
Abstract
Background
We have previously identified Urokinase Plasminogen Activator Receptor (PLAUR) as an asthma susceptibility gene. In the current study we tested the hypothesis that PLAUR single nucleotide polymorphisms (SNPs) determine baseline lung function and contribute to the development of Chronic Obstructive Pulmonary Disease (COPD) in smokers.
Methods
25 PLAUR SNPs were genotyped in COPD subjects and individuals with smoking history (n = 992). Linear regression was used to determine the effects of polymorphism on baseline lung function (FEV1, FEV1/FVC) in all smokers. Genotype frequencies were compared in spirometry defined smoking controls (n = 176) versus COPD cases (n = 599) and COPD severity (GOLD stratification) using logistic regression.
Results
Five SNPs showed a significant association (p < 0.01) with baseline lung function; rs2302524(Lys220Arg) and rs2283628(intron 3) were associated with lower and higher FEV1 respectively. rs740587(-22346), rs11668247(-20040) and rs344779(-3666) in the 5'region were associated with increased FEV1/FVC ratio. rs740587 was also protective for COPD susceptibility and rs11668247 was protective for COPD severity although no allele dose relationship was apparent. Interestingly, several of these associations were driven by male smokers not females.
Conclusion
This study provides tentative evidence that the asthma associated gene PLAUR also influences baseline lung function in smokers. However the case-control analyses do not support the conclusion that PLAUR is a major COPD susceptibility gene in smokers. PLAUR is a key serine protease receptor involved in the generation of plasmin and has been implicated in airway remodelling.
Similar content being viewed by others


Background
Chronic Obstructive Pulmonary Disease (COPD) and asthma are complex respiratory diseases involving both genetic and environmental factors (e.g. smoking exposure in COPD, allergen exposure in asthma) [1, 2]. Using 587 asthma families we have recently fine mapped a 14.4 Mb region on Chromosome 19q13 and identified the urokinase plasminogen activator receptor (PLAUR, plasminogen activator receptor, urokinase type, alternative symbols; UPAR and CD87) gene as an asthma susceptibility gene [3]. Importantly, we also demonstrated that polymorphisms spanning PLAUR predict decline in forced expiratory volume in 1 second (FEV1) in asthma subjects and determine plasma PLAUR levels [3]. PLAUR plays a key role in the formation of the serine protease plasmin by interacting with urokinase plasminogen activator (PLAU) [4] and has been implicated in many processes including; cell differentiation, proliferation and migration [5]. Plasminogen activator inhibitors (PAI-1 and PAI-2) regulate PLAUR activity [6].
While asthma and COPD are distinct clinical entities, a common feature of both diseases is airway remodelling, i.e. deposition of extracellular matrix (ECM) in the submucosa, thickening of the reticularis and smooth muscle hyperplasia [7, 8]. From the known biology of PLAUR, this protease receptor is potentially involved in these processes due to its role in matrix metalloproteinase (MMP) and transforming growth factor (TGF)β1 activation and tissue fibrosis [5].
Whole genome linkage analyses using the Boston early onset COPD family cohort has identified linkage to chromosome 19q13 for FEV1, FVC and FEV1/FVC, LOD 1.40 (59 cM), 1.20 (101 cM), 1.47 (61 cM) respectively [9]. Typing additional markers on 19q13 provided further support for this region i.e. FEV1 and FEV1/FVC LOD 1.73 (62 cM), 1.70 (62 cM) which was strengthened by selection of former/current smokers, LOD 3.30 (71 cM) and 1.96 (63 cM) respectively [10]. The PLAUR gene is located at 67 cM and based on genetic and biological evidence may be a COPD susceptibility gene. Smoking is associated with an increased decline in FEV1 and is a major risk factor for the development of COPD [11] therefore we investigated the role of PLAUR SNPs in smokers.
The aim of the current study was to test the hypotheses that a) polymorphisms spanning PLAUR contribute to COPD susceptibility and severity in smokers and b) that these polymorphisms influence baseline lung function (FEV1 and FEV1/FVC) in smokers. We have genotyped 27 SNPs spanning PLAUR in a cohort recruited for COPD or smoking history (n = 992 subjects) and completed a series of association analyses. We provide tentative evidence that polymorphisms spanning PLAUR influence baseline lung function but do not support the conclusion that PLAUR is a major COPD susceptibility gene in smokers.
Methods
Subjects and Clinical Assessment
The subjects were recruited from five UK centres for smoking history and/or COPD diagnosis (Table 1). 537 subjects were collected in Nottingham, Caucasian > 40 years and smoking >10 pack years or other centres (n = 455) recruited for spirometry defined COPD confirmed by a physician, Caucasian > 45 years old and smoking >10 pack years. The combined population (n = 992) recruited for smoking history or COPD diagnosis were stratified into healthy smokers (n = 176, post bronchodilator (BD, salbutamol) FEV1 > 80% and postBD FEV1/FVC > 0.7) and COPD subjects (n = 599, post BD FEV1 < 80% and postBD FEV1/FVC < 0.7). Subjects without data or not meeting these criteria were excluded from the case control analyses. COPD severity was investigated using post bronchodilator spirometry, i.e. GOLD classification [12] (n = 643, Additional file 1). Ethical approval was obtained from local ethics committees (Nottingham, Sheffield, Manchester, Leicester, Oxford). Informed Consent from all subjects was obtained.
SNP selection and Genotyping
The Human PLAUR gene has been sequenced in 46 Caucasian individuals (SeattleSNPs, http://pga.mbt.washington.edu/). SNPs were chosen for inferred function or their ability to tag Linkage Disequlibrium (LD) blocks ([3], Table 2). CEPH genotyping data from the HapMap project (B34) in conjunction with Haploview software (v3.3) was used to identify tagging SNPs within the gene (R2 0.8, Minor Allele Frequency (MAF) 0.1) or 5'distal region (R2 0.75, MAF 0.2) [13]. SNPs were genotyped by Kbiosciences (Hitchin, UK) using genomic DNA. Hardy-Weinberg Equilibrium was assessed using Haploview software [13].
Statistical Analyses
Using SPSS (version 15, SPSS Inc., Chicago, IL) logistic regression analyses were completed for dichotomous traits using additive (e.g. AA vs. AC vs. CC), recessive (AA/AC vs. CC) or dominant (AA vs. AC/CC) models. The COPD susceptibility analyses included, age, sex and pack years as covariates (Table 1) and the GOLD 1 versus 4 analyses included age as a covariate. Unadjusted contingency table analyses were completed using genotype or allele models (GraphPad Prism version 5, San Diego, CA). Linear regression determined the contribution of each SNP to baseline FEV1 (litres) or FEV1/FVC ratio using additive, recessive or dominant models including; age, sex, height and smoking pack years as covariates. Haploview software was used to identify PLAUR haplotypes. A p < 0.01 was considered significant for all analyses.
5'region analyses
Basal promoter activity of PLAUR has been mapped to 220 bp upstream of the start codon [14]. Using DNA from 31 Caucasian subjects a 4 kb promoter region was amplified and sequenced (Applied Biosystems, UK). SNPs were analysed for transcription factor (TF) binding site changes using online databases as described [15].
Results
Clinical Characteristics and Allele Frequencies
Baseline features of the COPD cases (n = 599) and controls (n = 176) are shown in Table 1. As anticipated baseline FEV1 and FEV1/FVC of the smoking controls and COPD cases are significantly different (p < 0.0001). Comparison of other baseline features between controls and cases identified significant differences for age, sex and pack years, therefore in subsequent analyses we adjusted for these variables. Also shown are the baseline features of male and female smokers which show that females have significantly less smoking exposure and increased lung function compared to male subjects (p < 0.001). SNPs spanning PLAUR were genotyped (Table 2). Two of the 27 SNPs, rs1994417 and rs4251831 showed deviation from Hardy-Weinberg equilibrium (p = 0.028 and p = 0.004 respectively) in the entire population (n = 992). These SNPs were removed from subsequent analyses.
Haplotype structure
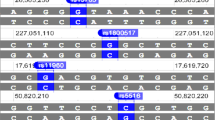
The haplotype structure of the PLAUR region generated using all data is shown in Figure 1. These data revealed that there is some redundancy in the genotyping (as expected) and that there is a block of Linkage Disequilibrium (LD) in the 3'region, a region of low LD spanning the gene and several blocks of LD in the 5'region (Figure 1).

Schematic representation of the PLAUR gene and haplotype block structure of PLAUR SNPs in smokers. (A) Schematic representation of the PLAUR gene on chromosome 19 illustrating the position of SNPs 1-13 (see Table 2). SNPs 14-25 located in the promoter region/5'region are omitted for clarity. The PLAUR gene is displayed in the reverse orientation to that observed on chromosome 19 (~40 kb). Exons are depicted as open boxes except for alternatively spliced exon 7 (VIIB, grey, see [34]). (B) Haplotype block structure of all 25 SNPs on Chromosome 19 in smokers (n = 992). The colour of shading represents R2 (a measure of LD) and numerical values are given (generated using Haploview software [13]). Haplotype blocks were defined using confidence intervals for strong LD D' 0.7-0.98.
PLAURSNPs and COPD susceptibility in smokers
Analyses of the smoking control cohort (n = 176) versus the COPD cohort (n = 599) identified only one SNP meeting statistical significance (p < 0.01), i.e. rs740587(-22346) showed a protective effect for COPD susceptibility (Table 3). Several other SNPs showed borderline significance including SNPs in intron 1 (rs2286960, protective(P)) and in the 5'region i.e. rs344779(-3666, P), rs11668247(-20040, P) and rs346054(-30147, P). These data provide limited evidence that PLAUR SNPs are associated with COPD susceptibility per se.
PLAURSNPs do not show association with spirometry defined COPD severity
Subjects were stratified according to GOLD criteria (Additional file 1) and unadjusted analyses comparing GOLD groups 1, 2, 3 and 4 and extreme severity (GOLD 1 versus 4, adjusted for age) were completed. GOLD Stage 1-4 analyses did not identify any SNPs showing significant association. Adjusted analyses (age) of GOLD Stage 1 and Stage 4 using logistic regression identified rs11668247(5'UTR-20040) as a protective allele (dominant model OR 0.32 CI 0.14-0.77, p = 0.011) (Additional file 1).
PLAURSNPs influence baseline lung function in smokers
In addition to dichotomous trait analyses based on post bronchodilator spirometry we also investigated the role of PLAUR SNPs in determining baseline lung function, FEV1 and FEV1/FVC ratio in the entire population (n = 992) (Figure 2 and Additional file 2). The FEV1 analyses identified two SNP associations; rs2302524(Lys220Arg) was associated with lower FEV1 and rs2283628 (intron 3) was associated with higher FEV1 (Figure 2). Both associations were driven by carriers of two variant alleles (recessive model p = 0.006 and p = 0.008 respectively).

PLAUR SNPs are associated with baseline FEV 1 and FEV 1 /FVC. Regression analysis was used to investigate the association between PLAUR SNPs and baseline FEV1 and FEV1/FVC ratio using the additive, recessive or dominant models. Data represent mean ± standard error. Covariates included in the model included age, gender, height and pack years. A = Additive, R = Recessive, D = Dominant models showing p < 0.01 are presented.
In the baseline FEV1/FVC analyses three SNPs showed association, i.e. rs740587(5'UTR-22346), rs11668247(5'UTR-20040) and rs344779(5'UTR-3666) in the 5'region were significantly associated with improved FEV1/FVC ratio (Figure 2, Additional file 2). Borderline significance was also observed for rs344781(5'UTR-466) and rs2283628(intron 3) for higher baseline FEV1/FVC values and rs346043(5'UTR-204590) and rs2356338(5'UTR-649) for reduced baseline FEV1/FVC values. These data highlight the potential importance of genetic determinants in the 5'region particularly for the FEV1/FVC phenotype. In order to determine if males or females are driving these associations with lung function we completed gender specific analyses for the key associated SNPs (Additional file 2). The baseline features of the male and female only cohorts are shown in Table 1. These data suggested that both the rs740587(5'UTR-22346) and rs344779(5'UTR-3666) associations with FEV1/FVC are driven by males as the allele dose effect observed in all subjects is present in male subjects and is lost in females e.g. rs344779 FEV1/FVC data for males; GG 51.8 ± 1.2 (185), GT 54.5 ± 1.0 (260), TT 59.1 ± 2.0 (66) p = 0.007 versus females; GG 56.8 ± 1.4 (146), GT 57.7 ± 1.2 (195), TT 59.5 ± 1.8 (66), p = 0.531.
PLAURpromoter analyses identifies novel SNPs with predicted function
As several associations were located in the 5'region we sequenced 4000 bps of the key promoter region (LD block 2, Figure 1) in 37 Caucasian subjects to identify novel variation. A further five novel and two reported polymorphism were identified (Additional file 3). The 12 polymorphisms were in high LD (data not shown) which suggested identification of causative polymorphisms will be difficult. Nonetheless, we conducted a bioinformatics analysis and three of these polymorphisms were found to generate binding sites for the E26 transforming sequence (ets) family transcription factor binding sites. Multiple potential changes in transcription factor (TF) binding sites were observed for SNPs in the 5' distal region e.g. alterations in hypoxia inducible factor 1 binding sites (Additional file 3).
Discussion
This study provides preliminary evidence that SNPs within PLAUR influence baseline lung function in smokers, however does not support the conclusion that PLAUR SNPs contribute significantly to the multiple genetic factors that predispose smokers to develop COPD.
We have previously identified PLAUR as an asthma and lung function associated gene and in particular identified the 3'region (rs4803648, rs4802189), intron 3 (rs2239372) and 5'region (rs2356338, rs4493171, rs346043) as determinants [3]. PLAUR SNPs predicted decline in FEV1 in asthma subjects and were associated with PLAUR levels in plasma [3] (see Table 4). These data and the emerging role of PLAUR in tissue remodelling suggested PLAUR may also influence COPD susceptibility in smokers. To test this hypothesis we genotyped 27 SNPs spanning PLAUR in a cohort of 992 smokers recruited for smoking history and/or COPD diagnosis, 25 SNPs passed quality control. Data from all subjects identified multiple 5'region LD blocks, a region of low LD spanning the gene and a 3'region LD block in keeping with our previous data [3].
In our first analyses we determined if PLAUR SNPs are risk factors for the development of spirometry defined COPD. These data identified only one SNP association (p < 0.01) i.e. rs740587 in the 5'region was protective however multiple borderline associations were observed particularly for additional SNPs in the promoter/5'region. We have previously shown that asthma risk alleles span the entire PLAUR gene including the 5'region, intron 3 and 3'region (Table 4, [3]). There was no direct concordance between asthma and COPD risk alleles and our data does not support the conclusion that PLAUR SNPs are major risk factors for the development of COPD in smokers.
The finding that the only signal for COPD susceptibility maps to the 5'region is of interest due to the observation that soluble PLAUR is elevated seven fold in the sputum of COPD patients [16] suggesting mechanisms underlying disease association may at least in part involve altered PLAUR transcription. In agreement with this hypothesis a recent study identified PLAUR mRNA expression in lung tissue as a marker of COPD and showed a significant correlation between FEF25-75(%Pred) and PLAUR mRNA expression (r = -0.44) [17].
COPD severity analyses based on GOLD classification did not identify any SNP showing association. Additional analysis of extremes of severity (GOLD 1 versus 4) again highlighted a role for rs11668247 in the 5'region in keeping with the COPD susceptibility analyses suggesting a modest influence of the distal 5'region SNPs.
In order to extend our case/control analyses we investigated the role of PLAUR SNPs in determining baseline lung function in this cohort. These analyses identified several significant associations; rs2302524(Lys220Arg) and rs2283628 (intron 3) were associated with lower and higher FEV1 respectively. rs740587(5'UTR-22346), rs11668247(5'UTR-20040) and rs344779(5'UTR-3666) in the 5'region were all associated with increased FEV1/FVC ratio. Interestingly, several SNPs showed very clear allele dose effects on FEV1/FVC ratio e.g. rs740587(5'UTR-22346), providing greater confidence in these data. The magnitude of effect was ~5% change in FEV1/FVC between genotypes and so potentially these effects are clinically relevant and surprising for a single SNP in a single gene. Interestingly, two of the SNPs associated with baseline FEV1/FVC were also associated with COPD susceptibility and severity respectively, i.e. rs740587(5'UTR-22346) and rs11668247(5'UTR-20040) in the 5'region. The rs2302524 (Lys220Arg) association with reduced baseline FEV1 values is of interest as this variant may be predicted to change the coding sequence of the PLAUR protein and this polymorphism has previously been associated with increased asthma risk and increased decline in FEV1 in asthma ([3], Table 4) The Lys220Arg is considered a conservative substitution, however the functional significance of this finding remains to be resolved. Interestingly, our data suggests a potential need for both alleles to be present prior to a physiological effect, i.e. a recessive effect.
It is also important to note that the single SNP associations reflect the linkage disequilibrium spanning the PLAUR gene e.g. rs11668247 and rs740587 are both associated with increased baseline FEV1/FVC and these SNPs are in high LD i.e. are inherited together more than by chance suggesting they both tag or contribute to the causation mechanism.
For the key associations with lung function we were also interested to determine if males or females were driving the associations and therefore completed gender specific analyses. All baseline features including mean age, smoking pack years and lung function (pre and post bronchodilator FEV1 and FEV1/FVC) were significantly different between the male and female smokers. These data are in keeping with a recent study that examined gender specific differences between subjects with severe emphysema and again identified less severe airway obstruction in females [18]. Our data suggest that the rs740587(5'UTR-22346) and rs344779(5'UTR-3666) associations with FEV1/FVC are driven by the male subjects as females do not exhibit the allele dose effect on FEV1/FVC ratio. Interesting, by selecting male subjects only the magnitude of effect was also increased i.e. the range of FEV1/FVC between genotypes for rs344779 for all subjects was 5.1% but for males was 7.3%, females 2.7%. This would suggest females do not demonstrate the lower FEV1/FVC ratio in carriers of the common alleles at these loci. These data are intriguing and potentially suggest that the effects of the polymorphisms in the 5'region are unmasked only in males i.e. gender specific transcriptional mechanisms. However, it is also feasible that the SNP effects are only identifiable in more severe airway obstruction i.e. in the males. This observation requires further investigation.
Cardiovascular disease including; ischemic heart diseases, hypertension and myocardial infarction have also been shown to be a common co-morbidity in COPD [11] and COPD may be an independent risk factor for the development of cardiovascular disease [19]. PLAUR levels have been associated with vascular remodelling [20] and monocyte adhesion in acute myocardial infarction [21]. Therefore it is feasible that in the COPD case control analyses we are really testing association with cardiovascular disease not COPD. While we have not formally tested the association between PLAUR SNPs and cardiovascular disease outcomes as we do not have this information, this seems unlikely. Our data suggests PLAUR SNPs influence baseline lung function in smokers irrespective of the presence or absence of COPD. Similarly, we have identified that PLAUR SNPs are determinants of baseline lung function in asthmatic children where clearly cardiovascular disease is not anticipated to confound the analyses [3].
In order to begin to investigate mechanisms underlying associations we have sequenced 4000 bp of the core promoter and identified a further five novel and two reported polymorphisms making a total of 12 polymorphisms in high LD (Additional file 3). Several of these SNPs generate ets family binding sites. c-ets-1 is of particular interest as this transcription factor is induced by oxidative stress e.g. H2O2 [22]. Importantly, H2O2 in breath condensate is elevated in COPD [23] and asthma [24]. These data potentially provide the link between the airway environment and expression of PLAUR determined by the presence/absence of SNPs. Interestingly, PLAU and PAI-1 expression are also elevated in sputum from asthma and COPD subjects [16] suggesting the plasminogen system may be augmented in these diseases. Importantly, PLAU and PAI1 transcription is also up regulated by ets-1 [25, 26]. Also of interest is the alteration in Hypoxia Inducible Factor (HIF) binding sites due to the presence of a polymorphism (e.g. SNP rs11668247(5'UTR-20040) that was associated with COPD severity and lung function generates a HIF1 site. PLAUR transcription has been shown to be increased by HIF1β under hypoxia conditions [27] which may be a mechanism involved in epithelial mesenchymal transition (EMT) [28].
The exact mechanism by which PLAUR contributes to the underlying pathobiology of the airways remains to be resolved, however its role in the generation of plasmin which subsequently leads to ECM degradation, MMP activation, TGFβ1 activation, 5-lipoxygenase activation and cell migration implicate tissue remodelling as a key function [29, 30]. The complex role of the PLAU-PLAUR pathway in the airways is illustrated in Figure 3. It is important to note that there is accumulating evidence that genetic polymorphism in genes encoding components of the PLAU-PLAUR regulatory network constitute risk factors for the development of COPD e.g. polymorphisms spanning Serpin peptidase inhibitor, clade E (SERPINE2) have shown significant association with COPD susceptibility, FEV1 and FEV1/FVC [31]. The SERPINE2 gene encodes a PLAU inhibitor providing further support for the role of the PLAU-PLAUR pathway in COPD pathogenesis.

The complex role of the PLAU-PLAUR pathway in the airways. PLAUR is a complex, multi-domain (D1, 2, 3) molecule and exists as a membrane bound GPI linked protein and in multiple soluble forms. The interaction between PLAU-PLAUR is critical for the conversion of plasminogen to plasmin and is regulated by a series of proteins including; PAI-1, PAI-2 and SERPINE2 which have previously been implicated in COPD pathogenesis [16, 31]. Plasmin has many downstream proteolytic effects including those common to remodelling of the airways e.g. MMP activation. In addition PLAUR interacts with several membrane receptors leading to the activation of signalling cascades resulting in alterations in; proliferation, migration, adhesion, endocytosis and cytoskeletal changes. PLAUR also exists in multiple soluble forms (sPLAUR) generated by splicing and/or proteolytic cleavage implicated in chemotaxis and interactions with the extracellular matrix [5, 35, 36].
Chromosome 19q13 has previously been investigated for genetic determinants influencing COPD susceptibility. In particular one of five tested SNPs spanning Transforming Growth Factor β1 TGFB1 (at 41.8 Mbp) was associated with severe COPD in case (n = 304) control (n = 441) analyses and airway obstruction in family (77 pedigrees) analyses (p < 0.05) [10]. Similarly, one of four tested SNPs within latent transforming growth factor beta binding protein 4, LTB4P (at 41.8 Mbp) was associated (p = 0.01) with a densiometric emphysema distribution phenotype (Basal 1/3 lower lobe) [32] but not lung function [33] in 282 and 304 subjects respectively from the National Emphysema Trial. Overall, therefore the evidence that TGFB1 and LTB4P underlie the linkage with FEV1 and FEV1/FVC observed to the region is underwhelming. Importantly, in our asthma analyses we excluded these regions as containing the main dominants of lung function using a combination of linkage and association analyses [3]. However, it is also important to note that in COPD the linkage peak may involve genetic determinants in multiple genes underlying the signal.
This study extends our asthma association and has several strengths including extensive evaluation of polymorphic variation spanning the PLAUR region to examine COPD susceptibility and the examination of dichotomous and continual lung function traits. However, the limitations of this study include the absence of an independent replication sample, and that some of the dichotomous analyses were based on small numbers. The low number of controls versus cases in the dichotomous COPD analysis may at least in part explain the lack of concordance in findings generated using the continual lung function traits which provided more clear evidence for association. It is also important to note that the current study examines the role of PLAUR SNPs in the development of airway obstruction in smokers and does not test the role of PLAUR polymorphisms in non smoking individuals.
Conclusion
We provide preliminary evidence that PLAUR SNPs influence baseline lung function in smokers. However, our data does not support a significant role for PLAUR SNPs contributing to the multiple genetic factors that predispose smokers to develop COPD.
References
Sayers I, Beghe B, Holloway J, Holgate S: Genetics of Asthma: what's new?. Asthma: Critical Debates. Edited by: Johnston SHS. 2002, Oxford: Blackwell Science Ltd, 138-168.
Wood AM, Stockley RA: The genetics of chronic obstructive pulmonary disease. Respir Res. 2006, 7: 130-10.1186/1465-9921-7-130.
Barton SJ, Koppelman GH, Vonk JM, Browning CA, Nolte IM, Stewart CE, Bainbridge S, Mutch S, Rose-Zerilli MJ, Postma DS, et al: PLAUR polymorphisms are associated with asthma, PLAUR levels and lung function decline. J Allergy Clin Immunol. 2009, 123: 1406-1415. 10.1016/j.jaci.2009.03.014.
Dear AE, Medcalf RL: The urokinase-type-plasminogen-activator receptor (CD87) is a pleiotropic molecule. Eur J Biochem. 1998, 252 (2): 185-193. 10.1046/j.1432-1327.1998.2520185.x.
Blasi F, Carmeliet P: uPAR: a versatile signalling orchestrator. Nat Rev Mol Cell Biol. 2002, 3 (12): 932-943. 10.1038/nrm977.
Kruithof EK: Plasminogen activator inhibitors--a review. Enzyme. 1988, 40 (2-3): 113-121.
Jeffery P: Structural alterations and inflammation of bronchi in asthma. Int J Clin Pract Suppl. 1998, 96: 5-14.
Hogg J: Peripheral lung remodelling in asthma and chronic obstructive pulmonary disease. Eur Respir J. 2004, 24 (6): 893-894. 10.1183/09031936.04.00110704.
Silverman EK, Palmer LJ, Mosley JD, Barth M, Senter JM, Brown A, Drazen JM, Kwiatkowski DJ, Chapman HA, Campbell EJ, et al: Genomewide linkage analysis of quantitative spirometric phenotypes in severe early-onset chronic obstructive pulmonary disease. Am J Hum Genet. 2002, 70 (5): 1229-1239. 10.1086/340316.
Celedon JC, Lange C, Raby BA, Litonjua AA, Palmer LJ, DeMeo DL, Reilly JJ, Kwiatkowski DJ, Chapman HA, Laird N, et al: The transforming growth factor-beta1 (TGFB1) gene is associated with chronic obstructive pulmonary disease (COPD). Hum Mol Genet. 2004, 13 (15): 1649-1656. 10.1093/hmg/ddh171.
Viegi G, Pistelli F, Sherrill DL, Maio S, Baldacci S, Carrozzi L: Definition, epidemiology and natural history of COPD. Eur Respir J. 2007, 30 (5): 993-1013. 10.1183/09031936.00082507.
Celli BR, MacNee W: Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004, 23 (6): 932-946. 10.1183/09031936.04.00014304.
Barrett JC, Fry B, Maller J, Daly MJ: Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005, 21 (2): 263-265. 10.1093/bioinformatics/bth457.
Soravia E, Grebe A, De Luca P, Helin K, Suh TT, Degen JL, Blasi F: A conserved TATA-less proximal promoter drives basal transcription from the urokinase-type plasminogen activator receptor gene. Blood. 1995, 86 (2): 624-635.
Smith N, Browning CA, Duroudier N, Stewart C, Peel S, Swan C, Hall IP, Sayers I: Salmeterol and cytokines modulate inositol-phosphate signalling in human airway smooth muscle cells via regulation at the receptor locus. Respir Res. 2007, 8: 68-10.1186/1465-9921-8-68.
Xiao W, Hsu YP, Ishizaka A, Kirikae T, Moss RB: Sputum cathelicidin, urokinase plasminogen activation system components, and cytokines discriminate cystic fibrosis, COPD, and asthma inflammation. Chest. 2005, 128 (4): 2316-2326. 10.1378/chest.128.4.2316.
Wang IM, Stepaniants S, Boie Y, Mortimer JR, Kennedy B, Elliott M, Hayashi S, Loy L, Coulter S, Cervino S, et al: Gene expression profiling in patients with chronic obstructive pulmonary disease and lung cancer. Am J Respir Crit Care Med. 2008, 177 (4): 402-411. 10.1164/rccm.200703-390OC.
Martinez FJ, Curtis JL, Sciurba F, Mumford J, Giardino ND, Weinmann G, Kazerooni E, Murray S, Criner GJ, Sin DD, et al: Sex differences in severe pulmonary emphysema. Am J Respir Crit Care Med. 2007, 176 (3): 243-252. 10.1164/rccm.200606-828OC.
Maclay JD, McAllister DA, Macnee W: Cardiovascular risk in chronic obstructive pulmonary disease. Respirology. 2007, 12 (5): 634-641. 10.1111/j.1440-1843.2007.01136.x.
Tkachuk V, Stepanova V, Little PJ, Bobik A: Regulation and role of urokinase plasminogen activator in vascular remodelling. Clin Exp Pharmacol Physiol. 1996, 23 (9): 759-765. 10.1111/j.1440-1681.1996.tb01177.x.
May AE, Schmidt R, Kanse SM, Chavakis T, Stephens RW, Schomig A, Preissner KT, Neumann F-J: Urokinase receptor surface expression regulates monocyte adhesion in acute myocardial infarction. Blood. 2002, 100 (10): 3611-3617. 10.1182/blood-2002-03-0778.
Wilson LA, Gemin A, Espiritu R, Singh G: ets-1 is transcriptionally up-regulated by H2O2 via an antioxidant response element. FASEB J. 2005, 19 (14): 2085-2087.
Dekhuijzen PN, Aben KK, Dekker I, Aarts LP, Wielders PL, van Herwaarden CL, Bast A: Increased exhalation of hydrogen peroxide in patients with stable and unstable chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1996, 154 (3 Pt 1): 813-816.
Loukides S, Bouros D, Papatheodorou G, Panagou P, Siafakas NM: The relationships among hydrogen peroxide in expired breath condensate, airway inflammation, and asthma severity. Chest. 2002, 121 (2): 338-346. 10.1378/chest.121.2.338.
Watabe T, Yoshida K, Shindoh M, Kaya M, Fujikawa K, Sato H, Seiki M, Ishii S, Fujinaga K: The Ets-1 and Ets-2 transcription factors activate the promoters for invasion-associated urokinase and collagenase genes in response to epidermal growth factor. Int J Cancer. 1998, 77 (1): 128-137. 10.1002/(SICI)1097-0215(19980703)77:1<128::AID-IJC20>3.0.CO;2-9.
Nakatsuka H, Sokabe T, Yamamoto K, Sato Y, Hatakeyama K, Kamiya A, Ando J: Shear stress induces hepatocyte PAI-1 gene expression through cooperative Sp1/Ets-1 activation of transcription. Am J Physiol Gastrointest Liver Physiol. 2006, 291 (1): G26-34. 10.1152/ajpgi.00467.2005.
Buchler P, Reber HA, Tomlinson JS, Hankinson O, Kallifatidis G, Friess H, Herr I, Hines OJ: Transcriptional regulation of urokinase-type plasminogen activator receptor by hypoxia-inducible factor 1 is crucial for invasion of pancreatic and liver cancer. Neoplasia (New York, NY). 2009, 11 (2): 196-206.
Lester RD, Jo M, Montel V, Takimoto S, Gonias SL: uPAR induces epithelial-mesenchymal transition in hypoxic breast cancer cells. The Journal of cell biology. 2007, 178 (3): 425-436. 10.1083/jcb.200701092.
Crippa MP: Urokinase-type plasminogen activator. Int J Biochem Cell Biol. 2007, 39 (4): 690-694. 10.1016/j.biocel.2006.10.008.
Kucharewicz I, Kowal K, Buczko W, Bodzenta-Lukaszyk A: The plasmin system in airway remodeling. Thromb Res. 2003, 112 (1-2): 1-7. 10.1016/j.thromres.2003.10.011.
Zhu G, Warren L, Aponte J, Gulsvik A, Bakke P, Anderson WH, Lomas DA, Silverman EK, Pillai SG: The SERPINE2 gene is associated with chronic obstructive pulmonary disease in two large populations. Am J Respir Crit Care Med. 2007, 176 (2): 167-173. 10.1164/rccm.200611-1723OC.
DeMeo DL, Hersh CP, Hoffman EA, Litonjua AA, Lazarus R, Sparrow D, Benditt JO, Criner G, Make B, Martinez FJ, et al: Genetic determinants of emphysema distribution in the national emphysema treatment trial. Am J Respir Crit Care Med. 2007, 176 (1): 42-48. 10.1164/rccm.200612-1797OC.
Hersh CP, Demeo DL, Lazarus R, Celedon JC, Raby BA, Benditt JO, Criner G, Make B, Martinez FJ, Scanlon PD, et al: Genetic association analysis of functional impairment in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006, 173 (9): 977-984. 10.1164/rccm.200509-1452OC.
Stewart CE, Sayers I: Characterisation of urokinase plasminogen activator receptor variants in human airway and peripheral cells. BMC Molecular Biology. 2009, 10: 75-10.1186/1471-2199-10-75.
Blasi F: uPA, uPAR, PAI-1: key intersection of proteolytic, adhesive and chemotactic highways?. Immunol Today. 1997, 18 (9): 415-417. 10.1016/S0167-5699(97)01121-3.
Blasi F: The urokinase receptor. A cell surface, regulated chemokine. Apmis. 1999, 107 (1): 96-101. 10.1111/j.1699-0463.1999.tb01531.x.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/10/112/prepub
Acknowledgements
This work is supported by the Medical Research Council (New Investigator Award to I. Sayers). We thank A. P. Henry for extracting DNA for the Nottingham populations and J. Blakey, A. Tulah, K. Al Balushi and A. P. Henry for generating the Nottingham phenotype database. We also thank Professor Moira White (University of Sheffield) for her contribution to the collection of COPD subjects and Sarah Mellor and Maureen Hill who helped to co-ordinate the COPD cohort DNA collection.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
IS designed the study, completed the statistical analyses and drafted the manuscript. CES completed the gene sequencing and promoter analyses. IPH, SGH, MFM, AJW, MJC and CR recruited and clinically characterised subjects. All authors contributed to the final version of the manuscript.
Electronic supplementary material
12881_2009_526_MOESM1_ESM.doc
Additional file 1: COPD Severity Analyses (GOLD classification). This file contains phenotypic characteristics of GOLD stratified COPD subjects and association analyses for PLAUR SNPs with disease severity. (DOC 156 KB)
12881_2009_526_MOESM2_ESM.doc
Additional file 2: Baseline Lung Function Analyses. This file contains details of linear regression analyses for baseline lung function (FEV1, FEV1/FVC) for PLAUR SNPs. (DOC 156 KB)
12881_2009_526_MOESM3_ESM.doc
Additional file 3: PLAUR 5'region sequencing and bioinformatics analyses. This file contains details of additional sequencing of the PLAUR promoter region including the identification of novel SNPs and a bioinformatics analysis to identify putative transcription factor changes resulting from SNPs. (DOC 68 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Stewart, C.E., Hall, I.P., Parker, S.G. et al. PLAURpolymorphisms and lung function in UK smokers. BMC Med Genet 10, 112 (2009). https://doi.org/10.1186/1471-2350-10-112
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-10-112