs with a lower allele frequency the power would be less. For example for a SNP with a minor allele frequency of 0.12 (such as rs6602024 in PFKP) we have 17.36% power to detect an odds ratio of 1.2, 31.42% power to detect an odds ratio of 1.3 and 48.05% power to detect an odds ratio of 1.4.
It is also possible that these genes may have more subtle effects and could affect a related phenotype, rather than be directly associated with SGA. Although beyond the scope of this paper it would be interesting to look at these SNPs/genes in relation such phenotypes e.g., catch up growth. Alternatively, the associations could reflect underlying LD with other markers in these genes. Further analysis in these genes with which we demonstrate an association with SGA is therefore required. Also, further investigation of the 36 genes for which we found no association should not be ruled out. The lack of association of these genes with SGA in our sample could be explained by a lack of power and we cannot rule out that we were unable to detect smaller effects of these variants. It is also possible that these obesity and/or diabetes genes may lead to small decreases in birthweight but do not result in the more severe SGA phenotype. Alternatively, it may be possible that any direct effects of susceptibility genes resulting in an individual being born SGA (by reduced insulin secretion) may be offset by an opposing effect from the maternal genotype (through the effects of the same variants on maternal glucose levels) [42]. Unfortunately, maternal DNA samples are not presently available from the ABC cohort and so we are unable to test this.
During revision of this manuscript Freathy and colleagues reported a meta-analysis of genome-wide association studies and followed up the top hits in 13 replication studies [43]. They identified two loci, in ADCY5 and near CCNL1, that are associated with birth weight and explain 0.3% and 0.1% of the variance in birth weight, respectively. Both loci were also associated with smallness for gestational age. SNPs in ADCY5 have recently been implicated in regulation of glucose levels and susceptibility to type 2 diabetes [44], providing further evidence that the association between lower birth weight and/or SGA and subsequent type 2 diabetes does indeed have a genetic component.
Conclusion
In conclusion, this study supports the concept that genetic factors associated with obesity and/or risk of type 2 diabetes are more prevalent in those born SGA compared to those born AGA. We have previously determined that maternal diet during pregnancy [15] and other environmental factors [14] are associated with differences in birthweight in the ABC study and now we have demonstrated a significant genetic contribution, suggesting that it is most likely that there is an interaction between the genetic determinants of birthweight, childhood growth and risk of adult metabolic diseases with both the intra- and extra-uterine environments.
Abbreviations
- ABC:
-
Auckland Birthweight Collaborative
- SGA:
-
Small for gestational age
- SNPs:
-
Single nucleotide polymorphisms
- ABC:
-
Auckland Birthweight Collaborative
- AGA:
-
Appropriate for gestational age
- CEPH:
-
Centre d'Etude du Polymorphisme Humain
- HWE:
-
Hardy Weinberg Equilibrium
- MALDI-TOF:
-
Matrix Assisted Laser Desorption/Ionization - Time of Flight.
References
Alkalay AL, Graham JM, Pomerance JJ: Evaluation of neonates born with intrauterine growth retardation: review and practice guidelines. J Perinatol. 1998, 18: 142-151.
Lee PA, Chernausek SD, Hokken-Koelega AC, Czernichow P, International Small for Gestational Age Advisory Board: International Small for Gestational Age Advisory Board: International Small for Gestational Age Advisory Board consensus development conference statement: management of short children born small for gestational age, April 24 - October 1, 2001. Pediatrics. 2001, 111: 1253-1261. 10.1542/peds.111.6.1253.
Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, Osmond C, Winter PD: Fetal and infant growth and impaired glucose tolerance at age 64. BMJ. 1991, 303: 1019-1022. 10.1136/bmj.303.6809.1019.
Barker DJ, Gluckman PD, Godfrey KM, Harding JE, Owens JA, Robinson JS: Fetal nutrition and cardiovascular disease in adult life. Lancet. 1993, 341: 938-941. 10.1016/0140-6736(93)91224-A.
Barker DJ, Hales CN, Fall CH, Osmond C, Phipps K, Clark PM: Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia. 1993, 36: 62-67. 10.1007/BF00399095.
Parsons TJ, Power C, Manor O: Fetal and early life growth and body mass index from birth to early adulthood in 1958 British cohort: longitudinal study. BMJ. 2001, 323 (7325): 1331-10.1136/bmj.323.7325.1331.
Singhal A, Wells J, Cole TJ, Fewtrell M, Lucas A: Programming of lean body mass: a link between birth weight, obesity, and cardiovascular disease?. Am J Clin Nutr. 2003, 77 (3): 726-30.
Hattersley AT, Tooke JE: The fetal insulin hypothesis: an alternative explanation of the association of low birthweight with diabetes and vascular disease. Lancet. 1999, 353: 1789-1792. 10.1016/S0140-6736(98)07546-1.
Barker DJ: The fetal and infant origins of adult disease. BMJ. 1990, 301: 1111-10.1136/bmj.301.6761.1111.
Barker DJP: Mothers, babies, and disease in later life. 1994, London, United Kingdom: BMJ Books
Barker DJP: Maternal nutrition, fetal nutrition and diseases in later life. Nutrition. 1997, 13: 807-10.1016/S0899-9007(97)00193-7.
Singhal A, Lucas A: Early origins of cardiovascular disease: is there a unifying hypothesis?. Lancet. 2004, 363: 1642-1645. 10.1016/S0140-6736(04)16210-7.
Kramer MS: Determinants of low birth weight: methodological assessment and meta analysis. Bull WHO. 1987, 65: 663-737.
Thompson JM, Clark PM, Robinson E, Becroft DM, Pattison NS, Glavish N, Pryor JE, Wild CJ, Rees K, Mitchell EA: Risk factors for small-for-gestational-age babies: The Auckland birthweight collaborative study. J Paediatr Child Health. 2001, 37: 369-375. 10.1046/j.1440-1754.2001.00684.x.
Mitchell EA, Robinson E, Clark PM, Becroft DM, Glavish N, Pattison NS, Pryor JE, Thompson JM, Wild CJ: Maternal nutritional risk factors for small for gestational age babies in a developed country: a case-control study. Arch Dis Child Fetal Neonatal Ed. 2004, 89 (5): F431-F435. 10.1136/adc.2003.036970.
Wang X, Zuckerman B, Coffman GA, Corwin MJ: Familial aggregation of low birth weight among whites and blacks in the United States. N Engl J Med. 1995, 333: 1744-1749. 10.1056/NEJM199512283332606.
Magnus P, Bakketeig LS, Hoffman H: Birth weight of relatives by maternal tendency to repeat small-for-gestational-age (SGA) births in successive pregnancies. Acta Obstet Gynecol Scand Suppl. 1997, 165: 35-38.
Infante-Rivard C: Studying genetic predisposition among small-for-gestational-age newborns. Semin Perinatol. 2007, 31 (4): 213-218. 10.1053/j.semperi.2007.05.001.
Howley EA, Walker M, Rodger MA: A systematic review of the association between factor V Leiden or prothrombin gene variant and intrauterine growth restriction. Am J Obstet Gynecol. 2005, 192: 694-708. 10.1016/j.ajog.2004.09.011.
Infante-Rivard C, Rivard GE, Guiguet M, Gauthier R: Thrombophilic polymorphisms and intrauterine growth restriction. Epidemiology. 2005, 16: 281-287. 10.1097/01.ede.0000158199.64871.b9.
Wang X, Zuckerman B, Pearson C, Kaufman G, Chen C, Wang G, Niu T, Wise PH, Bauchner H, Xu X: Maternal cigarette smoking, metabolic gene polymorphism, and infant birth weight. JAMA. 2002, 287: 195-202. 10.1001/jama.287.2.195.
Infante-Rivard C, Weinberg CR, Guiguet M: Xenobiotic-metabolizing genes and small-for-gestational-age births: interaction with maternal smoking. Epidemiology. 2006, 17: 38-46. 10.1097/01.ede.0000187669.34003.b1.
Infante-Rivard C, Lévy E, Rivard GE, Guiguet M, Feoli-Fonseca JC: Small babies receive the cardiovascular protective apolipoprotein 2 allele less frequently than expected. J Med Genet. 2003, 40: 626-629. 10.1136/jmg.40.8.626.
Akisu M, Balim Z, Cetin H, Kosova B, Yalaz M, Topcuoglu N, Kultursay N: The role of angiotensin-converting enzyme and apolipoprotein-E gene polymorphisms on lipid compositions in newborn infants with intrauterine growth restriction. Early Hum Dev. 2004, 78: 95-103. 10.1016/j.earlhumdev.2004.03.006.
Chen D, Hu Y, Chen C, Yang F, Fang Z, Wang L, Li J: Polymorphisms of the paraoxonase gene and risk of preterm delivery. Epidemiology. 2004, 15: 466-470. 10.1097/01.ede.0000129509.59912.b2.
Vaessen N, Janssen JA, Heutink P, Hofman A, Lamberts SW, Oostra BA, Pols HA, van Duijn CM: Association between genetic variation in the gene for insulin-like growth factor-I and low birthweight. Lancet. 2002, 359: 1036-1037. 10.1016/S0140-6736(02)08067-4.
Thompson JM, Mitchell EA, Borman B: Sex specific birthweight percentiles by gestational age for New Zealand. N Z Med J. 1994, 107: 1-3.
Jurinke C, van den Boom D, Cantor CR, Köster H: The use of massARRAY technology for high throughput genotyping. Adv Biochem Eng Biotechnol. 2002, 77: 57-74.
Storm N, Darnhofer-Patel B, van den Boom D, Rodi CP: MALDI-TOF mass spectrometry-based SNP genotyping. Methods Molecular Biology. 2003, 212: 241-262.
Ihaka R, Gentleman R: R: A language for data analysis and graphics. Journal of Computational and Graphical Statistics. 1996, 5: 299-314. 10.2307/1390807.
Weedon MN, Gloyn AL, Frayling TM, Hattersley AT, Davey Smith G, Ben-Shlomo Y: Quantitative traits associated with the Type 2 diabetes susceptibility allele in Kir6.2. Diabetologia. 2003, 46 (7): 1021-1023. 10.1007/s00125-003-1135-3.
Bennett AJ, Sovio U, Ruokonen A, Martikainen H, Pouta A, Hartikainen AL, Franks S, Elliott P, Järvelin MR, McCarthy MI: No evidence that established type 2 diabetes susceptibility variants in the PPARG and KCNJ11 genes have pleiotropic effects on early growth. Diabetologia. 2008, 51 (1): 82-85. 10.1007/s00125-007-0863-1.
Adair LS, Prentice AM: A critical evaluation of the fetal origins hypothesis and its implications for developing countries. J Nutr. 2004, 134: 191-193.
Shugart YY, Chen L, Day IN, Lewis SJ, Timpson NJ, Yuan W, Abdollahi MR, Ring SM, Ebrahim S, Golding J, Lawlor DA, Davey-Smith G: Two British women studies replicated the association between the Val66Met polymorphism in the brain-derived neurotrophic factor (BDNF) and BMI. Eur J Hum Genet. 2009, 17: 1050-1055. 10.1038/ejhg.2008.272.
Freathy RM, Bennett AJ, Ring SM, Shields B, Groves CJ, Timpson NJ, Weedon MN, Zeggini E, Lindgren CM, Lango H, Perry JR, Pouta A, Ruokonen A, Hyppönen E, Power C, Elliott P, Strachan DP, Järvelin MR, Smith GD, McCarthy MI, Frayling TM, Hattersley AT: Type 2 Diabetes Risk Alleles are Associated with Reduced Size at Birth. Diabetes. 2009, 58 (6): 1428-1433. 10.2337/db08-1739.
Zhao J, Li M, Bradfield JP, Wang K, Zhang H, Sleiman P, Kim CE, Annaiah K, Glaberson W, Glessner JT, Otieno FG, Thomas KA, Garris M, Hou C, Frackelton EC, Chiavacci RM, Berkowitz RI, Hakonarson H, Grant SF: Examination of type 2 diabetes loci implicates CDKAL1 as a birth weight gene. Diabetes. 2009, 58 (10): 2414-2418. 10.2337/db09-0506.
Pulizzi N, Lyssenko V, Jonsson A, Osmond C, Laakso M, Kajantie E, Barker DJ, Groop LC, Eriksson JG: Interaction between prenatal growth and high-risk genotypes in the development of type 2 diabetes. Diabetologia. 2009, 52 (5): 825-829. 10.1007/s00125-009-1291-1.
Cauchi S, Meyre D, Choquet H, Deghmoun S, Durand E, Gaget S, Lecoeur C, Froguel P, Levy-Marchal C: TCF7L2 rs7903146 variant does not associate with smallness for gestational age in the French population. BMC Med Genet. 2007, 8: 37-10.1186/1471-2350-8-37.
Mook-Kanamori DO, de Kort SW, van Duijn CM, Uitterlinden AG, Hofman A, Moll HA, Steegers EA, Hokken-Koelega AC, Jaddoe VW: Type 2 diabetes gene TCF7L2 polymorphism is not associated with fetal and postnatal growth in two birth cohort studies. BMC Med Genet. 2009, 10: 67-10.1186/1471-2350-10-67.
Freathy RM, Weedon MN, Bennett A, Hypponen E, Relton CL, Knight B, Shields B, Parnell KS, Groves CJ, Ring SM, Pembrey ME, Ben-Shlomo Y, Strachan DP, Power C, Jarvelin MR, McCarthy MI, Davey Smith G, Hattersley AT, Frayling TM: Type 2 diabetes TCF7L2 risk genotypes alter birth weight: a study of 24,053 individuals. Am J Hum Genet. 2007, 80: 1150-1161. 10.1086/518517.
Dupont WD, Plummer WD: Power and Sample Size Calculations for Studies Involving Linear Regression. Controlled Clinical Trials. 1998, 19: 589-601. 10.1016/S0197-2456(98)00037-3.
Hattersley AT, Beards F, Ballantyne E, Appleton M, Harvey R, Ellard S: Mutations in the glucokinase gene of the fetus result in reduced birth weight. Nature Genetics. 1998, 19: 268-270. 10.1038/953.
Freathy RM, Mook-Kanamori DO, Sovio U, et al: Variants in ADCY5 and near CCNL1 are associated with fetal growth and birth weight. Nature Genetics. 2010, 42: 430-435. 10.1038/ng.567.
Dupuis J, Langenberg C, Prokopenko I, et al: New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010, 42: 105-116. 10.1038/ng.520.
Zeggini E, Scott LJ, Saxena R, et al: Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nature Genetics. 2008, 40: 638-645. 10.1038/ng.120.
Wellcome Trust Case Control Consortium: Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature. 2007, 447: 661-678. 10.1038/nature05911.
Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, Timpson NJ, Perry JR, Rayner NW, Freathy RM, Barrett JC, Shields B, Morris AP, Ellard S, Groves CJ, Harries LW, Marchini JL, Owen KR, Knight B, Cardon LR, Walker M, Hitman GA, Morris AD, Doney AS, Wellcome Trust Case Control Consortium (WTCCC), McCarthy MI, Hattersley AT: Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007, 316: 1336-1341. 10.1126/science.1142364.
Scott LJ, Mohlke KL, Bonnycastle LL, et al: A genome wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007, 316: 1341-1345. 10.1126/science.1142382.
Deeb SS, Fajas L, Nemoto M, Pihlajamäki J, Mykkänen L, Kuusisto J, Laakso M, Fujimoto W, Auwerx J: A Pro12Ala substitution in PPARy2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nature Genetics. 1998, 20: 284-287. 10.1038/3099.
Altshuler D, Hirschhorn JN, Klannemark M, Lindgren CM, Vohl MC, Nemesh J, Lane CR, Schaffner SF, Bolk S, Brewer C, Tuomi T, Gaudet D, Hudson TJ, Daly M, Groop L, Lander ES: The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nature Genetics. 2000, 26: 76-80. 10.1038/79839.
Sandhu MS, Weedon MN, Fawcett KA, Wasson J, Debenham SL, Daly A, Lango H, Frayling TM, Neumann RJ, Sherva R, Blech I, Pharoah PD, Palmer CN, Kimber C, Tavendale R, Morris AD, McCarthy MI, Walker M, Hitman G, Glaser B, Permutt MA, Hattersley AT, Wareham NJ, Barroso I: Common variants in WFS1 confer risk of type 2 diabetes. Nature Genetics. 2007, 39: 951-953. 10.1038/ng2067.
Florez JC, Jablonski KA, McAteer J, Sandhu MS, Wareham NJ, Barroso I, Franks PW, Altshuler D, Knowler WC, Diabetes Prevention Program Research Group: Testing of diabetes-associated WFS1 polymorphisms in the diabetes prevention program. Diabetologica. 2008, 51: 451-457. 10.1007/s00125-007-0891-x.
Franks PW, Rolandsson O, Debenham SL, Fawcett KA, Payne F, Dina C, Froguel P, Mohlke KL, Willer C, Olsson T, Wareham NJ, Hallmans G, Barroso I, Sandhu MS: Replication of the association between variants in WFS1 and risk of type 2 diabetes in European populations. Diabetologica. 2008, 51: 458-463. 10.1007/s00125-007-0887-6.
Steinthorsdottir V, Thorleifsson G, Reynisdottir I, et al: A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nature Genetics. 2007, 39: 770-775. 10.1038/ng2043.
Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, Helgason A, Stefansson H, Emilsson V, Helgadottir A, Styrkarsdottir U, Magnusson KP, Walters GB, Palsdottir E, Jonsdottir T, Gudmundsdottir T, Gylfason A, Saemundsdottir J, Wilensky RL, Reilly MP, Rader DJ, Bagger Y, Christiansen C, Gudnason V, Sigurdsson G, Thorsteinsdottir U, Gulcher JR, Kong A, Stefansson K: Variant of transcription factor 7 like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nature Genetics. 2006, 38: 320-323. 10.1038/ng1732.
Helgason A, Pálsson S, Thorleifsson G, et al: Refining the impact of TCF7L2 gene variants on type 2 diabetes and adaptive evolution. Nature Genetics. 2007, 39: 218-225. 10.1038/ng1960.
Hani EH, Boutin P, Durand E, Inoue H, Permutt MA, Velho G, Froguel P: Missense mutations in the pancreatic islet beta cell inwardly rectifying K+ channel gene (KIR6.2/BIR): a meta-analysis suggests a role in the polygenic basis of type II diabetes mellitus in Caucasians. Diabetologia. 1998, 41: 1511-1515. 10.1007/s001250051098.
Gloyn AL, Weedon MN, Owen KR, Turner MJ, Knight BA, Hitman G, Walker M, Levy JC, Sampson M, Halford S, McCarthy MI, Hattersley AT, Frayling TM: Large scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes. 2003, 52: 568-572. 10.2337/diabetes.52.2.568.
Nielsen EM, Hansen L, Carstensen B, Echwald SM, Drivsholm T, Glümer C, Thorsteinsson B, Borch-Johnsen K, Hansen T, Pedersen O: The E23K variant of Kir6.2 associates with impaired post-OGTT serum insulin response and increased risk of type 2 diabetes. Diabetes. 2003, 52: 573-577. 10.2337/diabetes.52.2.573.
Prokopenko I, Langenberg C, Florez JC, et al: Variants in MTNR1B influence fasting glucose levels. Nature Genetics. 2009, 41 (1): 77-81. 10.1038/ng.290.
Lyssenko V, Nagorny CL, Erdos MR, et al: Common variant in MTNR1B associated with increased risk of type 2 diabetes and impaired early insulin secretion. Nature Genetics. 2009, 41 (1): 82-88. 10.1038/ng.288.
Bouatia-Naji N, Bonnefond A, Cavalcanti-Proença C, et al: A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nature Genetics. 2009, 41 (1): 89-94. 10.1038/ng.277.
Gudmundsson J, Sulem P, Steinthorsdottir V, et al: Two variants on chromosome 17 confer prostate cancer risk, and the one in TC2 protects against type 2 diabetes. Nature Genetics. 2007, 39: 977-983. 10.1038/ng2062.
Winckler W, Weedon MN, Graham RR, McCarroll SA, Purcell S, Almgren P, Tuomi T, Gaudet D, Boström KB, Walker M, Hitman G, Hattersley AT, McCarthy MI, Ardlie KG, Hirschhorn JN, Daly MJ, Frayling TM, Groop L, Altshuler D: Evaluation of common variants in the six known maturity-onset diabetes of the young (MODY) genes for association with type 2 diabetes. Diabetes. 2007, 56: 685-693. 10.2337/db06-0202.
Thorleifsson G, Walters GB, Gudbjartsson DF, et al: Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nature Genetics. 2009, 41: 18-24. 10.1038/ng.274.
Willer CJ, Speliotes EK, Loos RJ, et al: Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nature Genetics. 2009, 41 (1): 25-34. 10.1038/ng.287.
Scuteri A, Sanna S, Chen WM, Uda M, Albai G, Strait J, Najjar S, Nagaraja R, Orrú M, Usala G, Dei M, Lai S, Maschio A, Busonero F, Mulas A, Ehret GB, Fink AA, Weder AB, Cooper RS, Galan P, Chakravarti A, Schlessinger D, Cao A, Lakatta E, Abecasis GR: Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 2007, 3: e115-10.1371/journal.pgen.0030115.
Herbert A, Gerry NP, McQueen MB, Heid IM, Pfeufer A, Illig T, Wichmann HE, Meitinger T, Hunter D, Hu FB, Colditz G, Hinney A, Hebebrand J, Koberwitz K, Zhu X, Cooper R, Ardlie K, Lyon H, Hirschhorn JN, Laird NM, Lenburg ME, Lange C, Christman MF: A common genetic variant is associated with adult and childhood obesity. Science. 2006, 312: 279-283. 10.1126/science.1124779.
Liu YJ, Liu XG, Wang L, Dina C, Yan H, Liu JF, Levy S, Papasian CJ, Drees BM, Hamilton JJ, Meyre D, Delplanque J, Pei YF, Zhang L, Recker RR, Froguel P, Deng HW: Genome-wide association scans identified CTNNBL1 as a novel gene for obesity. Human Molecular Genetics. 2008, 17 (12): 1903-1813. 10.1093/hmg/ddn072.
Blakemore AI, Meyre D, Delplanque J, Vatin V, Lecoeur C, Marre M, Tichet J, Balkau B, Froguel P, Walley AJ: A rare variant in the visfatin gene (NAMPT/PBEF1) is associated with protection from obesity. Obesity. 2009, 17 (8): 1549-1553. 10.1038/oby.2009.75.
Yanagiya T, Tanabe A, Iida A, et al: Association of single-nucleotide polymorphisms in MTMR9 gene with obesity. Hum Mol Genet. 2007, 16 (24): 3017-3026. 10.1093/hmg/ddm260.
Meyre D, Delplanque J, Chèvre JC, et al: Genome-wide association study for early onset and morbid adult obesity identifies three new risk loci in European populations. Nature Genetics. 2009, 41 (2): 157-159. 10.1038/ng.301.
Dina C, Meyre D, Gallina S, Durand E, Körner A, Jacobson P, Carlsson LM, Kiess W, Vatin V, Lecoeur C, Delplanque J, Vaillant E, Pattou F, Ruiz J, Weill J, Levy-Marchal C, Horber F, Potoczna N, Hercberg S, Le Stunff C, Bougnères P, Kovacs P, Marre M, Balkau B, Cauchi S, Chèvre JC, Froguel P: Variation in FTO contributes to childhood obesity and severe adult obesity. Nature Genetics. 2007, 39: 724-726. 10.1038/ng2048.
Frayling TM, Timpson NJ, Weedon MN, et al: A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007, 316: 889-894. 10.1126/science.1141634.
Loos RJ, Lindgren CM, Li S, et al: Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nature Genetics. 2008, 40 (6): 768-775. 10.1038/ng.140.
Chambers JC, Elliott P, Zabaneh D, Zhang W, Li Y, Froguel P, Balding D, Scott J, Kooner JS: Common genetic variation near MC4R is associated with waist circumference and insulin resistance. Nature Genetics. 2008, 40 (6): 716-718. 10.1038/ng.156.
Freeman JV, Cole TJ, Chinn S, Jones PR, White EM, Preece MA: Cross-sectional stature and weight reference curves for the UK, 1990. Archives of Disease in Childhood. 1995, 73: 17-24. 10.1136/adc.73.1.17.
Cole TJ, Freeman JV, Preece MA: Body Mass Index reference curves for the UK, 1990. Archives of Disease in Childhood. 1995, 73: 25-29. 10.1136/adc.73.1.25.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/11/125/prepub
Acknowledgements
The initial study was funded by the Health Research Council of New Zealand. The 12 month postal questionnaire was funded by Hawkes Bay Medical Research Foundation. The 3.5 year follow-up study was funded by Child Health Research Foundation, Becroft Foundation and Auckland Medical Research Foundation. The 7 year follow-up study was funded by Child Health Research Foundation. The 11 year follow-up was funded by Child Health Research foundation and the Heart Foundation. The genetic component of this study was funded by Child Health Research Foundation.
EA Mitchell and JMD Thompson are supported by the Child Health Research Foundation. The 7 year follow-up study was conducted in the Children's Research Centre which is supported in part by the Starship Foundation and the Auckland District Health Board. We acknowledge the assistance of Gail Gillies, Barbara Rotherham and Helen Nagels for contacting or assessing the participants. We sincerely thank the parents and children for participating in this study.
AR Morgan, WJ Lam and LR Ferguson are supported by Nutrigenomics New Zealand which is a collaboration between AgResearch Ltd., Plant & Food Research and The University of Auckland with funding through the Foundation for Research Science and Technology.
We acknowledge use of genotype data from the British 1958 Birth Cohort DNA collection, funded by the Medical Research Council grant G0000934 and the Wellcome Trust grant 068545/Z/02.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
ARM made substantial contributions to this study. She identified genes to investigate, designed and carried out the genotyping experiments and was primary author of the manuscript. JMDT was responsible for the analysis and interpretation of data and contributed to the writing of the manuscript. He also helped in establishing the ABC cohort and its data collection. RM participated in the writing of the manuscript. PNB gave advice on study design and participated in the writing of the manuscript. WJL was responsible for DNA extraction and also assisted with the genotyping experiments. LRF participated in study design and coordination, and in editing of the manuscript. EAM conceived the study, and participated in its design and coordination. He also established the ABC cohort and its data collection. All authors read and approved the final manuscript.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Morgan, A.R., Thompson, J.M., Murphy, R. et al. Obesity and diabetes genes are associated with being born small for gestational age: Results from the Auckland Birthweight Collaborative study. BMC Med Genet 11, 125 (2010). https://doi.org/10.1186/1471-2350-11-125
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-11-125