
Abstract
Background
A recent genome-wide association (GWA) study of U.S. Caucasians suggested that eight single nucleotide polymorphisms (SNPs) in CTNNBL1 are associated with obesity and increased fat mass. We analysed the respective SNPs in data from our previously published GWA for early onset obesity (case-control design), in GWA data from a population-based cohort of adults, and in an independent family-based obesity study. We investigated whether variants in CTNNBL1 (including rs6013029) and in three other genes (SH3PXD2B, SLIT3 and FLJ42133,) were associated with obesity.
Methods
The GWA studies were carried out using Affymetrix® SNP Chips with approximately 500,000 markers each. In the families, SNP rs6013029 was genotyped using the TaqMan® allelic discrimination assay. The German case-control GWA included 487 extremely obese children and adolescents and 442 healthy lean individuals. The adult GWA included 1,644 individuals from a German population-based study (KORA). The 775 independent German families consisted of extremely obese children and adolescents and their parents.
Results
We found no evidence for an association of the reported variants in CTNNBL1 with early onset obesity or increased BMI. Further, in our family-based study we found no evidence for over-transmission of the rs6013029 risk-allele T to obese children. Additionally, we found no evidence for an association of SH3PXD2B, SLIT3 and FLJ42133 variants in our two GWA samples.
Conclusion
We detected no confirmation of the recent association of variants in CTNNBL1 with obesity in a population of Central European ancestry.
Similar content being viewed by others


Background
Obesity is a major health problem worldwide and results from an interplay of social, environmental and genetic factors [1]. Genome-wide association (GWA) studies have contributed to the identification of new polygenic variants contributing to inter-individual body mass index (BMI) differences [2–5]. Recently, Liu et al. [6] reported that variants in the beta catenin-like 1 gene (CTNNBL1) were associated with increased fat mass and obesity in a GWA conducted with 1,000 adult U.S. Caucasians. In the same report, this observation was validated in a French case-control sample (896 class III obese adults; BMI ≥ 40 kg/m2 and 2,916 normal weight controls; BMI < 25 kg/m2).
Our study had two objectives. First, we aimed to replicate the association of the obesity risk alleles (rs6013029 T-allele, rs16986921 T-allele, rs6020712 A-allele, rs6020846 G-allele, rs6020395 C-allele, rs16986890 G-allele, rs6096781 C-allele, and rs6020339 C-allele) of CTNNBL1 in two GWA data sets. Second, we explored three other genes SH3PXD2B (rs13356223, rs10077897 and rs13436547), SLIT3 (rs17734503 and rs12654448) and FLJ42133 (rs7363432 and rs6095722), also mentioned by Liu et al. [6] in our GWAs. We analysed three samples: (1) GWA data from 487 cases with early onset extreme obesity and 442 controls; (2) GWA data of 1,644 individuals from a population-based adult cohort and (3) genotyping data of the best CTNNBL1 SNP rs6013029 previously reported in [6] in a sample of 775 independent nuclear families each comprising one or more extremely obese offspring and both parents.
Methods
Participants and Genotyping
Case-control GWA
487 extremely obese children and adolescents (cases: mean age 14.38 ± 3.74; BMI = 33.40 ± 6.81; BMI Z-score = 4.63 ± 2.27; 42.9% male) and 442 healthy lean individuals (controls: mean age 26.07 ± 5.79; BMI = 18.31 ± 1.10; BMI Z-score = -1.38 ± 0.35; 38.7% male) from Germany. The use of lean adults who were never overweight or obese during childhood (assessed by interview [7]) as control group reduces the chances of misclassification compared to the use of lean children as controls that might become overweight in adulthood. This sample was genotyped using the Affymetrix® Genome-Wide Human SNP Array 5.0 with 440,794 markers. Details on this GWA have been reported elsewhere [7]. The study was approved by the local ethics committee.
Population-based GWA
KORA (Kooperative Gesundheitsforschung im Raum Augsburg, follow up of Survey 3 (F3); 'Cooperative Health Research in the Region of Augsburg') comprises 3,126 German adults representative of the population within the age range 25–74 years in Augsburg and surrounding areas (Bavaria, Germany). 1,644 probands (mean age 52.52 ± 10.09; BMI = 27.33 ± 4.12; 48.9% male) were genotyped using the Affymetrix® GeneChip® Human Mapping 500K Array Set (for details on the sample see [8]). The ethics committee of the Länderärztekammer for Bavaria approved the study.
Family-based study
775 German families comprising 1,058 extremely obese children and adolescents (775 index patients, 283 siblings; mean age 13.88 ± 3.69; BMI 31.12 ± 6.06 kg/m2; BMI Z-score = 3.91 ± 2.02, 45.8% male) and 1,550 parents (mean age 42.56 ± 5.95; BMI 30.37 ± 6.29 kg/m2; BMI Z-score = 1.68 ± 1.83) were recruited at the University of Marburg and the University Duisburg-Essen. Participants were genotyped for the SNP rs6013029 using the TaqMan® allelic discrimination assay (C_29958195_10 assay, Applied Biosystems, Germany); the call rate was 99.7%, with 100% concordance of duplicates. All individuals studied are Caucasians from Central Europe, with German ancestry. All studies were conducted in accordance with the guidelines of The Declaration of Helsinki.
Statistics
Prior to analysis, the genotype distributions of all three samples (case-control GWA sample, population based GWA sample, and the family sample) were tested for deviations from Hardy-Weinberg equilibrium using an exact two-sided test [9]. The association between increased BMI and CTNNBL1 polymorphisms in the KORA cohort was analysed using linear regression analysis adjusted for age and sex while logistic regression was used for data from the case-control GWA. In both cases we used an additive model for the risk allele as described in [6]. In our family-based study we tested for overtransmission of the rs6013029 "T" allele – reported in the original study as being the risk allele – to affected offspring with the Pedigree Disequilibrium Test (PDT-sum) [10] and generated a genotype relative risk estimates using conditional logistic regression.
Power calculations based on the effect of genetic variants in rs6013029 were performed for the case-control and the cohort using the program QUANTO Version 1.2.3 http://hydra.usc.edu/gxe and for the family-based sample using TDT Power Calculator 1.2.1 http://www.biostat.jhsph.edu/~wmchen/pc.html. For these calculations we assumed a minor allele frequency = 0.05 and genetic effect size of OR = 1.42 as estimated in [6] for the tests which used the case-control and family setting, while a true genetic effect of β = 0.1 (increase in mean BMI with each additional risk allele) was chosen for the cohort. In either case α = 0.05 (one-sided) was chosen.
Results and discussion
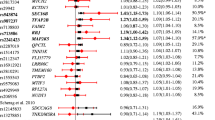
We analysed the data of both GWA studies on the SNPs previously reported in [6] of the CTNNBL1 gene. There was no indication of a deviation from Hardy-Weinberg equilibrium at any of these markers in either GWA sample or among the founders in the families based on the exact test described above (all p-values > 0.05). Furthermore, there was no evidence for an association of any of the SNPs in the CTNNBL1 gene with obesity in our data (Table 1). The strongest signal in the original report (rs6013029) achieved a two-sided p-value of 0.53 in our case-control GWA with an estimated odds ratio (OR) of 0.88 (95% confidence interval (CI) 0.60 – 1.30) for the risk-allele T. Even though there is some overlap in the confidence intervals when comparing our result to the results of the original report's French case-control validation sample with 1.42 (95% CI 1.14 – 1.77) the point estimators indicate different directions of the T-allele effect. Combined with the absence of an observed association of this marker with BMI in the KORA cohort a false positive initial observation is the most likely explanation (Table 1).
As rs6013029 was the main initial finding [6], we nevertheless decided to genotype this variant in 775 independent families ascertained for at least one obese offspring. We detected no evidence for an overtransmission of the T-allele – risk allele in the original study – to the obese offspring (two-sided p = 0.50), and an effect size estimate based on this sample of genotype relative risk (GRR) = 0.933 (95% CI 0.717 – 1.214) failed to exclude unity as well. The other CTNNBL1 SNPs previously described in [6] are displayed in Table 1. In addition, we also tried to explore the unvalidated results of SNPs from three additional genes (FLJ42133: rs7363432 and rs6095722; SH3PXD2B: rs13356223, rs10077897 and rs13436547; SLIT3: rs17734503 and rs12654448) which were also reported to be associated with increased fat mass (FLJ42133) or increased BMI (SH3PXD2B and SLIT3), respectively (Table 2). Once again, no evidence for an association in either of our GWAs was detected. In view of the requirement that replication studies need to be adequately powered, we assessed the power of each or our three samples based on the parameters listed above. For the given samples sizes our family-based replication study had a power > 80%, the cohort had a very limited power of about 10% while the case-control GWA had a slightly larger power of about 54%. If the initially reported values overestimate the true genetic effect, which is presumably quite often the case [11], our data nevertheless contribute to a more precise idea of the impact of CTNNBL1 variants on obesity.
In sum, our results underline the importance of replication of GWA results in independent samples even though independent validations may have been reported within the same initial study. While replication of association with obesity of intron 1 variants in FTO has been demonstrated robustly in almost all subsequent studies comprising obese adults and children [7, 12–15], the study by Liu et al. [6] was an exception as none of the intron 1 FTO SNPs showed evidence for a body weight-related association. Interestingly, however, the study did find some evidence for an association of variants in INSIG2 with obesity [5, 16–19]. Both examples underline the difficulties that arise when trying to validate, confirm and replicate associations with such complex traits as obesity. Our failure to replicate the initial findings [6] also does not appear to be a result of population stratification. All recruitment was done in Germany for which population stratification effects have shown to be of minor importance [20].
Another possible explanation for a lack of replication is that our results are mainly based on data for children and adolescents which are different from [6] where only adults were investigated. Again the example of FTO [6] highlights how validated associations found in adults with obesity may also be present in children with extreme obesity [3, 21]. Recently, two independent studies comprising more than 32,000 [22] and 14,000 [23] individuals also did not find significant association of the CTNNBL1 variant rs6013029 and obesity. Our study is a replication and validation attempt with sufficient combined power to independently replicate an initial finding [6], while also providing some evidence to support the decision not to follow-up variants that did not "survive" a validation within the same initial report [6]. Although we were not able to replicate the original findings, our data may be useful for a meta-analytical assessment of the association of CTNNBL1 variants and obesity. A retrospective look at the conflicting reports on INSIG2 and the recent reports on CTNNBL1 suggests that research on mediating and moderating variables to more comprehensively assess phenotype-genotype relationships is urgently needed.
Conclusion
We did not detect confirmation of association of variants in CTNNBL1 with obesity in a population of Central European ancestry. Further studies have to be performed to validate or not the initial findings about the association of CTNNBL1 variants and obesity.
References
Hebebrand J, Wulftange H, Goerg T, Ziegler A, Hinney A, Barth N, Mayer H, Remschmidt H: Epidemic obesity: are genetic factors involved via increased rates of assortative mating?. Int J Obes. 2000, 24: 345-353. 10.1038/sj.ijo.0801135.
Hinney A, Hebebrand J: Polygenic obesity in humans. Obesity Facts. 2008
Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JRB, Elliott KS, Lango H, Rayner NW, Shields B, Harries LW, Barett JC, Ellard S, Groves CJ, Knight B, Patch AM, Ness AR, Ebrahim S, Lawlor DA, Ring SM, Ben-Shlomo Y, Jarvelin MR, Sovio U, Bennet AJ, Melzer D, Ferrucci L, Loos RJF, Barroso I, Wareham NJ, Karpe F, Owen KR, Cardon LR, Walker M, Hitman GA, Palmer CNA, Doney ASF, Morris AD, Davey-Smith G, The Wellcome Trust Case Control Consortium, Hattersley AT, McCarthy MI: A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007, 316: 889-894. 10.1126/science.1141634.
Scuteri A, Sanna S, Chen WM, Uda M, Albai G, Strait J, Najjar S, Nagaraja R, Orrú M, Usala G, Dei M, Lai S, Maschio A, Busonero F, Mulas A, Ehret GB, Fink AA, Weder AB, Cooper RS, Galan P, Chakravarti A, Schlessinger D, Cao A, Lakatta E, Abecasis GR: Genome-Wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 2007, 3: e115-10.1371/journal.pgen.0030115.
Herbert A, Gerry NP, McQueen MB, Heid IM, Pfeufer A, Illig T, Wichmann HE, Meitinger T, Hunter D, Hu FB, Colditz G, Hinney A, Hebebrand J, Koberwitz K, Zhu X, Cooper R, Ardlie K, Lyon H, Hirschhorn JN, Laird NM, Lenburg ME, Lange C, Christman MF: A Common genetic variant is associated with adult and childhood obesity. Science. 2006, 312: 279-283. 10.1126/science.1124779.
Liu Y-J, Liu X-G, Wang L, Dina C, Yan H, Liu J-F, Levy S, Papasian CJ, Drees BM, Hamilton JJ, Meyre D, Delplanque J, Pei Y-F, Zhang L, Recker RR, Froguel P, Deng H-W: Genome-wide association scans identified CTNNBL1 as a novel gene for obesity. Hum Mol Genet. 2008, 17 (12): 1803-1813. 10.1093/hmg/ddn072.
Hinney A, Nguyen TT, Scherag A, Friedel S, Brönner G, Müller TD, Grallert H, Illig T, Wichmann HE, Rief W, Schäfer H, Hebebrand J: Genome Wide Association (GWA) Study for early onset extreme obesity supports the role of Fat Mass and Obesity Associated Gene (FTO) variants. PLoS ONE. 2007, 2 (12): e1361-10.1371/journal.pone.0001361.
Wichmann HE, Gieger C, Illig T, MONICA/KORA Study Group: KORA-gen-resource for population genetics, controls and a broad spectrum of disease phenotypes. Gesundheitswesen. 2005, 67 (Suppl 1): S26-S30.
Wigginton E, Cutler D, Abecasis G: A Note on Exact Tests of Hardy-Weinberg Equilibrium. Am J Hum Genet. 2005, 76: 887-883. 10.1086/429864.
Ioannidis JP, Trikalinos TA, Ntzani EE, Contopoulos-Ioannidis DG: Genetic associations in large versus small studies: an empirical assessment. Lancet. 2003, 361: 567-571. 10.1016/S0140-6736(03)12516-0.
Martin ER, Bass MP, Kaplan NL: Correcting for a potential bias in the pedigree disequilibrium test. Am J Hum Genet. 2001, 68 (4): 1065-1067. 10.1086/319525.
Jacobsson JA, Danielsson P, Svensson V, Klovins J, Gyllensten U, Marcus C, Schiöth HB, Fredriksson R: Major gender differences in association of FTO gene variant among severely obese children with obesity and obesity related phenotypes. Biochem Biophys Res Commun. 2008, 368 (3): 476-482. 10.1016/j.bbrc.2008.01.087.
Dina C, Meyre D, Gallina S, Durand E, Körner A, Jacobson P, Carlsson LMS, Kiess W, Vatin V, Lecoeur C, Delplanque J, Vaillant E, Pattou F, Ruiz J, Weill J, Levy-Marchal C, Horber F, Potoczna N, Hercberg S, Stunff CL, Bougenères P, Kovacs P, Marre M, Balkau B, Cauchi S, Chèvre JC, Froguel P: Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet. 2007, 39 (6): 724-726. 10.1038/ng2048.
Grant SFA, Li M, Bradfield JP, Kim CE, Annaiah K, Santa E, Glessner JT, Casalunovo T, Frackelton EC, Otieno FG, Shaner JL, Smith RM, Imielinski M, Eckert AW, Chiavacci RM, Berkowitz RI, Hakonarson H: Association analysis of the FTO gene with obesity in children of Caucasian and African ancestry reveals a common tagging SNP. PLos One. 2008, 3 (3): e1746-10.1371/journal.pone.0001746.
Marvelle AF, Lange LA, Qin L, Adair LS, Mohlke KL: Association of FTO with obesity-related traits in the Cebu Longitudinal Health and Nutrition Survey (CLHNS) cohort. Diabetes. 2008, 57 (7): 1987-1991. 10.2337/db07-1700.
Rosskopf D, Bornhorst A, Rimmbach C, Schwahn C, Kayser A, Krüger A, Tessmann G, Geissler I, Kroemer HK, Völzke H: Comment on "A common genetic variant is associated with adult and childhood obesity". Science. 2007, 315 (5809): 187-10.1126/science.1130571.
Hall DH, Rahman T, Avery PJ, Keavney B: INSIG-2 promoter polymorphisms and obesity related phenotypes: association study in 1428 members of 248 families. BMC Med Genet. 2006, 7: 83-10.1186/1471-2350-7-83.
Dina C, Meyre D, Samson C, Tichet J, Marre M, Jouret B, Charles MA, Balkau B, Froguel P: Comment on "A common genetic variant is associated with adult and childhood obesity". Science. 2007, 315 (5809): 187-10.1126/science.1129402.
Loos RJ, Barroso I, O'Rahilly S, Wareham NJ: Comment on "A common genetic variant is associated with adult and childhood obesity". Science. 2007, 315 (5809): 187-10.1126/science.1130012.
Steffens M, Lamina C, Illig T, Bettecken T, Vogler R, Entz P, Suk EK, Toliat MR, Klopp N, Caliebe A, König IR, Köhler K, Ludemann J, Diaz Lacava A, Fimmers R, Lichtner P, Ziegler A, Wolf A, Krawczak M, Nūrnberg P, Hampe J, Schreiber S, Meitinger T, Wichmann HE, Roeder K, Wienker TF, Baur MP: SNP-based analysis of genetic substructure in the German population. Hum Here. 2006, 62 (1): 20-9. 10.1159/000095850.
Loos RJ, Lindgren CM, Li S, Wheeler E, Zhao JH, Prokopenko I, Inouye M, Freathy RM, Attwood AP, Beckmann JS, Berndt SI, The Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial, Jacobs KB, Chanock SJ, Hayes RB, Bergmann S, Bennett AJ, Bingham SA, Bochud M, Brown M, Cauchi S, Connell JM, Cooper C, Smith GD, Day I, Dina C, De S, Dermitzakis ET, Doney AS, Elliott KS, Elliott P, Evans DM, Sadaf Farooqi I, Froguel P, Ghori J, Groves CJ, Gwilliam R, Hadley D, Hall AS, Hattersley AT, Hebebrand J, Heid IM, KORA, Lamina C, Gieger C, Illig T, Meitinger T, Wichmann HE, Herrera B, Hinney A, Hunt SE, Jarvelin MR, Johnson T, Jolley JD, Karpe F, Keniry A, Khaw KT, Luben RN, Mangino M, Marchini J, McArdle WL, McGinnis R, Meyre D, Munroe PB, Morris AD, Ness AR, Neville MJ, Nica AC, Ong KK, O'Rahilly S, Owen KR, Palmer CN, Papadakis K, Potter S, Pouta A, Qi L, Nurses' Health Study, Kraft P, Hankinson SE, Hunter DJ, Hu FB, Randall JC, Rayner NW, Ring SM, Sandhu MS, Scherag A, Sims MA, Song K, Soranzo N, Speliotes EK, Diabetes Genetics Initiative, Lyon HN, Voight BF, Ridderstrale M, Groop L, Syddall HE, Teichmann SA, Timpson NJ, Tobias JH, Uda M, The SardiNIA Study, Scheet P, Sanna S, Abecasis GR, Albai G, Nagaraja R, Schlessinger D, Ganz Vogel CI, Wallace C, Waterworth DM, Weedon MN, The Wellcome Trust Case Control Consortium, Willer CJ, FUSION, Jackson AU, Tuomilehto J, Collins FS, Boehnke M, Mohlke KL, Wraight VL, Yuan X, Zeggini E, Hirschhorn JN, Strachan DP, Ouwehand WH, Caulfield MJ, Samani NJ, Frayling TM, Vollenweider P, Waeber G, Mooser V, Deloukas P, McCarthy MI, Wareham NJ, Barroso I: Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat Genet. 2008, 40: 768-775. 10.1038/ng.140.
Willer CJ, Speliotes EK, Loos RJ, Li S, Lindgren CM, Heid IM, Berndt SI, Elliott AL, Jackson AU, Lamina C, Lettre G, Lim N, Lyon HN, McCarroll SA, Papadakis K, Qi L, Randall JC, Roccasecca RM, Sanna S, Scheet P, Weedon MN, Wheeler E, Zhao JH, Jacobs LC, Prokopenko I, Soranzo N, Tanaka T, Timpson NJ, Almgren P, Bennett A, Bergman RN, Bingham SA, Bonnycastle LL, Brown M, Burtt NP, Chines P, Coin L, Collins FS, Connell JM, Cooper C, Smith GD, Dennison EM, Deodhar P, Elliott P, Erdos MR, Estrada K, Evans DM, Gianniny L, Gieger C, Gillson CJ, Guiducci C, Hackett R, Hadley D, Hall AS, Havulinna AS, Hebebrand J, Hofman A, Isomaa B, Jacobs KB, Johnson T, Jousilahti P, Jovanovic Z, Khaw KT, Kraft P, Kuokkanen M, Kuusisto J, Laitinen J, Lakatta EG, Luan J, Luben RN, Mangino M, McArdle WL, Meitinger T, Mulas A, Munroe PB, Narisu N, Ness AR, Northstone K, O'Rahilly S, Purmann C, Rees MG, Ridderstråle M, Ring SM, Rivadeneira F, Ruokonen A, Sandhu MS, Saramies J, Scott LJ, Scuteri A, Silander K, Sims MA, Song K, Stephens J, Stevens S, Stringham HM, Tung YC, Valle TT, Van Duijn CM, Vimaleswaran KS, Vollenweider P, Waeber G, Wallace C, Watanabe RM, Waterworth DM, Watkins N, Wellcome Trust Case Control Consortium, Witteman JC, Zeggini E, Zhai G, Zillikens MC, Altshuler D, Caulfield MJ, Chanock SJ, Farooqi IS, Ferrucci L, Guralnik JM, Hattersley AT, Hu FB, Jarvelin MR, Laakso M, Mooser V, Ong KK, Ouwehand WH, Salomaa V, Samani NJ, Spector TD, Tuomi T, Tuomilehto J, Uda M, Uitterlinden AG, Wareham NJ, Deloukas P, Frayling TM, Groop LC, Hayes RB, Hunter DJ, Mohlke KL, Peltonen L, Schlessinger D, Strachan DP, Wichmann HE, McCarthy MI, Boehnke M, Barroso I, Abecasis GR, Hirschhorn JN, Genetic Investigation of ANthropometric Traits Consortium: Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009, 41 (1): 25-34. 10.1038/ng.287.
Meyre D, Delplanque J, Chèvre JC, Lecoeur C, Lobbens S, Gallina S, Durand E, Vatin V, Degraeve F, Proença C, Gaget S, Körner A, Kovacs P, Kiess W, Tichet J, Marre M, Hartikainen AL, Horber F, Potoczna N, Hercberg S, Levy-Marchal C, Pattou F, Heude B, Tauber M, McCarthy MI, Blakemore AIF, Montpetit A, Polychronakos C, Weill J, Coin LJM, Asher J, Elliott P, Järvelin MR, Visvikis-Siest S, Balkau B, Sladek R, Balding D, Walley A, Dina C, Froguel P: Genome-wide association study for early-onset and morbid adult obesity identifies three new risk loci in European populations. Nat Genet. 2009, 41 (2): 157-159. 10.1038/ng.301.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/10/14/prepub
Acknowledgements
We thank the probands for their participation. We thank the technical assistance of Jitka Andrä and Sieglinde Düerkop. This work was supported by grants from the German Ministry of Education & Research (BMBF, NGFN2 and NGFNplus: 01GS0820, 01GS0830 and 01GS0823), the German Research Foundation (DFG: HE 1446/4-1,2) and the European Union (FP6 LSHMCT-2003-503041).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
CIGV participated of the study design, perform the genotyping and drafted the manuscript; BG performed statistical analysis and drafted the manuscript; AS participated of the study design, performed secondary statistical analyses and drafted the manuscript; TM, SF, HG, IMH, TI, HEW and HS participated of the study design; JH and AH conceived the study, participated in its design and coordination and drafted the manuscript. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Vogel, C.I., Greene, B., Scherag, A. et al. Non-replication of an association of CTNNBL1polymorphisms and obesity in a population of Central European ancestry. BMC Med Genet 10, 14 (2009). https://doi.org/10.1186/1471-2350-10-14
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-10-14