
Abstract
Background
Autism, a heterogeneous disease, is described as a genetic psychiatry disorder. Recently, abnormalities at the synapse are supposed to be important for the etiology of autism.SHANK3 (SH3 and multiple ankyrin repeat domains protein) gene encodes a master synaptic scaffolding protein at postsynaptic density (PSD) of excitatory synapse. Rare mutations and copy number variation (CNV) evidence suggested SHANK3 as a strong candidate gene for the pathogenesis of autism.
Methods
We performed an association study between SHANK3 gene polymorphisms and autism in Chinese Han population. We analyzed the association between five single nucleotide polymorphisms (SNPs) of the SHANK3 gene and autism in 305 Chinese Han trios, using the family based association test (FBAT). Linkage disequilibrium (LD) analysis showed the presence of LD between pairwise markers across the locus. We also performed mutation screening for the rare de novo mutations reported previously.
Results
No significant evidence between any SNPs of SHANK3 and autism was observed. We did not detect any mutations described previously in our cohort.
Conclusion
We suggest that SHANK3 might not represent a major susceptibility gene for autism in Chinese Han population.
Similar content being viewed by others

Background
Autism is a pervasive developmental disorder mainly characterized by limited or absent verbal communication, lacking of reciprocal social interaction or responsiveness and restricted, stereotypical, and ritualized patterns of interests and behavior. Autism together with childhood disintegrative disorder, pervasive not otherwise specified (PDD-NOS, or atypical autism) and Asperger syndrome share the similar characteristics and are all included as autism spectrum disorder (ASD), also known as pervasive developmental disorder (PDD). Family and twin studies have conclusively described autism as a highly heritable neuropsychiatry disorder with heritability estimates of over 90% and the environmental factors contributing no more than 10% [1–3]. Nevertheless, autism is etiologically heterogeneous.
Shank3 (SH3 and multiple ankyrin repeat domains 3; also termed ProSAP2, proline-rich synapse-associated protein 2) is a master synaptic scaffolding protein[4, 5]. In rats and human beings, Shank3 is expressed preferentially in cerebral cortex and cerebellum[6, 7]. With its multiple protein interaction domains, this molecule directly or indirectly connects with neurotransmitter receptors and cytoskeleton proteins[8, 9]. It also participates in the formation, maturation and enlargement of dendritic spines and is essential for the formation of functional synapses[10].
Accumulating discoveries indicate that autism's cause may reside in abnormalities at the synapse[3]. Synapses are the physical sites through which neurons in the brain connect with each other into an integrated circuit. In 2003, the alterations in synaptic function was first proposed to be a possible cause of autism[11]. Neuroligins are a family of postsynaptic cell adhesion molecules and may be involved in the synaptogenesis[12]. Mutations of genes encoding neuroligins (NLGN3, NLGN4X) were supposed to be pathogenic for autism and Asperger syndrome[13]. Neuroligin-deficiency mouse models according to these findings exhibit some deficits that are reminiscent of ASD in human[14, 15]. The "neuroligin autim pathway" was postulated[3]. Shank3 acts as a binding partner for neuroligins(NLGNs)[16]. It was reported that rare mutations in SHANK3 may contribute to the pathogenesis of autism. Durand et al. reported two de novo alterations in SHANK3 in subjects with ASD but not in control individuals. One is a G insertion, and the other is a deletion of the terminal 22q13 with the breakpoint in intron8 of SHANK3[17]. Moessner et al. identified de novo variants with an A962G exchange in exon8 leading to a heterozygous Q321R substitution[18]. They also reported a heterozygous deletion encompassing SHANK3 in a female proband but in neither parent nor in two unaffected brothers[18]. Recently Gauthier et al. found a de novo deletion at an intronic splice site in their autistic patients, and this deletion will lead to aberrant splicing of the transcript[19]. SHANK3 could also belong to the "NLGN autism pathway"[1].
All these indicate that SHANK3 might be a strong candidate gene for autism. Resequencing has been applied to identify the rare mutations of this gene, but the linkage and association studies of SHANK3 are still insufficient. In this study, we attempted to investigate the association between the SHANK3 gene polymorphisms and autism in 305 Chinese Han trios on a population-based approach using the family-based association test (FBAT). We also performed mutation screen of this gene in probands with autism in order to detect the rare de novo mutations reported previously.
Methods
Subjects
The sample for this study consisted of 305 Chinese Han family trios (singleton autistic disorder patients and their unaffected biological parents). These families were recruited at the Institute of Mental Health, Peking University, China. Of the 305 autistic child probands, 281 were male and 24 were female. The mean age of the children at the time of testing was 11 years (range 3–25 years). Diagnoses of autism were established by senior psychiatrists. All patients fulfilled the DSM-IV criteria for autistic disorder. The cases were assessed using childhood autism rating scale (CARS)[20] and autism behavior checklist (ABC)[21]. Children with fragile × syndrome, tuberous sclerosis, a previously identified chromosomal abnormality, dysmorphic features, or any other neurological condition suspected to be associated with autism were excluded. All subjects provided written informed consent for participation in this study. The study was approved by the Ethics Committee of the Health Science Center, Peking University.
Genotyping and sequencing
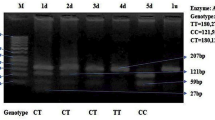
Genomic DNA was extracted from the blood using a Qiagen QIAamp DNA Mini Kit. We selected seven single nucleotide polymorphisms (SNPs) in the SHANK3 gene according to the dbSNP http://www.ncbi.nlm.nih.gov/SNP/ and the international HapMap project http://www.hapmap.org/, including rs9616915(missense polymorphism), rs2106112(synonymous polymorphism), rs6010065, rs13057681(missense polymorphism), rs2301584(3'UTR), rs41281537 and rs756638. Mutation screen for the rare de novo mutataion reported previously was performed by sequencing exon8 (for A962G[18]), exon21 (for G insertion[17]) and donor splice site G deletion of intron19[19] in all the 305 probands with autism. The SNPs with frequencies of minor allele frequency (MAF) in our sample greater than 5% were used as genetic markers in this study. Three SNPs (rs9616915, rs6010065, rs13057681) were analyzed by polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) analysis. Direct DNA sequencing was used for analyzing the other SNPs (rs2106112, rs2301584, rs41281537, rs756638) and detecting the rare de novo mutation reported previously. The information of primers and PCR-RFLP analysis is given in Table 1. The PCR amplification was performed in a 25 μl volume containing GC Buffer (TaKaRa), 200 mM of each dNTPs, 0.3 mM of each primer, 1 U of Taq DNA polymerase, and 40 ng of the genomic DNA. The conditions used for PCR amplification were an initial denaturation phase at 94°C for 5 min, followed by 36 cycles at 94°C for 30 sec, annealing at 60–65°C for 30–60 sec, and extension at 72°C for 30 sec, followed by a final extension phase at 72°C for 7 min. A 15 μL aliquot of the PCR product mixtures was completely digested with 3 units of restriction enzyme overnight. Digestion products were visualized through ethidium bromide staining after electrophoresis in 1%–3% agarose gels. The DNA sequencing was performed after cleaning the PCR product using a BigDye Terminator Cycle Sequencing Ready Reaction Kit with Ampli Taq DNA polymerase (PE Biosystem). The inner primers were used for the cycle-sequencing reaction, and the fragments were separated by electrophoresis on an ABI PRISM 377-96 DNA Sequencer (Applied Biosystem, Foster city, U.S.A).
Statistical analyses
Deviation from the Hardy-Weinberg equilibrium (HWE) for genotype frequency distributions was analyzed using the Chi-square goodness-of-fit test. To perform single- and multi-locus tests of association, we used the FBAT program (v. 1.5.1)[22]. The FBAT program uses a generalized score statistic to perform a variety of transmission disequilibrium tests, including haplotype analysis. Moreover, the FBAT program provides pairwise linkage disequilibrium (LD) analysis to detect an inter-marker relationship, using D' values. SNP pairs were considered to be in strong LD if D' > 0.70. The global haplotype tests of association were performed under "multiallelic" mode in haplotype FBAT. Meanwhile, the individual haplotype tests were conducted under "biallelic" mode in haplotype FBAT. Family-based association tests were performed under an additive model in the present study. The significance level for all statistical tests was two-tailed P < 0.05.
Results
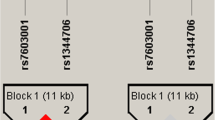
Seven SNPs in SHANK3 gene were genotyped in 305 Chinese Han autism trios. SNPs rs2106112 and rs13057681 were not polymorphic in our samples. They were not used as genetic markers for the association analyses. The other five SNPs (rs9616915, rs6010065, rs2301584, rs41281537, rs756638) were polymorphic with MAF > 5% and were then used as genetic markers for the association study. None of the genotype distributions of these five SNPs in parents or patients deviated from Hardy-Weinberg equilibrium (data not shown). Allele frequencies and the results of FBAT for single SNP analyses are shown in Table 2. None of the five SNPs was significant evidence (P < 0.05) for preferential transmission of an allele by FBAT in all samples. To further analyze the pattern of linkage disequilibrium (LD) in our sample, we computed pairwise LD for all possible combination of the five SNPs using D' values (Table 3). Four SNPs (rs6010065, rs2301584, rs41281537, rs756638) were found to be in strong LD with each other (D' > 0.7) except for rs9616915 (Table 3). To determine whether any specific haplotype would confer a higher risk for autism, we tested all specific and globe haplotype composed of these SNPs. Two haplotypes displayed weak association with autism. These are C-G-G-A (rs6010065- rs2301584- rs41281537- rs756638, P = 0.040) and G-A (rs41281537- rs756638, P = 0.047), respectively (Table 3). After 1,000 permutation tests the haplotype C-G-G-A was still associated with autism (P = 0.041), but no significance remained in the haplotype G-A (P = 0.050). No statistically significant association (P < 0.05) was observed for any other haplotypes in any set of families in HBAT analyses (data not shown). Moreover, the entire global test for haplotype transmission revealed a negative result (data not shown). To detect the de novo mutations reported before, we sequenced the exon8 (for A962G)[18], part of exon21 (for G insertion)[17], the donor splice site of intron19[19] of SHANK3 in 305 probands. However, we didn't find any genetic variants or the mutations reported previously.
The SHANK3 gene spans about 60 kb. In Affimatrix SNP 5.0 chip of 240 trios from this sample, it included 16 CNV probes covering SHANK3 gene and its flanking region. The smallest distance between two probes was less than 500 bp. In these probes, one CNV probe was located at intron8 which was reported as a breakpoint of a de novo deletion[17], and a few CNV probes were quite close to exon21 which was reported as a breakpoint of a de novo translocation in 22q13 deletion syndrome[7]. However, we didn't find any genomic imbalance in all these 240 trios or control (unpublished data).
Discussion
Shank3 is a master synaptic scaffolding protein, acting as the bridge of some neurotransmitter receptors and the downstream signal transduction. It has been postulated to perform important roles in excitatory synapse assembly [23–26], dendritic formation and maturation[10, 27, 28]. The de novo alterations of this gene and their roles in the pathogenesis of autism have been reported by some studies [17–19]. Based on this evidence, we hypothesized that the SHANK3 might be a strong candidate gene for autism. However, there was few evidence of association between SHANK3 polymorphisms and autism. In the present study, we investigated the association of SHANK3 polymorphisms and autism in 305 Chinese Han trios. We detected seven dbSNPs of SHANK3, including two nonpolymorphic SNPs rs2106112 (synonymous mutation) and rs13057681 (H1033D) in our samples. In a family-based association study for all the other five SNPs we found no evidence for transmission disequilibrium for any single marker (P > 0.05), even for the missense SNP rs9616915C>T which was reported as a non-synonymous variant identified in ASD[19]. In attempt to identify de novo genetic variants in SHANK3 that had been reported previously, we performed mutation screen of exon8, part of exon21, the donor slpice site of intron19 of SHANK3 in 305 probands with autism, and only identified one novel synonymous variant (T1231). In addition, the specific and global-haplotype FBAT tests of association were performed. Four SNPs were located in a block with high LD including rs6010065, rs2301584, rs41281537, rs756638. However, only C-G-G-A (rs6010065- rs2301584- rs41281537- rs756638, P = 0.040) exhibited a weak association with autism in our sample. We didn't detect any genomic imbalance of SHANK3 and its flanking region in our sample using Affimetrix 5.0 chip either. Our finding suggested that SHANK3 doesn't represent a major susceptibility gene for autism in the autism families ascertained from Chinese Han population.
Several potential reasons might explain the difference among various reports about SHANK3 gene and autism. One might be the racial and ethnic differences in genotype distribution and association with autism risk. For example, the allele frequency of the SHANK3 non-synonymous SNP rs9616915 was obviously different between Chinese and the other populations in the National Center for Biotechnology Information (NCBI) gene database. For European (CEU), and Nigeria population, the frequencies of allele C are 0.533 and 0.383 respectively, but for Chinese Han population (CHB) it is 0.044. Further, the variety of the investigated samples should be noticed. The previous evidence about SHANK3 was always from subjects with ASD, including autism, Asperger syndrome and PDD-NOS. Our autism cases only include patients with infantile autism, and not other cases of the etiologically more heterogeneous ASD. The heterogeneity of the investigated samples combined with the heterogeneity of neurodevelopmental disorders might hamper the understanding of genetic factors associated with autism. In order to enhance the possibility of finding relevant genetic cause, the phenotype variability in sample should be reduced[29]. In the present study we examined a relatively homogenous sample of autism to reduce the heterogeneity. The location of SHANK3 is at 22q13.3, which is a critical region for 22q13 deletion syndrome that also has autistic behavior. There are strong evidence that haploinsufficiency of SHANK3 plays a major role in 22q13 deletion syndrome[30]. SHANK3 is very likely to be involved in the pathogenesis of some mutual phenotypes of ASD and 22q13 deletion syndrome, such as delay of expressive speech. Furthermore, 22q13 deletion syndrome has a clinical phenotype overlapping in part the ASD phenotype. So subjects with 22q13 deletion may be included in ASD samples. It is really critical for the researches on autism to exclude the 22q13 deletion syndrome. We didn't detect any genomic imbalance of 22q13 in our samples using Affimetrix SNP 5.0 chip. Moreover, although the disruption of SHANK3 seems to be associated with 22q13 deletion syndrome, there is also contrary evidence that the haploinsufficiency for 22q13 genes other than SHANK3 have major effects[31]. The present study indicates that SHANK3 may not be a critical gene for the etiology of infantile autism in Chinese Han population. As autism is a heterogeneous disease, the rare mutations of SHANK3 gene seem to explain the etiology of only a small proportion of cases with autism. Sykes et al. reported recently that they didn't find any CNV or SNP association of SHANK3 within their ASD sample, although they didn't sequence the gene[32]. Their suggestion that SHANK3 deletions may be limited to a portion of autism was coincident with ours.
There was few association or linkage study for SHANK3 and autism. Our family-based association study provided an indication that SHANK3 was not critical for the pathogenesis of autism in Chinese Han population or only account for a small proportion of autism individuals. In addition, our results also reinforce the need for the detailed LD mapping, mutation screening and CNV analysis of SHANK3 in different population or other neurodevelopmental disorders.
Conclusion
The present study did not find strong evidence of SHANK3 polymorphisms and autism or identify any described non-synonymous mutations in our cohort. These might indicate that SHANK3 doesn't represent a major susceptibility gene for autism in the autism families ascertained from Chinese Han population.
References
Persico AM, Bourgeron T: Searching for ways out of the autism maze: genetic, epigenetic and environmental clues. Trends Neurosci. 2006, 29: 349-358. 10.1016/j.tins.2006.05.010.
Abrahams BS, Geschwind DH: Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008, 9: 341-355. 10.1038/nrg2346.
Garber K: Neuroscience. Autism's cause may reside in abnormalities at the synapse. Science. 2007, 317: 190-191. 10.1126/science.317.5835.190.
Ehlers MD: Molecular morphogens for dendritic spines. Trends Neurosci. 2002, 25: 64-67. 10.1016/S0166-2236(02)02061-1.
Sheng M, Kim E: The Shank family of scaffold proteins. J Cell Sci. 2000, 113 (Pt 11): 1851-1856.
Lim S, Naisbitt S, Yoon J, Hwang JI, Suh PG, Sheng M, Kim E: Characterization of the Shank family of synaptic proteins. Multiple genes, alternative splicing, and differential expression in brain and development. J Biol Chem. 1999, 274: 29510-29518. 10.1074/jbc.274.41.29510.
Bonaglia MC, Giorda R, Borgatti R, Felisari G, Gagliardi C, Selicorni A, Zuffardi O: Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. Am J Hum Genet. 2001, 69: 261-268. 10.1086/321293.
Boeckers TM, Bockmann J, Kreutz MR, Gundelfinger ED: ProSAP/Shank proteins – a family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. J Neurochem. 2002, 81: 903-910. 10.1046/j.1471-4159.2002.00931.x.
Ehlers MD: Synapse structure: glutamate receptors connected by the shanks. Curr Biol. 1999, 9: R848-850. 10.1016/S0960-9822(00)80043-3.
Roussignol G, Ango F, Romorini S, Tu JC, Sala C, Worley PF, Bockaert J, Fagni L: Shank expression is sufficient to induce functional dendritic spine synapses in aspiny neurons. J Neurosci. 2005, 25: 3560-3570. 10.1523/JNEUROSCI.4354-04.2005.
Zoghbi HY: Postnatal neurodevelopmental disorders: meeting at the synapse?. Science. 2003, 302: 826-830. 10.1126/science.1089071.
Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann K, Zhang W, Sudhof TC, Brose N: Neuroligins determine synapse maturation and function. Neuron. 2006, 51: 741-754. 10.1016/j.neuron.2006.09.003.
Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, et al: Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003, 34: 27-29. 10.1038/ng1136.
Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM, Sudhof TC: A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. 2007, 318: 71-76. 10.1126/science.1146221.
Jamain S, Radyushkin K, Hammerschmidt K, Granon S, Boretius S, Varoqueaux F, Ramanantsoa N, Gallego J, Ronnenberg A, Winter D, et al: Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc Natl Acad Sci USA. 2008, 105: 1710-1715. 10.1073/pnas.0711555105.
Meyer G, Varoqueaux F, Neeb A, Oschlies M, Brose N: The complexity of PDZ domain-mediated interactions at glutamatergic synapses: a case study on neuroligin. Neuropharmacology. 2004, 47: 724-733. 10.1016/j.neuropharm.2004.06.023.
Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsater H, et al: Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007, 39: 25-27. 10.1038/ng1933.
Moessner R, Marshall CR, Sutcliffe JS, Skaug J, Pinto D, Vincent J, Zwaigenbaum L, Fernandez B, Roberts W, Szatmari P, et al: Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet. 2007, 81: 1289-1297. 10.1086/522590.
Gauthier J, Spiegelman D, Piton A, Lafreniere RG, Laurent S, St-Onge J, Lapointe L, Hamdan FF, Cossette P, Mottron L, et al: Novel de novo SHANK3 mutation in autistic patients. Am J Med Genet B Neuropsychiatr Genet. 2009, 150B930: 421-4.
Schopler E, Reichler RJ, DeVellis RF, Daly K: Toward objective classification of childhood autism: Childhood Autism Rating Scale (CARS). J Autism Dev Disord. 1980, 10: 91-103. 10.1007/BF02408436.
Krug DA, Arick J, Almond P: Behavior checklist for identifying severely handicapped individuals with high levels of autistic behavior. J Child Psychol Psychiatry. 1980, 21: 221-229. 10.1111/j.1469-7610.1980.tb01797.x.
Rabinowitz D, Laird N: A unified approach to adjusting association tests for population admixture with arbitrary pedigree structure and arbitrary missing marker information. Hum Hered. 2000, 50: 211-223. 10.1159/000022918.
Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, Doan A, Aakalu VK, Lanahan AA, Sheng M, et al: Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron. 1999, 23: 583-592. 10.1016/S0896-6273(00)80810-7.
Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, Sheng M: Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron. 1999, 23: 569-582. 10.1016/S0896-6273(00)80809-0.
Qualmann B, Boeckers TM, Jeromin M, Gundelfinger ED, Kessels MM: Linkage of the actin cytoskeleton to the postsynaptic density via direct interactions of Abp1 with the ProSAP/Shank family. J Neurosci. 2004, 24: 2481-2495. 10.1523/JNEUROSCI.5479-03.2004.
Proepper C, Johannsen S, Liebau S, Dahl J, Vaida B, Bockmann J, Kreutz MR, Gundelfinger ED, Boeckers TM: Abelson interacting protein 1 (Abi-1) is essential for dendrite morphogenesis and synapse formation. Embo J. 2007, 26: 1397-1409. 10.1038/sj.emboj.7601569.
Boeckers TM, Liedtke T, Spilker C, Dresbach T, Bockmann J, Kreutz MR, Gundelfinger ED: C-terminal synaptic targeting elements for postsynaptic density proteins ProSAP1/Shank2 and ProSAP2/Shank3. J Neurochem. 2005, 92: 519-524. 10.1111/j.1471-4159.2004.02910.x.
Baron MK, Boeckers TM, Vaida B, Faham S, Gingery M, Sawaya MR, Salyer D, Gundelfinger ED, Bowie JU: An architectural framework that may lie at the core of the postsynaptic density. Science. 2006, 311: 531-535. 10.1126/science.1118995.
Wermter AK, Kamp-Becker I, Strauch K, Schulte-Korne G, Remschmidt H: No evidence for involvement of genetic variants in the X-linked neuroligin genes NLGN3 and NLGN4X in probands with autism spectrum disorder on high functioning level. Am J Med Genet B Neuropsychiatr Genet. 2008, 147B: 535-537. 10.1002/ajmg.b.30618.
Wilson HL, Wong AC, Shaw SR, Tse WY, Stapleton GA, Phelan MC, Hu S, Marshall J, McDermid HE: Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J Med Genet. 2003, 40: 575-584. 10.1136/jmg.40.8.575.
Wilson HL, Crolla JA, Walker D, Artifoni L, Dallapiccola B, Takano T, Vasudevan P, Huang S, Maloney V, Yobb T, et al: Interstitial 22q13 deletions: genes other than SHANK3 have major effects on cognitive and language development. Eur J Hum Genet. 2008, 16: 1301-1310. 10.1038/ejhg.2008.107.
Sykes NH, Toma C, Wilson N, Volpi EV, Sousa I, Pagnamenta AT, Tancredi R, Battaglia A, Maestrini E, Bailey AJ, et al: Copy number variation and association analysis of SHANK3 as a candidate gene for autism in the IMGSAC collection. Eur J Hum Genet. 2009 in press.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/10/61/prepub
Acknowledgements
We thank all the patients and their families for their support and participation. This work was supported by National High Technology Research and Development Program of China (2006AA02Z195), the National Natural Science Foundation of China (30870897), and the Beijing Municipal Natural Science Foundation (7081005).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
JQ carried out sequencing and PCR-RFLP analysis, and contributed to manuscript writing. MJ performed the statistical analysis and most of the sample collection. LW aided in statistical analysis. TL and YR aided in sample storage and preparations. JL, YG, JZ and XY contributed to the sample collection. WY and DZ participated in the design of the study and manuscript writing.
Jian Qin, Meixiang Jia contributed equally to this work.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Qin, J., Jia, M., Wang, L. et al. Association study of SHANK3 gene polymorphisms with autism in Chinese Han population. BMC Med Genet 10, 61 (2009). https://doi.org/10.1186/1471-2350-10-61
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-10-61