
Abstract
Background
Cellulose acetate phthalate (CAP), a promising candidate microbicide for prevention of sexual transmission of the human immunodeficiency virus type 1 (HIV-1) and other sexually transmitted disease (STD) pathogens, was shown to inactivate HIV-1 and to block the coreceptor binding site on the virus envelope glycoprotein gp120. It did not interfere with virus binding to CD4. Since CD4 is the primary cellular receptor for HIV-1, it was of interest to study CAP binding to HIV-1 complexes with soluble CD4 (sCD4) and its consequences, including changes in the conformation of the envelope glycoprotein gp41 within virus particles.
Methods
Enzyme-linked immunosorbent assays (ELISA) were used to study CAP binding to HIV-1-sCD4 complexes and to detect gp41 six-helix bundles accessible on virus particles using antibodies specific for the α-helical core domain of gp41.
Results
1) Pretreatment of HIV-1 with sCD4 augments subsequent binding of CAP; 2) there is synergism between CAP and sCD4 for inhibition of HIV-1 infection; 3) treatment of HIV-1 with CAP induced the formation of gp41 six-helix bundles.
Conclusions
CAP and sCD4 bind to distinct sites on HIV-1 IIIB and BaL virions and their simultaneous binding has profound effects on virus structure and infectivity. The formation of gp41 six-helical bundles, induced by CAP, is known to render the virus incompetent for fusion with target cells thus preventing infection.
Similar content being viewed by others


Background
Cellulose acetate phthalate (CAP) is a promising microbicide candidate for prevention of infection by sexually transmitted disease (STD) pathogens, including HIV-1 [1–7]. CAP inactivates HIV-1 and blocks the coreceptor binding site on the virus envelope glycoprotein gp120, while leaving the site for the primary cellular receptor CD4 accessible [8, 9] Soluble CD4 (sCD4) was shown to inhibit HIV-1 infection by two mechanisms: reversible blockage of virus binding to receptors, and irreversible inactivation of virus infectivity [10]. Since CAP and sCD4 bind to distinct domains on the HIV-1 envelope, it was of interest to determine whether or not these two ligands affect virus infectivity synergistically as do other combinations of anti-HIV-1 drugs and sCD4 [11, 12] Binding of sCD4 leads to conformational changes in gp120 [13–17]. Binding of gp120 to coreceptors CXCR4 and CCR5, respectively, triggers additional conformational changes in HIV-1 envelope glycoproteins [18, 19] For these reasons it was of interest to determine whether a) pretreatment of HIV-1 with sCD4 would affect subsequent binding of CAP to virus particles, and b) CAP binding to virus particles in the presence or absence of sCD4 would elicit conformational changes which could affect HIV-1 infectivity. Such studies were expected to elucidate further the mechanisms involved in the antiviral/virucidal activity of CAP and to contribute to the potential development of microbicides combining two or more anti-HIV-1 compounds with distinct target sites.
Methods
Reagents
The following monoclonal antibodies (mAbs) were used: NC-1, a mouse mAb raised against the gp41 six-helix bundle from HIV-1 IIIB [20]; and anti-p24 mAb (ImmunoDiagnostics, Inc., Woburn, MA). Rabbit antibodies against the gp41 six-helix bundle were prepared as described [21]. Rabbit antiserum against HIV-1 IIIB gp120 was prepared as described [22] and shown to cross-react with HIV-1 BaL (own unpublished data). Recombinant soluble CD4 (sCD4) was from Genentech Inc., South San Francisco, CA. Recombinant HIV-1 IIIB gp120, biotinylated gp120 and biotinylated sCD4 were from ImmunoDiagnostics, Inc., Woburn, MA. Purified recombinant protein A/G was from Pierce, Rockford, IL. Pelletted, 1000-fold concentrates of HIV-1 IIIB (6.8 × 1010 virus particles/ml) and BaL (2.47 × 1010 virus particles/ml) [23] were from Advanced Biotechnologies, Inc., Columbia, MD. Biotin labeled goat anti-mouse IgG and anti-rabbit IgG were from Roche Diagnostics Corporation, Indianapolis, IN. Chicken serum was from OEM Concepts, Toms River, NJ. Antiserum to phthalate was prepared by immunization of rabbits with phthalic anhydride treated rabbit serum albumin [24]. Horseradish peroxidase (HRP) labeled streptavidin was from Zymed, South San Francisco, CA. HRP was quantitated using a kit from Kirkegaard & Perry Laboratories, Inc., Gaithersburg, MD. Enzyme linked immunosorbent assay (ELISA) kits for the HIV-1 p24 antigen were from Beckman Coulter, Inc., Miami, FL. The tyrosine-sulfated peptide from CCR5 [25]; {S-peptide; MDYQVSSPIYDINYYTSEPSQK; (Y = sulfotyrosine)} was from American Peptide, Sunnyvale, CA. The corresponding control peptide with tyrosines instead of sulfotyrosines, and N36 (SGIVQQQNNLLRAIEAQQHLLQLTVWGIKQLQARVL) and C34 (WIEWDREINNYTSIIYSLIEESQNQQEKNEQELL) peptide constituents of the gp41 core [20, 21] of HIV-1 BaL were from AnaSpec, Inc., San Jose, CA. CAP was a gift from Eastman Chemical Company, Kingsport, TN. H9 cells chronically infected with HIV-1 IIIB, and PM1 cells were obtained from the AIDS Research and Reference Reagent Program contributed by Drs. R. Gallo, P. Lusso and M. Reitz, respectively.
Inhibition of HIV-1 infection
HIV-1 IIIB (100 TCID50) in the presence or absence of graded concentrations of virus inhibitors, CAP and sCD4, respectively, in RPMI-1640 medium containing 10% fetal bovine serum (FBS) were mixed with MT-2 cells (104 cells/well) and placed into 96-well polystyrene plates. The mixtures were incubated at 37°C overnight. On the 2nd day, culture supernatants were removed from each well and fresh medium was added. On the 4th day, culture supernatants were collected and tested for p24 antigen by ELISA. Similar experiments were done with HIV-1 BaL (2.5 × 105 virus particles), except that PM1 cells [26] were used instead of MT-2 cells.
The inhibitory activity of CAP and sCD4 in combination, against HIV-1 infection was determined as described above. The CAP:sCD4 weight ratios in the combinations were 10:1 and 2:1 for HIV-1 IIIB and HIV-1 BaL, respectively. The 50% inhibitory concentrations (IC50) and the combination index values (CI) were calculated as previously described [27] using a computer program (CalcuSyn) kindly provided by Dr. T. C. Chou (Sloan-Kettering Cancer Center, New York). A CI value < 1 indicates synergy and that of > 1 indicates antagonism. The compound dose reductions were calculated based on the IC50 value for CAP and sCD4 used alone or in combination [27].
To measure the virucidal activity of CAP/sCD4 combinations, treated and untreated purified HIV-1 IIIB (3.4 × 108 virus particles) (unlike in the experiments described in an earlier report [8] where infectious tissue culture medium was used instead), were serially diluted 2- to 256-fold in RPMI-1640 medium containing 10% FBS, mixed with MT-2 cells and placed into 96-well polystyrene plates. Virus replication was monitored by measuring p24 antigen as described above. Similar experiments were done with HIV-1 BaL (2.5 × 108 virus particles), except that PM1 cells were used instead of MT-2 cells. The percentage of residual infectivity after CAP/sCD4 treatment was calculated from calibration curves relating absorbance to virus dilutions of untreated viruses.
Enzyme-linked immunosorbent assays (ELISA)
For virus capture assays, wells of 96-well polystyrene plates (Immulon II; Dynatech Laboratories Inc., Chantilly, VA) were coated either with CAP (1 μg/well). or with mAb NC-1 [20, 21]. For coating with CAP, a solution of CAP (100 μl; 10 μg/ml in 0.05 M acetate pH 6.0) was added to the wells. After incubation overnight at 4°C, the wells were washed and postcoated for 1 h at 20°C with bovine serum albumin (BSA) and gelatin (1 and 0.1 mg/ml in 0.05 M acetate pH 6.0). For coating with antibodies, wells were first coated with protein A/G (1 μg/well) in 0.1 M Tris buffer, pH 8.8 for 2 h at 20°C, followed by mAb NC-1 or normal mouse IgG (= control wells) {1 μg/well; diluted in phosphate buffered saline (PBS)} for 1 h at 20°C. Coating with the N36/C34 peptide complexes (0.01 to 10 μM) was done under conditions described for protein A/G. Subsequently the wells were washed and postcoated with BSA and gelatin as described above, except that these proteins were dissolved in 0.14 M NaCl, 0.01 M Tris, pH 7.0 (TS). Chicken serum (10%) in PBS (Ch-PBS) was used instead in experiments with HIV-1 BaL to suppress binding of this virus to control wells. The wells were washed with TS and stored at 4°C. HIV-1 virus particles suspended in diluents and treated as indicated in legends to Figs. 2, 3, 5 were added to the wells for 5 h at 4°C. Subsequently the wells were washed 10× with ice cold PBS or 1:50 anti-p24 mAb in Ch-PBS for HIV-1 BaL to minimize the contribution of p24 antigen not associated with virus particles to absorbance readings corresponding to p24 antigen released from detergent treated virus. The washed wells with bound virus particles were then treated with lysis buffer (1% Nonidet P40 {NP40}, 100 μg/ml BSA in PBS) for 30 min at 37°C. The supernatants were removed and tested for p24 antigen using ELISA kits from Beckman Coulter, Inc. following the manufacturer's protocol.

Binding of biotinyl-gp120, biotinyl-sCD4 and biotinyl-gp120-sCD4 complexes to CAP coated wells. Graded quantities of biotinyl-gp120 (indicated on the abscissa) and of biotinyl-gp120-sCD4 complexes were added to CAP coated and control wells. The complexes were prepared by mixing 1 μg of biotinyl-gp120 with 500 ng of sCD4 for 30 min at 20°C in PBS. To determine the effect of sCD4/gp120 ratios on gp120 binding to CAP, constant amounts (500 ng) of biotinyl-gp120 were mixed with graded quantities of sCD4 (0 to 1,000 ng) (insert) and the mixtures were further handled as described above. Binding of biotinyl-gp120 did not increase at sCD4/biotinyl-gp120 weight ratios >1 (data not shown). In control experiments, graded quantities of biotinyl-sCD4 in the absence of gp120 were added to the wells. After incubation at 4°C overnight, the wells were washed and bound biotinylated proteins were detected from subsequent binding of HRP-streptavidin. Absorbance corresponding to biotinylated proteins bound to control wells was in the range of 0–0.026 and was substracted from the absorbance corresponding to biotinylated proteins bound to CAP coated wells.

Binding of HIV-1 and HIV-1-CD4 complexes to CAP coated wells. Suspensions (50 μl) of purified HIV-1 IIIB (6.8 × 109 virus particles/ml) and of HIV-1 BaL (2.5 × 109 virus particles/ml), respectively, in 0.1 M sodium acetate pH 7.0 were mixed with 5 μg of sCD4 and incubated for 30 min at 20°C. sCD4 was not added to control virus preparations. The suspensions were cooled on ice and polyethylene glycol 6000 (PEG) was added to a final concentration of 3% to separate HIV-1 from sCD4 (which does not precipitate in 3% PEG). After 90 min at 4°C, the mixtures were centrifuged at 10,000 rpm, the supernatant fluids removed and the pellets washed once with 3% PEG in PBS containing 10 mg/ml BSA. The pellets were resuspended in sodium acetate buffer pH 7.0 containing 25 μg/ml BSA, and serial dilutions (indicated on the abscissa) were added to CAP coated and control wells, respectively. After incubation for 5 h at 4°C, the supernatant fluids were removed, the wells washed, and the bound virus quantitated by ELISA for p24 antigen. The amount of HIV-1 IIIB and HIV-1 BaL bound to control wells was ≤ 2.1% and ≤ 13%, respectively, of that bound to CAP coated wells. All experiments were done at least in triplicate.

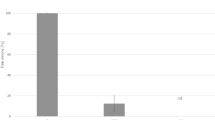
Synergism between CAP and sCD4 for virucidal activity against HIV-1 IIIB. sCD4 was added to 50 μl of HIV-1 IIIB (6.8 × 109 virus particles/ml) in PBS. After 30 min at 20°C, CAP was added to final concentrations between 0 and 1250 μg/ml and the mixtures were incubated for 5 min at 37°C. CAP at the same concentrations described above was also added to virus not pretreated with sCD4. Virus suspensions were put on ice and PEG was added to a final concentration of 3% to separate HIV-1 from CAP and sCD4. After 90 min at 4°C, the mixtures were centrifuged at 10,000 rpm, the supernatant fluids removed and the pellets washed twice with 3% PEG in PBS containing 10 mg/ml BSA. The final washed pellets were resuspended in tissue culture medium, and serial 2-fold dilutions in the same medium were added to MT-2 cells. Infection was monitored by ELISA for p24 antigen. The gray horizontal line corresponds to virus inactivation (80.2 ± 1.3% of residual infectivity) caused by treatment with sCD4 in the absence of CAP. All experiments were done at least in triplicate.

Immunological cross-reactivity between six-helix bundle structures derived from HIV-1 IIIB and BaL. Serial two-fold dilutions of mAb NC-1, raised against HIV-1 IIIB gp41 six-helix bundles, were added to wells coated with the respective N36/C34 peptide complexes [20, 21] and binding of the IgG antibodies was determined as described in Methods. Normal mouse IgG was used in control experiments instead of mAb NC-1. Dilutions of the respective gp41 six-helix bundles were also tested in a sandwich ELISA (insert). The absorbance for controls in the absence of gp41 bundles was 0.

Treatment of HIV-1 with CAP enhances the expression of binding sites for mAb NC-1. Purified HIV-1 IIIB (6.8 × 109 virus particles) and HIV-1 BaL (2.5 × 109 virus particles), respectively, each in 100 μl PBS were incubated for 30 min at 20°C in the presence (5 μg) or absence of sCD4. Each sample was divided into two equal portions. CAP in 0.1 M acetate pH 6.0 was added to one aliquot (final concentration 5 mg/ml). An equivalent amount of acetate without CAP was added to the second aliquot. After incubation for 5 min at 37°C, the suspensions were cooled on ice and PEG was added to a final concentration of 3% to separate HIV-1 from sCD4 and CAP. After 90 min at 4°C, the mixtures were centrifuged at 10,000 rpm, the supernatant fluids removed and the pellets washed once with 3% PEG in PBS containing 10 mg/ml BSA. The final washed pelleted viruses were suspended in PBS containing 100 μg/ml of each BSA and gelatin, and dilutions (indicated on the abscissa) in the same buffer were added to mAb NC-1 coated and control wells. After 5 h at 4°C, the wells were washed and bound virus was quantitated by ELISA for p24 antigen. The amount of HIV-1 IIIB and HIV-1 BaL, respectively, bound to control wells was ≤ 2.1% and ≤ 6.1% of that bound to mAb NC-1 coated wells. All experiments were done at least in triplicate.
To measure the binding of biotinyl-sCD4 and biotinyl-gp120 (in the presence and absence of sCD4), respectively, to CAP, the biotinylated proteins were added to CAP and control postcoated wells without CAP, respectively, at dilutions shown in Fig. 1. The binding of these biotinylated proteins to the wells, after washing with TS, was determined by adding HRP-streptavidin (1 μg/ml) in TS containing 0.25% gelatin and 0.05 % Tween 20 for 30 min at 37°C. The wells were washed and bound HRP was detected using the test kit from Kirkegaard & Perry following the manufacturer's protocol, and the absorbance was read at 450 nm.
The sandwich ELISA for the gp41 six-helix bundle was performed as described [21]. Treated and control virus preparations were incubated with lysis buffer for 30 min at 20°C and then added to wells coated with rabbit polyclonal antibodies to the gp41 core. In control experiments, CAP alone (5 mg/ml in lysis buffer) was added to the wells. After incubation at 4°C overnight, binding of six-helix bundles was determined from subsequent binding of mAb NC-1, which was added at 1 μg/ml in PBS/1% BSA/1% gelatin (100 μl/well) for 1 h at 37°C. Subsequently the wells were washed three times with PBS/0.05% Tween 20 and biotin labeled anti-mouse IgG (100 μl/well; 125 ng/ml diluted in PBS containing 1% dry fat-free milk) was added. After incubation for 1 h at 37°C, the wells were washed as described above and HRP-streptavidin (125 ng/ml in PBS containing 10% goat serum; 100 μl/well) was added. After incubation for 1 h at 37°C, the wells were washed six times with PBS/0.05% Tween 20. HRP was quantitated as described above.
To exclude the remote possibility that mAb NC-1 reacted with CAP, serial two-fold dilutions (0.25 to 8 μg/ml) of the mAb and of control mouse IgG, respectively (each at 16 μg/ml in PBS-BG) were added to CAP coated or CAP-gp120 coated wells for 1 h at 37°C. The wells were washed and bound IgG was quantitated as described above for the sandwich ELISA. CAP-gp120 wells were prepared by coating first with HIV-1 IIIB gp120 under conditions described above for protein A/G, except that the pH was 8.0 instead of 8.8. Subsequently, CAP was added to the wells as described above. In control experiments, serial dilutions (1/200 to 1/1,600) of rabbit anti-phthalate antiserum and of normal rabbit serum, respectively (each diluted 100-fold in PBS-BG) were added to the wells. Bound rabbit IgG was quantitated using biotinylated goat anti-rabbit IgG.
Shedding of gp120 from virus particles
Preparations of purified HIV-1 IIIB and BaL, respectively, were incubated for 5 min at 37°C in the presence or absence of CAP (final concentration 5 mg/ml). Control preparations were not exposed to 37°C. Virus particles and released gp120 were separated by centrifugation as described [28]. The virus containing pellets and supernatant fluids were assayed by an ELISA allowing gp120 determination in the presence of CAP. Wells of polystyrene plates were coated with protein A/G, followed by polyclonal rabbit anti-gp120 (diluted 500-fold) under conditions described above for virus capture assays. Serial twofold dilutions in PBS-BG of preparations containing gp120 were added to the wells. After 4 h at 20°C, the wells were washed and bound gp120 was detected by addition of biotinyl-sCD4 (1 μg) followed by HRP-streptavidin as described above. The amount of gp120 in the virus pellets and the supernatant fluids was calculated from calibration curves relating absorbance readings to gp120 dilutions. All determinations were done in triplicate.
Molecular modeling: Docking of CAP on the gp41core structure
The acetylated and phthaloylated cellotetraose unit (CTAP) composed of four 1,4-linked β-D-glucose units, which is a representative part of CAP was created in Quanta 2000 [29] as described before [8]. CTAP was minimized by the steepest descent method followed by the adopted basis Newton-Raphson (ABNR) method. The energy difference of 0.05 Kcal/mol between two successive structures during both minimization steps was used as the termination criterion.
The docking simulations of CTAP were performed using the DockVision program [30] on the entire surface of the gp41 core structure {The X-ray crystal structure of the gp41 core, 1aik, was retrieved from the protein databank http://www.rcsb.org}. A grid box (125 × 125 Å × 125 Å) was created to cover the entire gp41 core surface for CTAP to dock. The default forcefield (Research Potential Function) was used to perform 1000 Monte Carlo runs for docking simulations. Both CTAP and the gp41 core structure were kept rigid during docking. Intermolecular energy criteria were used to select the lowest energy docked CTAP.
Electrostatic potential maps of gp120 and the gp120-CD4 complex
Electrostatic potentials were calculated using a Poisson-Boltzmann solver included in the GRASP program [31]. All default parameters were used. The electrostatic potential maps are shown on the accessible surface of gp120 and the gp120-CD4 complex.
Results
Enhancement of CAP-HIV-1 binding by CD4
Earlier studies [8] indicated that CAP binding to the envelope glycoprotein gp120 and to HIV-1 virus particles, respectively, did not interfere with their subsequent association with sCD4. Thus, it would be expected that CAP would not inhibit the attachment of HIV-1 to target cells and would block only subsequent steps obligatory for HIV-1 infection initiated by engagement of CXCR4 and CCR5 coreceptors, respectively. The latter event was shown to be blocked by CAP [8] and has been considered the basis for the virus inhibitory and virus inactivating properties of CAP at neutral pH. However, it has not been determined whether or not occupancy of CD4 binding sites within gp120 would affect subsequent interactions with CAP. To answer this question, the binding of gp120 and gp120-sCD4 complexes, respectively, to immobilized CAP was studied. Results shown in Fig. 1 indicated that sCD4 enhanced gp120-CAP binding. Maximum enhancement was observed at sCD4/gp120 weight ratios of ≥ 0.6 (Fig. 1 insert), corresponding to a molar ratio of ≥ 1.2 [32], in agreement with the observation that gp120-CD4 complexes consist of one molecule each of CD4 and gp120 [16]. Biotinyl-sCD4 in the absence of gp120 did not bind to CAP. Similarly, pretreatment of HIV-1 with sCD4 resulted in subsequent increased binding of virus with CAP, the effect being much more pronounced with HIV-1 BaL in comparison with HIV-1 IIIB (Fig. 2).
Synergism between CAP and sCD4 in inhibiting HIV-1 infection
Since HIV-1 can bind CAP and sCD4 simultaneously, and the binding of CAP is enhanced in the presence of sCD4, it was of interest to determine whether these two ligands act on HIV-1 cooperatively, resulting in synergism of their antiviral effects. This indeed was observed (Table 1 and 2, Fig. 3). CAP and sCD4 synergistically inhibited infection by HIV-1 IIIB (Table 1) and HIV-1 BaL (Table 2). A similar synergism was observed for virucidal activity against HIV-1 IIIB (Fig. 3) but only additive effects were found for HIV-1 BaL (in the absence of sCD4, ED50 for CAP = 1.49 ± 0.38 mg/ml; in the presence of sCD4 [100 μg/ml], which caused an 1.85-fold decrease of infectivity, ED50 = 1.39 ± 0.18 mg/ml for residual infectivity).
Treatment of HIV-1 with CAP leads to induction of gp41 six-helix bundles
Earlier studies [8], in which the binding of CAP treated and untreated virus with antibodies specific for distinct regions on the envelope glycoproteins gp120 and gp41 was studied, indicated that CAP had either no effect or caused decreased binding with antibodies against several peptides from gp120 but only against a single peptide, 557–586, from gp41 (see Discussion). It was intended to expand these studies to mAb NC-1, specific for the gp41 six-helix bundle [20]. Results of preliminary studies indicated that CAP did not interfere with the six-helix bundle formation from constituent peptides derived from near the N- and C-terminus of the gp41 ectodomain [20]; (own unpublished data). Surprisingly, and unlike with mAb 2F5 [8] specific for the C-terminal region of the gp41 ectodomain [33], CAP treatment enhanced the binding of both HIV-1 IIIB and HIV-1 BaL to mAb NC-1, reacting with gp41 six-helix bundles from both HIV-1 IIIB and BaL (Fig. 4), suggesting the formation of these structures within virus particles as a result of CAP treatment (Fig. 5). Treatment with both CAP and sCD4 further enhanced the expression of the six-helix bundles in the case of HIV-1 Bal (Fig 5B), but not in the case of HIV-1 IIIB (Fig 5A). sCD4 alone was less effective in enhancing the expression of the six-helix bundles. Treatment of neither recombinant gp160 or the gp41 C-peptide with CAP resulted in generation of epitopes recognized by mAb NC-1 (data not shown).
To exclude the remote possibility that mAb NC-1 reacted with CAP or protein-bound CAP, and that this, rather than binding to six-helix bundles, would lead to results shown in Fig. 5, the binding of the mAb to CAP coated wells was investigated. No significant binding of mAb NC-1 and of control mouse IgG, respectively, to wells coated by CAP directly or to CAP bound to HIV-1 IIIB gp120 was observed. On the other hand, anti-phthalate antibodies reacted with both forms of immobilized CAP (Fig. 6). These results support the conclusions from results in Fig. 5.

Monoclonal antibody NC-1 does not bind to CAP. For experimental design see Methods section.
Engagement of the gp120 coreceptor binding site with a tyrosine sulfated peptide from the N-terminus of the coreceptor CCR5 [25], shown to inhibit infection by CCR5-dependent, but not CXCR4-dependent, HIV-1 isolates, was sufficient to increase the expression of the gp41 six-helix bundles in HIV-1 BaL virus particles (Fig. 7). A CCR5 control peptide the tyrosines of which were not sulfated did not have this effect. Analogous experiments with HIV-1 IIIB were not performed since there are no published data concerning the biological properties of tyrosine sulfated peptides from the N-terminus of CXCR4.

Effect of a tyrosine sulfated CCR5 peptide [25, 56, 57] on expression on HIV-1 BaL of binding sites for mAb NC-1. Purified HIV-1 BaL (quantity indicated in the legend for Fig. 2) was mixed with the tyrosine sulfated S peptide and a control non-sulfated peptide, respectively, from CCR5 for 5 min at 37°C, followed by 30 min at 4°C. Control virus was incubated under the same conditions in the absence of the peptide. Serial dilutions (indicated on the abscissa) of the treated and untreated virus were added to mAb NC-1 and control normal mouse IgG coated wells, respectively, and the bound virus was quantitated by ELISA for p24. The quantity of virus bound to control wells was ≤ 2.3% of that bound to mAb NC-1 coated wells. All experiments were done at least in triplicate.
In the experiments described above evidence for the formation of gp41 six-helix bundles was obtained from capture of CAP, CD4 and CCR5 S-peptide treated virus particles, respectively, onto wells coated with mAb NC-1. This assay is equivalent to an immunoprecipitation assay with solid phase mAb NC-1. In order to provide further evidence for the induction of the gp41 six-helix bundles by the distinct ligands binding to the HIV envelope, the newly formed structures in virus lysates were also quantitated by a sandwich ELISA [21]. The results not only unequivocally confirmed the induction of gp41 six-helix bundle structures by sCD4, CAP and the S-peptide from CCR5, but also provided evidence that these structures were undetectable in untreated virus particles (Fig. 8). CAP (0.078 to 10 mg/ml) in the absence of virus particles provided negative results in this assay. The CCR5 peptide lacking sulfated tyrosines did not induce the six-helix bundles. Thus, it seems likely that the detection of gp41 six-helix bundles in untreated HIV-1 using the virion capture assay was due to their spontaneous formation during prolonged incubation of HIV-1 in these tests. The six-helix bundles were also induced by heating (10 min at 60°C) HIV-1 virus particles (Fig. 8).

Induction of gp41 six-helix bundles by HIV-1 treatment with sCD4, CAP, CCR5 S-peptide and heat. HIV-1 IIIB and HIV-1 BaL, respectively, were treated with CAP and sCD4 as described in the legend for Fig. 5 or exposed to 60°C for 10 min. HIV-1 BaL was also treated with the S-peptide and a control non-sulfated peptide from CCR5 as described in the legend for Fig. 7. The treated virus preparations and untreated control virus were treated with lysis buffer (see Methods) for 30 min at 20°C. CAP in lysis buffer (0.078 to 10 mg/ml) was also tested; the results for a 5 mg/ml concentration are shown (identical results were obtained for all other concentrations). The lysates were tested by a sandwich ELISA for the gp41 six-helix bundle (see Methods). All experiments were done at least in triplicate.
In summary, blocking by CAP of the coreceptor binding sites on the virus envelope glycoprotein gp120 within HIV-1 virus particles appears to induce conformational changes in gp41 leading to the formation of six-helix bundle structures.
Shedding of gp120 from virus particles
It was reported that treatment of HIV-1 gp120/gp41 envelope glycoprotein oligomers with sCD4 lead to shedding of gp120-sCD4 complexes concomitant with increased exposure of some cryptic epitopes on gp41 [13, 34, 35]. Therefore, it was of interest to determine whether or not shedding of gp120 from virus particles was required for the CAP induced exposure of binding sites for mAb NC-1 on gp41. Treatment of HIV-1 IIIB and BaL with CAP did not decrease the level of virus-associated gp120 in comparison with control virus preparations (Table 3). The results agree with the half life of 40 h for virus associated gp120 in the course of spontaneous gp120 shedding from HIV-1 HXB3 at 37°C [36] and the small sCD4 induced release of gp120 within 5 min at 37°C [37]. Thus gp120 shedding was not a prerequisite for the formation of gp41 six-helix bundles.
Discussion
Earlier studies [8] indicated that CAP treated HIV-1 particles have their coreceptor, i.e. CXCR4 and CCR5, binding sites obstructed, while sites involved in association with CD4 appeared unaffected. This suggested that HIV-1 could bind CD4 and CAP at the same time, independently. Since association with CD4 induces conformational changes in the HIV-1 envelope glycoprotein gp120 [14, 16, 17], it was of interest to determine: (a) whether CD4 binding to HIV-1 would affect subsequent association of the virus with CAP and (b) the consequences of both CD4 and CAP binding to virus particles. First, it was found that pretreatment of gp120 with sCD4 enhanced subsequent binding with CAP (Fig. 1). This could be ascribed to conformational changes in gp120, to the concealment of surface areas with the greatest negative charge on gp120 by CD4 [16], (Fig. 9), which could diminish the electrostatic attraction between gp120 and negatively charged CAP, or to additional CAP binding sites on the CD4 portion of gp120-sCD4 complexes. The binding of HIV-1 with CAP was similarly enhanced by pretreatment with sCD4 (Fig. 2), the effect being much more pronounced with the R5 virus, HIV-1 BaL, in comparison with the X4 virus, HIV-1 IIIB. These observations may be related to the recognized role of CD4 in inducing conformational changes in gp120 that contribute to the exposure of binding sites for CXCR4 and CCR5 [16, 17, 38].

Electrostatic potential maps of gp120 and the gp120-CD4 complex. Electrostatic potentials are shown for the solvent accessible surfaces. (A) The electrostatic potential map on gp120. Blue indicates electropositive areas whereas red represents electronegative areas. (B) After CD4 binds to gp120, the electropositive surface area (blue) increases markedly while the most negatively charged (red) area on gp120 (arrow) becomes blocked by sCD4.
Furthermore, it seemed important to determine whether simultaneous CD4 and enhanced CAP binding to HIV-1 would result in synergistic effects for inhibition of HIV-1 infection. Evidence for such synergism was indeed established (Table 1 and 2, Fig. 3).
Earlier studies [8], in which the binding of CAP treated and untreated virus to antibodies specific for distinct regions on the envelope glycoproteins gp120 and gp41 was studied, indicated that CAP caused decreased binding with antibodies against several peptides from gp120 but only with a single antibody against peptide 557–586 from gp41 (for numbering of amino acid residues see our earlier publication [39]). Thus, there are fewer binding sites for CAP on gp41 than on gp120. Interestingly, molecular docking studies revealed that phthalic and acetic acid anhydride modified cellotetratose, a subunit of CAP, docked to a single site on the gp41 core structure overlapping the peptide 557–586 (Fig. 10). This region is in the vicinity of the most prominent positively charged areas on the surface of the gp41 core which has an overall negative charge. Since CAP blocks coreceptor binding sites on gp120 [8] it was of interest to determine whether this blockade would lead to conformational changes in HIV-1 gp41, similar to those elicited by CD4 or coreceptor binding to gp120.

Docking of a cellotetraose acetate phthalate (CTAP) unit of CAP to the gp41 core structure. (A) Docking of CTAP on the gp41 core. The inner N-peptide coiled-coiled trimer is represented in green whereas the outer C-peptide helices are represented in yellow. Residues on the gp41 core interacting with CTAP (gray and red) are color-labeled. The brown residues are hydrophobic whereas blue residues are positively charged. One of the negatively charged groups from the phthalic acid moieties of CTAP docked near an R579 residue of the gp41 core. Two of the CTAP phenyl groups have hydrophobic contact with gp41 W571. The peptide segment 557–586 {[8, 39], corresponding to residues 550–571 in the X-ray crystal structure} antibodies to which are prevented by CAP from binding to gp41, is indicated in light blue in one of the three inner helices. (B) Electrostatic potential surface of the gp41 core created by the GRASP [31] program. The CTAP molecule docked near a relatively electropositive site on gp41. Most of the surface is highly electronegative.
The occupancy of CD4 and coreceptor binding sites by their respective ligands elicits downstream conformational changes in the envelope glycoprotein gp41, rendering it competent for fusion between virus and target cell membranes [18, 19, 40–43]. Shedding of gp120 from virus particles is not required for subsequent membrane fusion events [44]. The induction of gp41 six-helix bundles, detectable by mAb NC-1, by CAP in the absence of gp120 shedding is consistent with this conclusion. The conformational changes lead to the formation of a coiled-coil in gp41, consisting of three NH2-terminal leucine/isoleucine zipper regions, each contributed by one of the three subunits of the envelope glycoprotein trimer. In the presence of target cell membranes, the NH2-terminal fusion peptide is displaced in the direction of the target cell membrane, into which it inserts. Thus, the HIV-1 envelope glycoprotein gp41 becomes an integral component of two membranes, the viral membrane and the cellular membrane. The outer surface of the coiled-coil contains grooves into which three heptad repeat regions from the C-terminal part of the gp41 ectodomain pack, resulting in a stable six-helix bundle [42, 45–54]. The six-helix bundle structure can be detected by specific antibodies [14, 20, 43, 55].
Results presented here indicate that purified HIV-1 particles do not contain detectable six-helix bundle structures. Their expression is induced by CAP treatment of the virus (Fig. 5 and 8). Prior engagement of CD4 binding sites is not required for the induction of the six-helix bundles by CAP, but increases their expression (Fig. 5 and 8). The apparent cooperativity between CAP and sCD4 in induction of the gp41 six-helix bundle structures may possibly be related to the observed synergism between these two ligands for inhibition of HIV-1 infection (Table 1 and 2, Fig. 3). The hypothesis that engagement of coreceptor binding sites on gp120 by CAP leads to the expression of gp41 six-helix bundle structures is supported by the finding that a tyrosine sulfated S-peptide, but not the non-sulfated peptide, from the N-terminus of CCR5 [25, 56, 57] has an effect similar to that of CAP (Fig. 7). The helix-bundles were also induced by heating HIV-1 virus particles at 60°C, in agreement with the irreversible induction of the fusogenic conformation in influenza virus hemagglutinin by heat [58, 59]
In summary, the results presented here suggest that treatment of HIV-1 with CAP leads to conformational changes in the envelope glycoproteins, ultimately resulting, in the absence of target cell membranes, in the formation of gp41 six-helix bundles. These structures are extremely stable and represent a terminal, functionally inactive viral constituent [54, 60]; (Fig. 11), analogous to that of inactivated influenza virus hemagglutinin HA2 exposed to low pH in the absence of cell membranes [59, 61–64].

Proposed model for CAP induced formation of the gp41 six-helix bundles within HIV-1 particles. In the native state ((1)), HIV-1 envelope glycoprotein spikes correspond to a trimeric gp120(pink)/gp41(yellow) complex, the fusion peptide from the gp41-N terminus not being shown. The relative positions of the C-helix (yellow) and N-helix in the native envelope spike are not known and only the C-helix is depicted. Following interaction of the gp120 portion of the spikes with cellular receptors or CAP, the gp120/gp41 trimeric complex undergoes a conformational change, in which the fusion peptide (red) becomes exposed and the N-terminal half of gp41 (green) becomes a trimeric coiled coil. In this configuration ((2)) the C-peptide portion of gp41 (yellow) is not yet associated with the N-peptide coiled coil. This intermediate conformation is converted into a six-helix bundle when the C-peptide region binds to the N-peptide coiled coil region. This structure ((3)), in the absence of target cell membranes, corresponds to a nonfunctional spike, which cannot initiate anymore fusion with target cells. The position of fusion peptides in the model for "dead-end" spikes is not shown.
Conclusions
Earlier studies describing the underlying molecular mechanisms involved in the HIV-1 inhibitory effect of the candidate microbicide CAP indicated that this compound remains bound to HIV-1, impairing virus infectivity by blockade of binding sites for cellular coreceptors CXCR4 and CCR5 [8]. Results reported here further extend these findings and show that: 1) there is synergism between sCD4 and CAP for inhibition of virus infectivity; 2) CAP binding to HIV-1 leads to conformational changes in viral envelope glycoproteins resulting in the expression of functionally inert six-helix bundle structures.
To the best of our knowledge, results reported here and earlier [8] represent the most detailed study on the mechanism of action of a polymeric anti-HIV-1 compound and offer new opportunities for microbicide research, including the design of combined microbicides with distinct target sites on HIV-1 and acting synergistically.
Abbreviations
- CAP:
-
cellulose acetate phthalate
- STD:
-
sexually transmitted disease
- ELISA:
-
enzyme-linked immunosorbent assay
- mAbs:
-
monoclonal antibodies
- FBS:
-
fetal bovine serum
- PEG 6000:
-
polyethylene glycol 6000
- HIV-1:
-
human immunodeficiency virus type 1
- BSA:
-
bovine serum albumin
- PBS:
-
phosphate buffered saline
- Ch-PBS:
-
chicken serum (10%) in PBS
- PBS-BG:
-
1% BSA/1% gelatin in PBS
- HRP:
-
horseradish peroxidase
- sCD4:
-
soluble CD4
- TS:
-
0.14 M NaCl, 0.01 M Tris, pH 7.0
- CTAP:
-
cellotetraose acetate phthalate
- pdb:
-
Protein Data Bank
- ED50 :
-
effective dose for 50 % inhibition.
References
Neurath AR, Strick N, Li Y-Y, Lin K, Jiang S: Design of a "microbicide" for prevention of sexually transmitted diseases using "inactive" pharmaceutical excipients. Biologicals. 1999, 27: 11-21. 10.1006/biol.1998.0169.
Gyotoku T, Aurelian L, Neurath AR: Cellulose acetate phthalate (CAP): an 'inactive' pharmaceutical excipient with antiviral activity in the mouse model of genital herpesvirus infection. Antiviral Chem Chemother. 1999, 10: 327-332.
Neurath AR, Li Y-Y, Mandeville R, Richard L: In vitro activity of a cellulose acetate phthalate topical cream against organisms associated with bacterial vaginosis. J Antimicrob Chemother. 2000, 45: 713-714. 10.1093/jac/45.5.713.
Neurath AR: Microbicide for prevention of sexually transmitted diseases using a pharmaceutical excipient. AIDS Patient Care STDS. 2000, 14: 215-219. 10.1089/108729100317830.
Neurath AR, Stick N, Li Y-Y, Radigan L, Jiang S: A microbicide for the third millenium (cellulose acetate phthalate). XIII International AIDS Conference. 2000, 713-717.
Manson KH, Wyand MS, Miller C, Neurath AR: The effect of a cellulose acetate phthalate topical cream on vaginal transmission of simian immunodeficiency virus in rhesus monkeys. Antimicrob Agents Chemother. 2000, 44: 3199-3202. 10.1128/AAC.44.11.3199-3202.2000.
Kawamura T, Cohen SS, Borris DL, Aquilino EA, Glushakova S, Margolis LB, Orenstein JM, Offord RE, Neurath AR, Blauvelt A: Candidate microbicides block HIV-1 infection of human immature Langerhans cells within epithelial tissue explants. J Exp Med. 2000, 192: 1491-1500. 10.1084/jem.192.10.1491.
Neurath AR, Strick N, Li YY, Debnath AK: Cellulose acetate phthalate, a common pharmaceutical excipient, inactivates HIV-1 and blocks the coreceptor binding site on the virus envelope glycoprotein gp120. BMC Infect Dis. 2001, 1: 17-10.1186/1471-2334-1-17.
Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, et al: Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996, 381: 661-666. 10.1038/381661a0.
Orloff SL, Kennedy MS, Belperron AA, Maddon PJ, McDougal JS: Two mechanisms of soluble CD4 (sCD4)-mediated inhibition of human immunodeficiency virus type 1 (HIV-1) infectivity and their relation to primary HIV-1 isolates with reduced sensitivity to sCD4. J Virol. 1993, 67: 1461-1471.
Johnson VA, Barlow MA, Chou TC, Fisher RA, Walker BD, Hirsch MS, Schooley RT: Synergistic inhibition of human immunodeficiency virus type 1 (HIV-1) replication in vitro by recombinant soluble CD4 and 3'-azido-3'-deoxythymidine. J Infect Dis. 1989, 159: 837-844.
Pan X-Z, Qiu Z-D, Baron PA, Gold JWM, Polsky B, Chou T-C, Armstrong D: Three-drug synergistic inhibition of HIV-1 replication in vitro by 3'-fluoro-3'-deoxythymidine, recombinant soluble CD4, and recombinant interferon-alpha. AIDS Res Hum Retroviruses. 1992, 8: 589-595.
Sattentau QJ, Moore JP: Conformational changes induced in the human immunodeficiency virus envelope glycoprotein by soluble CD4 binding. J Exp Med. 1991, 174: 407-415. 10.1084/jem.174.2.407.
Gorny MK, Zolla-Pazner S: Recognition by human monoclonal antibodies of free and complexed peptides representing the prefusogenic and fusogenic forms of human immunodeficiency virus type 1 gp41. J Virol. 2000, 74: 6186-6192. 10.1128/JVI.74.13.6186-6192.2000.
Wyatt R, Kwong PD, Desjardins E, Sweet RW, Robinson J, Hendrickson WA, Sodroski JG: The antigenic structure of the HIV gp120 envelope glycoprotein. Nature. 1998, 393: 705-711. 10.1038/31514.
Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA: Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998, 393: 648-659. 10.1038/31405.
Wyatt R, Sodroski J: The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science. 1998, 280: 1884-1888. 10.1126/science.280.5371.1884.
Jones PL, Korte T, Blumenthal R: Conformational changes in cell surface HIV-1 envelope glycoproteins are triggered by cooperation between cell surface CD4 and co-receptors. J Biol Chem. 1998, 273: 404-409. 10.1074/jbc.273.1.404.
Dimitrov AS, Xiao X, Dimitrov DS, Blumenthal R: Early intermediates in HIV-1 envelope glycoprotein-mediated fusion triggered by CD4 and co-receptor complexes. J Biol Chem. 2001, 276: 30335-30341. 10.1074/jbc.M103788200.
Jiang S, Lin K, Lu M: A conformation-specific monoclonal antibody reacting with fusion-active gp41 from the human immunodeficiency virus type 1 envelope glycoprotein. J Virol. 1998, 72: 10213-10217.
Jiang S, Lin K, Zhang L, Debnath AK: A screening assay for antiviral compounds targeted to the HIV-1 gp41 core structure using a conformation-specific monoclonal antibody. J Virol Methods. 1999, 80: 85-96. 10.1016/S0166-0934(99)00041-5.
Neurath AR, Strick N, Li Y-Y, Jiang S: Improbability of harmful autoimmune responses resulting from immunization with HIV-1 envelope glycoproteins. AIDS Res Hum Retroviruses. 1993, 9: 1195-1208.
Gartner S, Markovits P, Markovitz DM, Kaplan MH, Gallo RC, Popovic M: The role of mononuclear phagocytes in HTLV-III/LAV infection. Science. 1986, 233: 215-219.
Neurath AR, Debnath AK, Strick N, Li Y-Y, Lin K, Jiang S: Blocking of CD4 cell receptors for the human immunodeficiency virus type 1 (HIV-1) by chemically modified bovine milk proteins: potential for AIDS prophylaxis. J Mol Recognition. 1995, 8: 304-316.
Farzan M, Vasilieva N, Schnitzler CE, Chung S, Robinson J, Gerard NP, Gerard C, Choe H, Sodroski J: A tyrosine-sulfated peptide based on the N terminus of CCR5 interacts with a CD4-enhanced epitope of the HIV-1 gp120 envelope glycoprotein and inhibits HIV-1 entry. J Biol Chem. 2000, 275: 33516-33521. 10.1074/jbc.M007228200.
Lusso P, Cocchi F, Balotta C, Markham PD, Louie A, Farci P, Pal R, Gallo RC, Reitz MS: Growth of macrophage-tropic and primary human immunodeficiency virus type 1 (HIV-1) isolates in a unique CD4+ T-cell clone (PM1): Failure to downregulate CD4 and to interfere with cell-line-tropic HIV-1. J Virol. 1995, 69: 3712-3720.
Chou T-C, Talalay P: Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984, 22: 27-55. 10.1016/0065-2571(84)90007-4.
Willey RL, Theodore TS, Martin MA: Amino acid substitutions in the human immunodeficiency virus type 1 gp120 V3 loop that change viral tropism also alter physical and functional properties of the virion envelope. J Virol. 1994, 68: 4409-4419.
Quanta 2000. 9685 Scranton Road, San Diego, CA. 92121, Accelrys, Inc.
Hart TN, Ness SR, Read RJ: Critical evaluation of the research docking program for the CASP2 challenge. Proteins. 1997, Suppl 1: 205-209. 10.1002/(SICI)1097-0134(1997)1+<205::AID-PROT27>3.3.CO;2-P.
Nicholls A, Sharp KA, Honig B: Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins: Structure, Function, and Genetics. 1991, 11: 281-296.
Moebius U: Cluster report: CD4. In Leucocyte Typing IV: White Cell Differentiation Antigens. Edited by: Knapp W, Dörken B, Gilks WR, Rieber EP, Schmidt RE, Stein H, von dem Borne AEGKr. 1989, Oxford, Oxford University Press, 314-316.
Parker CE, Deterding LJ, Hager-Braun C, Binley JM, Schulke N, Katinger H, Moore JP, Tomer KB: Fine definition of the epitope on the gp41 glycoprotein of human immunodeficiency virus type 1 for the neutralizing monoclonal antibody 2F5. J Virol. 2001, 75: 10906-10911. 10.1128/JVI.75.22.10906-10911.2001.
Sattentau QJ, Moore JP, Vignaux F, Traincard F, Poignard P: Conformational changes induced in the envelope glycoproteins of the human and simian immunodeficiency viruses by soluble receptor binding. J Virol. 1993, 67: 7383-7393.
Sattentau QJ, Zolla-Pazner S, Poignard P: Epitope exposure on functional, oligomeric HIV-1 gp41 molecules. Virol. 1995, 206: 713-717.
Layne SP, Merges MJ, Dembo M, Spouge JL, Conley SR, Moore JP, Raina JL, Renz H, Gelderblom HR, Nara PL: Factors underlying spontaneous inactivation and susceptibility to neutralization of human immunodeficiency virus. Virol. 1992, 189: 695-714. 10.1016/0042-6822(92)90593-E.
McKeating JA, McKnight A, Moore JP: Differential loss of envelope glycoprotein gp120 from virions of human immunodeficiency virus type 1 isolates: Effects on infectivity and neutralization. J Virol. 1991, 65: 852-860.
Littman DR: Chemokine receptors: keys to AIDS pathogenesis?. Cell. 1998, 93: 677-680. 10.1016/S0092-8674(00)81429-4.
Neurath AR, Strick N, Jiang S: Synthetic peptides and anti-peptide antibodies as probes to study interdomain interactions involved in virus assembly: The envelope of the human immunodeficiency virus (HIV-1). Virol. 1992, 188: 1-13. 10.1016/0042-6822(92)90729-9.
Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, Rothman JE: SNAREpins: minimal machinery for membrane fusion. Cell. 1998, 92: 759-772. 10.1016/S0092-8674(00)81404-X.
Munoz-Barroso I, Durell S, Sakaguchi K, Appella E, Blumenthal R: Dilation of the human immunodeficiency virus-1 envelope glycoprotein fusion pore revealed by the inhibitory action of a synthetic peptide from gp41. J Cell Biol. 1998, 140: 315-323. 10.1083/jcb.140.2.315.
Furuta RA, Wild CT, Weng Y, Weiss CD: Capture of an early fusion-active conformation of HIV-1 gp41. Nat Struct Biol. 1998, 5: 276-279. 10.1038/871.
de Rosny E, Vassell R, Wingfield PT, Wild CT, Weiss CD: Peptides corresponding to the heptad repeat motifs in the transmembrane protein (gp41) of human immunodeficiency virus type 1 elicit antibodies to receptor-activated conformations of the envelope glycoprotein. J Virol. 2001, 75: 8859-8863. 10.1128/JVI.75.18.8859-8863.2001.
Thali M, Furman C, Helseth E, Repke H, Sodroski J: Lack of correlation between soluble CD4-induced shedding of the human immunodeficiency virus type 1 exterior envelope glcoprotein and subsequent membrane fusion events. J Virol. 1992, 66: 5516-5524.
Gallo SA, Puri A, Blumenthal R: HIV-1 gp41 six-helix bundle formation occurs rapidly after the engagement of gp120 by CXCR4 in the HIV-1 env-mediated fusion process. Biochemistry. 2001, 40: 12231-12236. 10.1021/bi0155596.
Kliger Y, Shai Y: Inhibition of HIV-1 entry before gp41 folds into its fusion-active conformation. J Mol Biol. 2000, 295: 163-168. 10.1006/jmbi.1999.3368.
Chan DC, Kim PS: HIV entry and its inhibition. Cell. 1998, 93: 681-684. 10.1016/S0092-8674(00)81430-0.
Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC: Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997, 387: 426-430. 10.1038/387426a0.
Tan K, Liu J, Wang J, Shen S, Lu M: Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc Natl Acad Sci U S A. 1997, 94: 12303-12308. 10.1073/pnas.94.23.12303.
Chan DC, Fass D, Berger JM, Kim PS: Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997, 89: 263-273. 10.1016/S0092-8674(00)80205-6.
Lu M, Ji H, Shen S: Subdomain folding and biological activity of the core structure from human immunodeficiency virus type 1 gp41: implications for viral membrane fusion. J Virol. 1999, 73: 4433-4438.
Weissenhorn W, Wharton SA, Calder LJ, Earl PL, Moss B, Aliprandis E, Skehel JJ, Wiley DC: The ectodomain of HIV-1 env subunit gp41 forms a soluble, α-helical, rod-like oligomer in the absence of gp120 and the N-terminal fusion peptide. EMBO J. 1996, 15: 1507-1514.
LeDuc DL, Shin YK: Insights into a structure-based mechanism of viral membrane fusion. Biosci Rep. 2000, 20: 557-570. 10.1023/A:1010463005396.
Doms RW, Moore JP: HIV-1 membrane fusion: targets of opportunity. J Cell Biol. 2000, 151: F9-14. 10.1083/jcb.151.2.F9.
Chen CH, Greenberg ML, Bolognesi DP, Matthews TJ: Monoclonal antibodies that bind to the core of fusion-active glycoprotein 41. AIDS Res Hum Retroviruses. 2000, 16: 2037-2041. 10.1089/088922200750054765.
Cormier EG, Persuh M, Thompson DAD, Lin SW, Sakmar TP, Olson WC, Dragic T: Specific interaction of CCR5 amino-terminal domain peptides containing sulfotyrosines with HIV-1 envelope glycoprotein gp120. Proc Natl Acad Sci U S A. 2000, 97: 5762-5767. 10.1073/pnas.97.11.5762.
Cormier EG, Tran DN, Yukhayeva L, Olson WC, Dragic T: Mapping the determinants of the CCR5 amino-terminal sulfopeptide interaction with soluble human immunodeficiency virus type 1 gp120-CD4 complexes. J Virol. 2001, 75: 5541-5549. 10.1128/JVI.75.12.5541-5549.2001.
Carr CM, Kim PS: A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell. 1993, 73: 823-832. 10.1016/0092-8674(93)90260-W.
Carr CM, Chaudhry C, Kim PS: Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc Natl Acad Sci U S A. 1997, 94: 14306-14313. 10.1073/pnas.94.26.14306.
Melikyan GB, Markosyan RM, Hemmati H, Delmedico MK, Lambert DM, Cohen FS: Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J Cell Biol. 2000, 151: 413-423. 10.1083/jcb.151.2.413.
Chen J, Wharton SA, Weissenhorn W, Calder LJ, Hughson FM, Skehel JJ, Wiley DC: A soluble domain of the membrane-anchoring chain of influenza virus hemagglutinin (HA2) folds in Escherichia coli into the low-pH-induced conformation. Proc Natl Acad Sci U S A. 1995, 92: 12205-12209.
Korte T, Ludwig K, Booy FP, Blumenthal R, Herrmann A: Conformational intermediates and fusion activity of influenza virus hemagglutinin. J Virol. 1999, 73: 4567-4574.
Bentz J: Membrane fusion mediated by coiled coils: a hypothesis. Biophys J. 2000, 78: 886-900.
Skehel JJ, Wiley DC: Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem. 2000, 69: 531-569. 10.1146/annurev.biochem.69.1.531.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2334/2/6/prepub
Acknowledgements
We thank Ms. Hong Lu for technical assistance, Ms. V. Kuhlemann for preparation of the manuscript and figures. This study was supported by NIH grants (P01 HD41761 and R01 AI46221) and the Marilyn M. Simpson Charitable Trust. A. K. Debnath had support from Philip Morris Companies, Inc., and Johnson & Johnson, Inc.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
None declared
Authors' contributions
Author 1 ARN developed the concepts representing the basis of the manuscript and designed most experiments. Author 2 NS carried out most experiments and contributed to the development of experimental techniques. Author 3 SJ developed monoclonal antibody NC-1 and was involved in studies on the gp41 six helix bundles and on synergism between CAP and sCD4 for inhibition of HIV-1 infectivity. Author 4 YYL did most tissue culture work and infectivity assays. Author 5 AKD did all the molecular modeling studies.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Neurath, A.R., Strick, N., Jiang, S. et al. Anti-HIV-1 activity of cellulose acetate phthalate: Synergy with soluble CD4 and induction of "dead-end" gp41 six-helix bundles. BMC Infect Dis 2, 6 (2002). https://doi.org/10.1186/1471-2334-2-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2334-2-6