
Abstract
Background
The pathogenesis of severe acute respiratory disease syndrome (SARS) is not fully understood. One case-control study has reported an association between susceptibility to SARS and mannan-binding lectin (MBL) in China. As the downstream protein of MBL, variants of the MBL-associated serine protease-2 (MASP2) gene may be associated with SARS coronavirus (SARS-CoV) infection in the same population.
Methods
Thirty individuals with SARS were chosen for analysis of MASP2 polymorphisms by means of PCR direct sequencing. Tag single nucleotide polymorphisms (tagSNPs) were chosen using pairwise tagging algorithms. The frequencies of four tag SNPs (rs12711521, rs2261695, rs2273346 and rs7548659) were ascertained in 376 SARS patients and 523 control subjects, using the Beckman SNPstream Ultra High Throughput genotyping platform.
Results
There is no significant association between alleles or genotypes of the MASP2 tagSNP and susceptibility to SARS-CoV in both Beijing and Guangzhou populations. Diplotype (rs2273346 and rs12711521)were analyzed for association with susceptibility to SARS, no statistically significant evidence of association was observed. The Beijing and Guangzhou sample groups were homogeneous regarding demographic and genetic parameters, a joined analysis also showed no statistically significant evidence of association.
Conclusion
Our data do not suggest a role for MASP2 polymorphisms in SARS susceptibility in northern and southern China.
Similar content being viewed by others


Background
Severe acute respiratory disease syndrome (SARS), a new and highly infectious disease that is caused by a previously undescribed coronavirus in humans, has created a major public health threat in many countries [1–3]. Progress has been made in understanding SARS coronavirus (SARS-CoV) and the epidemiology, clinical manifestations, laboratory findings and radiological features of this disease have all been studied in detail [4, 5]. However, its pathogenesis is still not fully understood. It has been reported that diabetes mellitus and heart disease are risk factors for adverse prognosis of SARS [6], however, little is known about the contribution of genetic factors.
SARS has been found to have a profoundly adverse effect on the immune system [7]. Variation in host immunity may be one of the important factors that determine susceptibility to SARS. A few case-control studies have reported an association between SARS susceptibility and human leucocyte antigen (HLA) and MBL [8–11]. Deficiency of MBL, a key component of the innate immune system, has been detected in SARS patients. Such a deficiency may increase susceptibility to SARS infection. Three papers reported the association of HLA and susceptibility to SARS [8–10]. Of these, two reported the association of HLA with susceptibility and resistance to the development of SARS [8, 9]. In a more recent paper, no statistical significance was found with the susceptibility and severity of the disease [10].
MBL is a member of the collectin family and plays an important role in innate immunity [12, 13]. MBL and ficolins distinguish between self, non-self and altered-self by recognizing patterns of ligands on the surface of microorganisms or aberrant cells. When this happens, MBL-associated serine protease-2 (MASP-2) is activated and cleaves complement factors to initiate the antibody-independent pathway of the complement system, thus starting inflammatory reactions.
MBL-associated serine proteases interact with MBL via the collagenous region of larger MBL oligomers. Four related proteins derived from two genes have been reported; namely MASP1, its alternative splicing variant MASP3, and MASP2 with its alternative splicing variant Map19 [14–16]. MASP2 activates the complement system by cleaving complement proteins C4 and C2. MASP2 is an essential component of the lectin pathway of complement activation.
MASP2 deficiency is observed because of genetic polymorphisms. It is known that a MASP2 polymorphism, namely D120G has a functional effect on the protein, and does not allow the formation of an active MBL-MASP complex. This variant has been described for the first time in a patient with an inherited deficiency of MASP2, who was characterized by augmented susceptibility to infection and development of immunological disease.
With regard to SARS-CoV infection, the codon 54 variant of the MBL gene has been shown to be associated with infection susceptibility but not with disease severity [11]. As the downstream protein of MBL, variants of the MASP2 gene may be associated with SARS-CoV infection. To examine the hypothesis that polymorphisms of the MASP2 gene in SARS patients are genetic factors that influence infection susceptibility, we studied MASP2 gene polymorphisms in DNA from two groups of Chinese SARS patients, and compared these with normal blood donors from the same region.
Methods
Recruitment of subjects for case-control study
This study was performed with the approval of the Ethical Committee of the Chinese Human Genome Center. A total of 376 SARS patients, who were diagnosed based on WHO criteria, were recruited during the 2003 outbreak. All SARS patients selected for study were unrelated and were shown to be seropositive by anti-SARS-N protein ELISA and immunofluorecent assay (IFA). The specificity of the ELISA assay is more than 99% [17, 18]. Of these, 272 were from the wards at Xiaotangshan Hospital, Beijing, China, and the remaining 104 were from the Eighth People's Hospital of Guangzhou in southern China. A total of 523 age, sex, and ethnicity-matched healthy, genetically unrelated and seronegative (confirmed by anti-SARS N protein antibody ELISA and SARS antibody IFA) adults were recruited as control subjects. We extracted genomic DNA from peripheral blood leukocytes of affected individuals and controls using the QIAamp DNA Mini Kit (Qiagen, Valencia, CA). DNA was quantified using standardized fluorometric reading on a DU 650 spectrophotometer (Beckman Coulter, Fullerton, CA). Each sample was diluted to a final concentration of 10 ng/ul.
Discovery of polymorphisms
Genomic DNA from 30 individuals with SARS was chosen for analysis of MASP2 gene polymorphisms. The sample included 60 chromosomes, which provided a 95% confidence level to detect alleles with a frequency >5% [19]. With a candidate-gene strategy, we screened for polymorphisms in all exons (~2.0 kb), 5'- and 3'-flanking regions (lengths of 2.5 kb), untranslated regions (~0.3 kb), and about 2.5 kb intronic sequences. DNA sequence spanning the MASP2 gene was obtained from the National Center for Biotechnology Information (NCBI) website (available from http://www.ncbi.nlm.nih.gov/; NT_021937.18). Primers were designed using the Premier program (Primer Premier 5). PCR primer sequences see Additional file 1. DNA samples from the 30 individuals were amplified and purified. The PCR reaction volume was 50 μl, containing 10–50 ng of DNA,10 pmol of each forward and reverse primer, 0.2 mmol/L of each dNTP,0.3 U of DNA polymerase(Tiangen Biotec Co, Beijing, China),10 mmol/L of Tris-HCl, 1.5 mmol/L of MgCl2, 50 mmol/L of KCl, and 0.1% Triton X-100. Amplication was carried out in a GeneAmp PCR system 9700(ABI) with cycle parameters of 5 min 94°C (initial denaturation), 32 rounds of 94°C 30 s, 54–55°C 30 s, and 72°C 45 s, and a final extention for 5 min at 72°C. PCR products were sequenced using an ABI PRISM Dye Terminator Sequencing Kit with AmpliTaq Gold DNA Polymerase (Applied Biosystems Division/Perkin-Elmer, Foster City, CA) and loaded onto an ABI 3100 sequencer. Polymorphism candidates were identified by the PolyPhred program. (P. Green, personal communication) SNP genotypes were verified by manual evaluation of the individual sequence traces.
Genotyping
Genotyping was completed using SNPstream Ultra High Throughput genotyping system (Beckman Coulter, Fullerton, CA) according to the manufacture's instructions. Briefly, the method combines solution-phase multiplex single nucleotide extension (SNE) with a solid-phase sorting of labeled SNE primers by hybridization to a chip that contains 384 4 × 4 arrays of 12 oligo-nucleotide tags and 4 oligo-nucleotides for positive and negative controls. Each SNE primer contained 1 of the 20 oligonucleotide tags at its 5' end, and the SNE reactions were performed in 12-plex. The microarray plate was imaged by the SNPscope reader (Beckman Coulter, Fullerton, CA). The two-color system allowed the detection of the SNP by comparing signals from the two fluorescent dyes. The image signals were then transferred to genotyping software that translated the images of the arrays into genotype calls. The error rate based upon sequencing for 10% of the SNPs examined in the present study was 0–1.2%.
Statistical analysis
The Exact Test was calculated to evaluate if the population was in Hardy-Weinberg equilibrium by the Markov chain method, taking P < 0.05 as the cutoff for assessing significance. The tagSNPs were chosen on a pairwise basis and linkage disequilibrium (LD) calculation was performed on the confidence interval basis using Haploview 3.2 software.
Genotype and alleles frequencies for each polymorphism were determined by gene counting. Diplotypes of each individual were inferred using the algorithm developed by Stephens et al. (2001) (PHASE). The chi square test was used to determine whether allele frequencies differed between SARS cases and controls. Binary logistic regression was used to analyze the association between a single locus and SARS susceptibility, adjusted for sex and age status, and odds ratio and 95% CI were used to measure strength of association in a genetic risk association study. These statistical analyses were implemented in Statistical Package for the Social Sciences 13.0 software (SPSS Inc, Chicago, Illinois, USA).
Results
Demographic characteristics of the population
Demographic characteristics are shown in Table 1. The age and sex distribution of the patients and controls were not significantly different, which indicated that the matching of controls to cases was adequate.
Screening MASP2for polymorphisms
Sequencing of the 11 exons of MASP2, the 5' and 3' regions of the gene, and some intronic sequences in 30 individuals with SARS identified 17 polymorphisms (Table 2). Eleven of the SNPs have been published in the dbSNP database http://www.ncbi.nlm.nih.gov/SNP/index.html. Allele and genotype frequencies were consistent with those expected under Hardy-Weinberg equilibrium. Nine SNPs (allele frequency >5%) were chosen with Haploview for assessment. The SNPs were contained in two blocks of LD (Fig. 1), as defined by Lewontin's| D'|. SNPs including rs7548659, rs2273347 and rs6695096 were located outside of the defined LD blocks. Four tag-SNPs were chosen with the pairwise tagging algorithm implemented in the Tagger program of Haploview: rs7548659, rs12711521, rs2273346 and rs2261695 (r2 threshold was 0.8). Genotype frequencies of the tag-SNP except rs2261695 in other populations that have been published in the HapMap database were shown in table 3 as control. http://www.hapmap.org/cgi-perl/gbrowse/hapmap3_B36/. The four TagSNPs were genotyped in SARS patients and controls with an average success rate of 96%.

Gene content of NC_000001.9in chromosome 1p36, discovered SNPs and LD of MASP2. (a) Genomic structure of genes in this region. (b) Exons of MASP2 and the position of SNPs discovered. (c) Pairwise LD between SNPs (MAF >0.05) at this gene. The value within each diamond represents the pairwise correction between SNPs (measured as D') defined by the upper left and upper right sides of the diamond. The diamond without a number corresponds to D' = 1. Shading represents the magnitude and significance of pairwise LD, with a red-to-white gradient reflecting higher to lower LD values.
MASP2polymorphisms and susceptibility to SARS-CoV
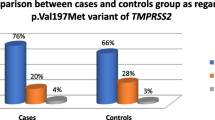
Genotype data and diplotype (rs2273346 and rs12711521) were analyzed for association with susceptibility to SARS, in Beijing and Guangzhou populations, using Binary logistic regression for overall genotypic association (Tables 4 and 5). No statistically significant evidence of association was observed. The Beijing and Guangzhou sample groups were homogeneous regarding demographic and genetic parameters, so a joined analysis was done, No statistically significant evidence of association was observed (Table 6).
Discussion
Possible contribution of host genetic factors to the susceptibility and outcome of SARS-CoV infection has been investigated through several association studies [20–23], MBL deficiency because of polymorphisms in the MBL gene has been shown to be involved in SARS-CoV infection. As the downstream protein of MBL, improper MASP activity can interfere with complement functions [24]. there is an association between the genetics of MASP2 and its serum level [24–27]. Thiel et al. [24] have analyzed the mutation of p.156_159-dupCHNH and SNPs p.R99Q, p.R118C, p.D120G, p.P126L and p.V377A in four populations: Africans from Zambia, Hong Kong Chinese, Brazilian Amerindians and Danish Caucasians. p.156_159-dupCHNH was only found in Chinese (gene frequency 0.26%), associated with low levels of MASP2, and p.D120G was found only in Caucasians. p.P126L and p.R99Q were present in Africans and Amerindians only. MASP2 levels were reduced in individuals with p.V377A (rs2273346). Therefore, we chose 30 individuals with SARS from Beijing for analysis of MASP2 gene polymorphisms. The sample included 60 chromosomes, which provided a 95% confidence level to detect alleles with a frequency >5%. However, we only observed the SNP rs2273346 (p.V377A) among those mentioned in the study of Thiel et al. We analyzed four tagSNPs in the MASP2 gene in two different SARS patients and controls, searching for a possible genotype-phenotype correction, but no statistically significant difference was found for any polymorphisms between the different groups genotyped, while the possible role of the rare variants is to be determined.
Conclusion
Our analysis indicates that MASP2 polymorphisms is not directly related to SARS-CoV susceptibility in northern and southern Chinese.
References
Peiris JS, LAI ST, Poon LL, Guan Y, Yam LY, Lim W, Nicholls J, Yee WK, Yan WW, Cheung MT, Cheng VC, Chan KH, Tsang KN, Yung RW, Ng IK, Yuen KY: Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003, 361: 1319-1325. 10.1016/S0140-6736(03)13077-2.
Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong S, Urbani C, Comer JA, Lim W, Rollin PE, Dowell SF, Ling AE, Humphrey CD, Shieh WJ, Guarner J, Paddock CD, Rota P, Fields B, eRisi JD, Yang JY, Cox N, Hughes JM, LeDuc JW, Bellini WJ, Anderson LJ: A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003, 348: 1953-1966. 10.1056/NEJMoa030781.
Drosten C, Gunther S, Preiser W, Werf van der S, Brodt HR, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RA, Berger A, Burguiere AM, Cinatl J, Eickmann M, Escriou N, Grywna K, Kramme S, Manuguerra JC, Muller S, Rickerts V, Sturmer M, Vieth S, Klenk HD, Osterhaus AD, Schmitz H, Doerr HW: Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003, 348: 1967-1976. 10.1056/NEJMoa030747.
Chan JW, Ng CK, Chan YH, Mok TY, Lee S, Chu SY, Law WL, Lee MP, Li PC: Short term outcome and risk factors for adverse clinical outcomes in adults with severe acute respiratory syndrome(SARS). Thorax. 2003, 58: 686-689. 10.1136/thorax.58.8.686.
World Health Organization: Consensus document on theepidemiology of severe acute respiratory syndrome (SARS). WHO/CDS/CSR/GAR/2003. Geneva. 2003, 11-
Booth CM, Matukas LM, Tomlinson GA: Clinical features and short-term outcomes of 144 patients with SARS in the greater Toronto area. JMAM. 2003, 289: 686-689. 10.1001/jama.289.6.686.
Guo Y, Christine K, McNutt AM, Gu J: Pathogenetic mechanisms of severe acute respiratory syndrome. Virus Res. 2008, 133: 4-12. 10.1016/j.virusres.2007.01.022.
Lin M, Tseng HK, Trejaut JA, Lee HL, Loo JH, Chu CC, Chen PJ, Su YW, Lim KH, Tsai ZU, Lin RY, Lin RS, Huang CH: Association of HLA class I with severe acute respiratory syndrome coronavirus infection. BMC Med Genet. 2003, 4: 9-15. 10.1186/1471-2350-4-9.
Ng MH, Lau KM, Li L, Cheng SH, Chan WY, Hui PK, Zee B, Leung CB, Sung JJ: Association of human-leukocyteantigen class I (B*0703) and class II (DRB1*0301) genotypes with susceptibility and resistance to the development of severe acute respiratory syndrome. J Infect Dis. 2004, 190: 515-518. 10.1086/421523.
Xiong P, Zeng X, Song MS, Jia SW, Zhong MH, Xiao LL, Lan W, Cai C, Wu XW, Gong FL, Wang W: Lack of association between HLA-A, -B and-DRB1 alleles and thedevelopment of SARS: a cohort of 95 SARS-recovered individuals in a population of Guangdong southern China. Int J Immunogenet. 2008, 35: 69-74.
Yuan FF, Tanner J, Chan PK, Biffin S, Dyer WB, Geczy AF, Tang JW, Hui DS, Sung JJ, Sullivan JS: Influence of FcgammaRIIA and MBL polymorphisms on severe acute respiratory syndrome. Tissue Antigens. 2005, 66: 291-296. 10.1111/j.1399-0039.2005.00476.x.
Zhang H, Zhou G, Zhi L, Yang H, Zhai Y, Dong X, Zhang X, Gao X, Zhu Y, He F: Association between mannan-binding lectin genepolymorphisms and susceptibility to severe acute respiratory syndromecoronavirus infection. J Infect Dis. 2005, 192: 1355-1361. 10.1086/491479.
Ihara I, Harada Y, Ihara S, Kawakami M: A newcomplement-dependent bactericidal factor found in nonimmune mouse sera: specific binding to polysaccharide of Ra chemotype Salmonella. J Immunol. 1982, 128: 1256-1260.
Dahl MR, Thiel S, Matsushita M, Fujita T, Willis AC, Christensen T, Vorup-Jensen T, Jensenius JC: MASP-3 and its association with distinct complexes of the mannan-binding lectin complement activation pathway. Immunity. 2001, 15: 127-135. 10.1016/S1074-7613(01)00161-3.
Thiel S, Vorup-Jensen T, Stover CM, Schwaeble W, Laursen SB, Poulsen K, Willis AC, Eggleton P, Hansen S, Holmskov U, Reid KB, Jensenius JC: A second serine protease associated with mannan-binding lectin that activates complement. Nature. 1997, 386: 506-510. 10.1038/386506a0.
Stover CM, Thiel S, Thelen M, Lynch NJ, Vorup-Jensen T, Jensenius JC, Schwaeble WJ: Two constituents of the initiation complex of the mannan-binding lectin activation pathway of complement are encoded by a single structural gene. J Immunol. 1999, 162: 3481-3490.
Liu Xuan, Shi Yulin, Li Ping, Li Linhai, Yi Yanping, Ma Qingjun, Cao Cheng: Profile of antibodies to the nucleocapsid protein of the Severe Acute Respiratory Syndrome (SARS)-associated coronavirus in probable SARS patients. Clin Diagn Lab Immunol. 2004, 11: 227-228.
Shi Y, Yi Y, Li P, Kuang T, Li L, Dong M, Ma Q, Cao C: Diagnosis of severe acute respiratory syndrome (SARS) by detection of SARS coronavirus nucleocapsid antibodies in an antigen-capturing enzyme-linked immunosorbent assay. J Clin Microbiol. 2003, 41: 5781-5782. 10.1128/JCM.41.12.5781-5782.2003.
Kruglyak L, Nickerson DA: Variation is the spice of life. Nat Genet. 2001, 27: 234-6. 10.1038/85776.
Itoyama S, Keicho N, Quy T, Phi NC, Long HT, Ha le D, Ban VV, Ohashi J, Hijikata M, Matsushita I, Kawana A, Yanai H, Kirikae T, Kuratsuji T, Sasazuki T: ACE1 polymorphism and progression of SARS. Biochem Biophys Res Commun. 2004, 323: 1124-1129. 10.1016/j.bbrc.2004.08.208.
Ng MW, Zhou G, Chong WP, Lee LW, Law HK, Zhang H, Wong WH, Fok SF, hai YZ, Yung RW, Chow EY, Au KL, Chan EY, Lim W, Peiris JS, He F, Lau YL: The association of RANTES polymorphism with severe acute respiratory syndrome in Hong Kong and Beijing Chinese. BMC Infect Dis. 2007, 7: 50-57. 10.1186/1471-2334-7-50.
He J, Feng D, de Vlas SJ, Wang H, Fontanet A, Zhang P, Plancoulaine S, Tang F, Zhan L, Yang H, Wang T, Richardus JH, Habbema JD, Cao W: Association of SARS susceptibility with single nucleic acid polymorphisms of OAS1 and MxA genes: a case-control study. BMC Infect Dis. 2006, 6: 106-112. 10.1186/1471-2334-6-106.
Chan VS, Chan KY, Chen Y, Poon LL, Cheung AN, Zheng B, Chan KH, Mak W, Ngan HY, Xu X, Screaton G, Tam PK, Austyn JM, Chan LC, Yip SP, Peiris M, Khoo US, Lin CL: Homozygous L-SIGN (CLEC4M) plays a protective role in SARS coronavirus infection. Nat Genet. 2006, 38: 38-46. 10.1038/ng1698.
Thiel S, Steffensen R, Christensen IJ, Ip WK, Lau YL, Reason IJ, Eiberg H, Gadjeva M, Ruseva M, Jensenius JC: Deficiency of mannan-binding lectin associated serine protease-2 due to missense polymorphisms. Genes Immun. 2007, 8: 154-163. 10.1038/sj.gene.6364373.
Sørensen R, Thiel S, Jensenius JC: Mannan-binding-lectin-associated serine proteases, characteristics and disease associations. Springer Semin Immunopathol. 2005, 27: 299-319. 10.1007/s00281-005-0006-z.
Lozano F, Suárez B, Muñoz A, Jensenius JC, Mensa J, Vives J, P J: Horcajada. Novel MASP2 variants detected among North African and Sub-Saharan individuals. Tissue Antigens. 2005, 66: 131-135. 10.1111/j.1399-0039.2005.00436.x.
Sorensen GL, Petersen I, Thiel S, Fenger M, Christensen K, Kyvik KO, Sørensen TI, Holmskov U, Jensenius JC: Genetic influences on mannan-binding lectin (MBL) and mannan-binding lectin associated serine protease-2 (MASP-2) activity. Genet Epidemiol. 2007, 31: 31-41. 10.1002/gepi.20187.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2334/9/51/prepub
Acknowledgements
This investigation was supported by grant 2005AA218080 and 2006AA022203 awarded by "863" High-Tech Development Program of China. The authors thank Dr. Wuchun Cao and Wei Liu for their help in the coordination of this study in China.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
WY have contributed in the preparation of the manuscript and the overall study; YJW and LP performed SNP discovery and data analyses; SYL, YRF and WXY collected and managed DNA samples and clinical information; YX and ZLN have contributed to genotyping; CC, LCX and MQJ performed the data analyses, prepared the manuscript and supervised this study. All authors contribute to writing of the final manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wang, Y., Yan, J., Shi, Y. et al. Lack of association between polymorphisms of MASP2and susceptibility to SARS coronavirus infection. BMC Infect Dis 9, 51 (2009). https://doi.org/10.1186/1471-2334-9-51
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2334-9-51