
Abstract
Background
Constitutive expression and localization of antimicrobial human β-defensin-1 (HBD-1) in human kidneys as a potential mechanism of antimicrobial defense has been previously reported. Inducible expression of human β-defensin-2 (HBD-2) has been described in various epithelial organs but not for the urogenital tract.
Methods
We investigated the gene- and protein expression of HBD-1 and HBD-2 by reverse transcriptase-polymerase chain reaction, and immunohistochemistry in 15 normal human kidney samples and 15 renal tissues with chronic bacterial infection. Additionally, cell culture experiments were performed to study HBD gene expression by real-time RT-PCR in response to inflammatory cytokines TNFα and IL-1β as well as lipopolysaccharide from Gram-negative bacteria.
Results
Constitutive HBD-1 gene- and protein expression was detected in normal renal tissue and kidneys with chronic infection. As a novel finding, inducible HBD-2 gene- and protein expression was demonstrated in tubulus epithelia with chronic infection but not in normal renal tissue. In pyelonephritic kidneys HBD-1 and HBD-2 expression showed a similar pattern of localizaton in distal tubules, loops of Henle and in collecting ducts of the kidney. Furthermore, real-time RT-PCR of kidney derived cell lines stimulated with inflammatory agents TNF-α, IL-1β and LPS revealed a strong increase in relative HBD-2 transcription level and also a slight increase in relative HBD-1 transcription level.
Conclusions
Upregulated HBD-2 expression in renal tubulus epithelium indicates a role of a wider range of human defensins for antimicrobial host defense in the urogenital tract than previously recognized.
Similar content being viewed by others


Background
Defensins are a family of cationic proteins (3–4 kD) typically containing 6 cystein residues. Expression of at least two subfamilies of defensins (α- and β-defensins) has been shown in insects, plants and humans in respect to a difference in the position and connectivity of cystein disulfide bonds [1, 2]. The characteristic fold of the protein molecules is assumed to exercise antimicrobial activity by selective disruption of microbial membranes. Multimeric pore formation in the cell membrane of pathogens by antimicrobial protein activity is held responsible for the killing of microorganisms with successive efflux of cellular contents and breakdown of intracellular homeostasis. Mammalian cells are thought to be more resistent to this effect because of a higher content of cholesterol and phospholipids in their membrane. Remarkably, it seems to be difficult for microorganisms to acquire resistance against this antimicrobial mechanism, assigning these proteins a putative role for future therapeutic applications [3].
At least four β-defensins (HBD-1, HBD-2, HBD-3, HBD-4) have been characterized in humans. HBD-1 was first isolated from the hemofiltrate of patients with end stage kidney disease undergoing dialysis, locating renal epithelia as the major source for HBD-1 [4]. HBD-2 was initially purified from lesional psoriatic scale extracts by bacteria affinity chromatography and revealed antimicrobial activity against Gram negative bacteria and yeast like candida [5]. Other localizations of HBD-2 expression include nasal [6], and oral mucosa [7, 8], human airway [9–11], nasolacrimal duct [26], and ocular surface epithelium [13] as well as intestinal epithelium [14] in response to infection and inflammation. Expression of HBD-3 in psoriatic scales and human keratinocytes [15] as well as in other epithelial and non epithelial tissues [12] have been reported recently. Finally, HBD-4 was identified by screening of genomic sequences and subsequent functional analysis with gene expression recognized in testis, stomach, uterus, neutrophils, thyroid, lung, and kidney [16].
Former studies have been presented on the expression, localization and function of HBD-1 in the human urogenital system [17–20]. Gene expression of HBD-1 was found by in situ hybridization in the columnar epithelial layers of the distal tubules, loops of Henle, and collecting ducts [17]. In contrast immunohistochemical methods stained only intratubular hyalin substance of the kidney.
At present, only a single investigation mentioned noticeable HBD-2 mRNA expression in one sample of human renal tissue by dot blot hybridization [9]. However, investigation of HBD-2 gene expression as well as protein expression in normal human kidneys and in kidneys with chronic bacterial infection has not yet been systematically investigated. The present study explores gene expression and immunohistochemical localization of HBD-1 and HBD-2 in extirpated kidneys with chronic pyelonephritis and in normal renal tissue obtained from tumor-bearing kidneys. Furthermore, in vitro gene expression of HBD-1 and HBD-2 in kidney derived cell lines under proinflammatory challenging was investigated.
Methods
Tissue samples
Samples of renal tissue from nephrectomy specimens were obtained immediately in the operating room and stored in liquid nitrogen under protocols approved by the institutional ethics committee. Fifteen renal tissue specimens were obtained from fifteen patients (mean age 60 years – range 12 months to 75 years, 8 female and 7 male patients) with chronic upper urinary tract infections undergoing nephrectomy. Clinical isolates from preoperatively collected urine samples of the 15 patients with chronic upper urinary tract infection revealed Escherichia coli in eleven, Klebsiella species in two and Pseudomonas aeruginosa in two further patients as causative agents. The etiology responsible for recurrent infection and decline of renal function were vesicoureteral reflux or obstruction of the upper urinary tract. The history of recurrent bacterial infection of the urinary tract usually preceded for a minimum of 10 months with subsequent decline of renal function before surgical removal of the affected organ was performed.
Furthermore, histologically confirmed samples of normal renal tissues were collected from patients who underwent nephrectomy for a kidney neoplasia (mean age of patients 65 years – range 47 years to 75 years, 8 female and 7 male patients). In most cases parenchymal architecture of the kidney was preserved and tissue was dissected into cortex and medulla before storage in liquid nitrogen.
Corresponding samples of normal and infected renal tissue samples preserved as neutral formalin-fixed and paraffin-embedded sections were prepared by the Department of Pathology, University of Kiel. The histopathological diagnosis of the tissue samples was independently confirmed by two pathologists.
Cell culture
The established human kidney carcinoma cell lines CAKI-1 and CAL-54 were purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany). Cell lines were cultured as a monolayer in RPMI 1640 medium (Biochrom, Berlin, Germany) supplemented with 10% heat-inactivated fetal calf serum (FCS), 2 mmol/L glutamine and 1 mmol/L pyruvate. Cultures were maintained at a temperature of 37°C in a humidified 5% CO2 atmosphere. All cell lines were routinely tested for mycoplasma contamination using a PCR mycoplasma detection kit (TaKaRa, Biomedical Europe S.A., Genevilliers, France) and contaminated cell cultures were discarded.
Proinflammatory in vitro challenging
Confluent cultures were split using trypsin/EDTA and cells were seeded in 6-well cell culture plates with 9,6 cm2 growth area per well (BD Primaria™, Becton Dickinson, Heidelberg, Germany). For cell challenging experiments, cells were grown in FCS free medium for at least 24 hours to exclude the influence of unknown inhibitory or stimulatory FCS compounds on HBD gene expression. Cells were exposed to inflammatory agents for at least 6 hours. Inflammatory challenging of the cells was performed using tumor-necrosis-factor alpha (TNFα, Clontech Laboratories GmbH, Heidelberg, Germany) or interleukin-1-beta (IL-1β, Biozol GmbH, Eching, Germany) at concentrations between 0.1 and 10 ng/ml or lipopolysaccharide from Escherichia coli (LPS, Sigma-Aldrich Chemie GmbH, Deisenhof, Germany) at concentrations between 0.1 and 100 μg/ml. At the end of the experiment the supernatant medium was removed and subsequent extraction of total RNA of the stimulated cells was performed.
Isolation of total RNA
Total RNA was isolated from cell lines and tissue samples based on the guanidium-thiocyanate-phenol-chloroform single-step isolation method [21]. Renal tissue samples (100–500 mg) were pulverized by mortar and pestel while being immersed in liquid nitrogen. Total RNA from tumor cell lines was extracted using RNA-Clean™ (AGS-Chemie Heidelberg, Germany) and total RNA from frozen tissue specimens was extracted by RNAzol™ (WAK-Chemie Medical GmbH, Bad Homburg, Germany) as recommended by the suppliers. To optimize the yield of RNA extraction, 5 μg of glycogen (Roche Boehringer, Mannheim, Germany) were added to the aqueous phase before precipitation with isopropanol. The RNA preparations were dissolved in 40 μl RNase free water. RNA recovery and purity were controlled by absorption measurement at 260 nm and 280 nm (Biophotometer, Eppendorf, Hamburg, Germany). Samples of 2 μg of the extracted total RNA were separated in 1% agarose gel (Small DNA agarose; Biozym, Hessisch Oldendorf, Germany). The integrity of total RNA was determined by visualising 28S and 18S ribosomal RNA. Samples with evidence of ribosomal RNA degradation were discarded.
RT-PCR of tissue samples
An aliquot of 2 μg of total RNA of each tissue sample was dissolved in a volume of 10 μl redestillated sterile water, denatured for 10 min at 70°C and then quickly chilled on ice. The cDNA was synthesized in a total volume of 20 μl containing 4 μl 5× first-strand buffer, 2 mM dithiothreitol (DTT), 200 U of SuperScript II (all Life Technologies, Eggenstein, Germany), 20 U of RNase inhibitor, 5 μM random hexamers, 1 mM deoxynucleotide triphosphate (dNTP) mix (all Applied Biosystems GmbH, Weiterstadt, Germany). The reaction mix was incubated for 10 min at 24°C and 60 min at 42°C. Reverse transcriptase was then inactivated for 3 min at 94°C.
HBD-1 and HBD-2 PCR of tissue samples
Primers for HBD-1 sense, 5'-ATGAGAACTTCCTACCTTCTGCT-3', and HBD-1 antisense, 5'-CTCTGTAACAGGTGCC-3', resulted in a 184-bp fragment. Primers for HBD-2 sense, 5'-CCAGCCATCAGCCATGAGGGT-3', and HBD-2 antisense, 5'-GGAGCCCTTTCTGAATCCGCA-3', resulted in a 255-bp fragment. Primers for the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) sense, 5'-CCAGCCGAGCCACATCGCTC-3' and GAPDH anti-sense, 5'-ATGAGCCCCAGCCTTCTCCAT-3' were added to each specific human β-defensin PCR reaction mix to monitor correct cDNA synthesis. GAPDH PCR resulted in a 359-bp fragment (GAPDH primer-sequences kindly provided by Dr. C. Schlüter, Department of Dermatology, University of Kiel, Germany). All primers were commercially synthesized (MWG-Biotech, Ebersberg, Germany).
For specific HBD-1 or HBD-2 RT-PCRs 5 μl of the cDNA synthesis mix were used in 50 μl of the final reaction mix, which contained 5 μl of 10× tricine buffer III, 200 μM deoxynucleotide triphosphate mixture, 1.5 μM of human β-defensin sense and 1.5 μM of human β-defensin antisense primer as well as 0.15 μM of GAPDH sense and 0.15 μM of GAPDH antisense primer. One unit of Taq-DNA-polymerase (Gibco BRL Life Technologies, Eggenstein, Germany) was added to the reaction mix after denaturing cDNA at 95°C for 5 min (Table 1) in a thermocycler (Biometra, Göttingen, Germany). Thirty PCR cycles were performed.
Two μg of total RNA from nasal epithelium which has been stimulated by bacterial lipopolysaccharide in vitro was used as a positive control for HBD-1 and HBD-2 RT-PCR. The absence of contamination was routinely checked by RT-PCR assays of negative samples, in which RNA samples were replaced with sterile water (water control) and by omitting SuperScript II. All RT-PCR products were separated by electrophoresis in a 2% agarose gel (Small DNA agarose, Biozym, Hessisch Oldendorf, Germany) in Tris-acetate-EDTA (TAE)-buffer and visualized by ethidium-bromide staining. Molecular weights were determined using a DNA molecular-weight marker (100-bp DNA ladder, Life Technologies, Eggenstein, Germany).
Representative fragments of specific HBD-1 and HBD-2 RT-PCRs were extracted from agarose gels by QIAEX II Gel Extraction-Kit (QIAGEN GmbH, Hilden, Germany). The isolated cDNA product was sequenced by an automatic sequencer ALFexpress (Pharmacia, Freiburg, Germany).
Real-time RT-PCR of stimulated cell lines
Real-time RT-PCR analyses were performed in a fluorescence temperature cycler (LightCycler, Roche Molecular Biochemicals) according to the manufacturer's instructions. This technique continuously monitors the cycle-by-cycle accumulation of fluorescently labeled PCR product. Briefly, total RNA from cultured epithelial cells was isolated and reverse transcribed as described above. The cDNA corresponding to 20 ng RNA served as a template in a 20 μl reaction containing 4 mM MgCl2, 0.5 μM of each primer and 1× LightCycler-FastStart DNA Master SYBR Green I mix (Roche Molecular Biochemicals). Samples were loaded into capillary tubes and incubated in the fluorescence thermocycler (LightCycler) for an initial denaturing at 95°C for 10 min followed by 45 cycles, each cycle consisting of 95°C for 15 s, 60°C (touchdown of -1°C/cycle from 66°C to 60°C) for 5 s and 72°C for 10 s. Cycle-to-cycle fluorescence emission readings were monitored at 72°C at the end of each cycle and analyzed using LightCycler Software (Roche Molecular Biochemicals). Melting curves were generated after each run (cooling the sample to 65°C for 15 s and then heating slowly at 0.2°C/s up to 95°C with continuous measurement of fluorescence) to confirm amplification of specific transcripts.
Amplification was carried out using gene specific, intron-spanning primers for HBD-1 and HBD-2 (HBD-1 sense: 5'-TTGTCTGAGATGGCCTCAGGTGGTAAC-3', HBD-1 antisense: 5'-ATACTTCAAAAGCAATTTTCCTTTAT-3'; HBD-2 sense: 5'-ATCAGCCATGAGGGTCTTGT-3', HBD-2 antisense: 5'-GAGACCACAGGTGCCAATTT-3'). Amplification of the housekeeping-gene GAPDH was performed with each cDNA in a separate PCR-reaction using intron-spanning primers GAPDH sense: 5'-CCAGCCGAGCCACATCGCTC-3' and GAPDH antisense: 5'-ATGAGCCCCAGCCTTCTCCAT-3'. Relative HBD-1 and HBD-2 transcript levels were corrected by normalization based on the GAPDH transcript levels. Specificity of HBD-1 and HBD-2 encoding PCR products was verified by agarose gel electrophoresis and by sequencing of representative samples.
HBD-1 and HBD-2 immunohistochemistry
All sections were cut from formalin-fixed and paraffin-embedded specimens at a thickness of 5 μm and mounted on adhesive slides (HistoBond slides, Marienfeld Glassware, Bad Mergentheim, Germany). The sections were deparaffinized (twice in Xylol, twice in 100%, 96%, and 70% Ethanol, twice in redestillated sterile water for 15 min each).
Antigenic epitopes were demasked by boiling sections for 2 min in citratbuffer (2.1 g citrat in 1 liter of redestillated sterile water at pH 6) in a pressure cooker. Sections were rinsed with redestillated sterile water and then treated with 0.5% hydrogen peroxide in methanol for 30 min to block endogenous peroxidase activity. Sections were rehydrated and washed in phosphate-buffered saline (PBS) before blocking with 5% of normal goat serum (Dako, Hamburg, Germany) in PBS and then incubating with avidin and biotin for 15 min. HBD-1 and HBD-2 polyclonal rabbit anti-serum (kindly provided by Tom Ganz, Erika Valore and Lide Liu, UCLA, USA) were diluted 1:500 in PBS. Sections were incubated for 1 h at room temperature and then washed several times in PBS and treated with biotinylated anti-goat immunoglobuline G (Dako, Hamburg, Germany). Sections were further incubated with solution of avidin-biotinylated peroxidase complex (Vector laboratories Inc., Burlingame, USA). Reactions were developed with 0.02% hydrogen peroxide and 0.1% diaminobenzdine tetrahydrochloride for 5–10 min. Sections were then counterstained with Harris modified hematoxylin (Merck, Darmstadt, Germany) for 1–3 min. Negative control sections were incubated with preimmune serum from the same rabbit as the polyclonal antibody was generated from. The immunoreactivity was graded based on the staining intensities above that seen on the negative controls. (1+ weak; 2+ moderate; 3+ strong). Staining of moderate and strong intensity were considered positive. Representative areas were evaluated estimating the percentage of histoanatomical structures stained and results were confirmed independently by two pathologists.
Results
RT-PCR of HBD-1 and HBD-2 in tissue samples
HBD-1 RT-PCR was positive in all 30 investigated samples of normal and chronically infected renal tissues showing a distinct band migrating as 184 bp product on 2% agarose gel electrophoresis. HBD-2 RT-PCR was positive in 14 out of 15 samples extracted from renal tissues with chronic bacterial infection yielding a 255 bp band. In contrast, HBD-2 RT-PCR was negative in all 15 normal kidney tissue extracts without infection from tumor bearing organs. The control for correct cDNA synthesis performed as GAPDH RT-PCR was positive in all samples as well as in HBD-1 and HBD-2 expressing positive controls of lipopolysaccharide stimulated nasal epithelium (Fig. 1).

HBD-1 and HBD-2 specific RT-PCR of normal renal parenchyma and renal tissues with chronic infection. HBD-1 (Fig. 1a) specific RT-PCR (PCR product: 184 bp), HBD-2 (Fig. 1a) specific RT-PCR (PCR product: 255 bp). Samples were separated by 2% agarose gel electrophoresis and stained with ethidium bromide. Representative data from a panel of 30 specimens are presented. Lanes 1 to 10 represent renal tissue extracts with chronic infection. Lanes 11–20 show RT-PCR results of normal renal tissue extracts from tumor-bearing organs. Outer lanes (M) represent a 100 bp-ladder molecular-weight marker. The control reaction for correct cDNA synthesis in all samples was performed as RT-PCR for GAPDH (glyceraldehyde-3-phosphate dehydrogenase) yielding a 359 bp product. Correct positive and negative controls including amplification without reverse transcription or cDNA sample replaced by sterile water were performed with each original experiment but are not shown due to limited gel space.
Immunolocalization of HBD-1 and HBD-2 peptide in renal tissue sections
Immunohistochemical staining with polyclonal rabbit antiserum against HBD-1 and HBD-2 was performed on formalin-fixed and paraffin-embedded renal tissue sections with chronic bacterial infection and normal renal parenchyma from tumor bearing organs. Bright-field microscopy demonstrated HBD-1 protein expression in all investigated specimens. There was no obvious difference noted in amount or intensity of staining in sections of normal and chronically infected kidneys. About 20 to 30 percent of distal tubules, loops of Henle and collecting ducts in all investigated tissue sections displayed generally strong HBD-1 cytoplasmatic staining (Fig. 2a). Positively stained structures were scattered across renal cortex and medulla localized right next to entirely negative nephrons. Generally, all glomerula and proximal tubules were negative for HBD-1 immunostaining. Negative controls incubated with preimmune serum showed no staining reaction (data not shown). In contrast, HBD-2 polyclonal antibody stained only sections from chronically infected tissue samples but no samples of normal renal tissue. In chronically infected samples HBD-2 antiserum stained distal tubules, loops of Henle and collecting ducts throughout cortex and medulla (Fig. 2b). Similar to HBD-1 staining patterns HBD-2 immunostaining were scattered across the section often localized next to corresponding parts of the nephron with no staining. Glomerula and proximal tubules were generally negative. Tissue sections from normal kidney samples showed no immunostaining with HBD-2 antiserum (Fig. 2c).

Immunostaining of formalin-fixed and paraffin-embedded sections of chronically infected kidneys with polyclonal rabbit anti-serum for HBD-1 (Fig. 2a) and HBD-2 (Fig. 2b). Strong cytoplasmatic staining by HBD-1 immunohistochemistry was experienced in 20 to 30 percent of distal tubules, loops of Henle and collecting ducts in all normal and chronically infected tissue sections. All glomerula and proximal tubules were negative for HBD-1 immunostaining. HBD-2 polyclonal antibody stained only sections from chronically infected tissue samples with moderate to strong intensity in distal tubules, loops of Henle and collecting ducts (Fig. 2b). Glomerula as well as proximal tubules showed no staining. Fig. 2c shows negative staining with preimmune rabbit serum.
HBD-1 and HBD-2 real-time RT-PCR in stimulated cell lines
As an unexpected finding, proinflammatory agents led to an increase of the relative transcription levels of HBD-1 which were corrected by normalization based on the GAPDH transcript levels (Fig. 3a,3b,3c). A 15-fold relative transcription level was noted when incubating CAKI-1 or CAL-54 with 10 ng/ml of TNFα compared to 1-fold in unstimulated controls or 3-fold when incubating with 0.1 ng/ml of TNFα. This observation was also confirmed for IL-1β and LPS stimulation to a lesser extent (Fig. 3b,3c).

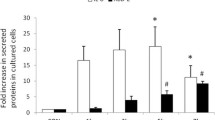
As expected relative HBD-2 transcript levels increased strongly by addition of TNFα, IL-1β, and moderated increase was noted under LPS stimulation (Fig. 4a,4b,4c). The highest relative transcription level (476×) was noted when incubating CAL-54 cells for 6 hours with 10 ng/ml of IL-1β (Fig. 4b), while incubating with the same amount of TNFα (Fig. 4a) resulted in a relative transcription level of (82×). A concentration of 100 μg/ml of LPS from Escherichia coli led to a slight increase in relative transcription levels (15×) when compared to negative controls (Fig. 4c).

Cultured CAL-54 cells were stimulated with increasing TNFα (Fig. 4a), IL-1β (Fig. 4b), and LPS (Fig. 4c) concentrations for 6 hours in FCS-free medium and HBD-2 mRNA expression was analyzed by real-time RT-PCR. Bars represent the relative HBD-2 transcript levels normalized to GAPDH transcript levels.
Sequencing of representative fragments of HBD-1 and HBD-2 specific RT-PCR proved 100% identity with known sequences from the GenBank nucleotide database (GenBank Accession No. X92744 and Z71389).
Discussion
Antimicrobial proteins are widely distributed constituents of innate host defense in animals and plants. Of the many known characteristic defensins which exert antimicrobial activity, β-defensins have been found only in vertebrates including mammals and birds [3]. Three authors have studied profoundly the expression and functional aspects of HBD-1 in urine and normal human kidneys [17, 19, 20]. In agreement with Valore's et al. investigation on HBD-1 gene expression we found constitutive HBD-1 gene expression in all examined tissue samples of normal kidneys as well as chronically infected renal specimens by RT-PCR. Albeit, in our immunostaining experiments we were able to visualize about 30 percent of positive distal tubules, Henle's loops and collecting ducts, contradicting Valore's immunostaining studies where only intraluminal hyalin substance in the renal tubulus would stain with HBD-1 antiserum [17]. Comparison of immunostaining and in situ hybridization results using digoxigenin labeled cDNA probes (data not shown) revealed corresponding patterns of positive protein and gene expression in histoanatomical structures such as distal tubules, Henle's loops and collecting ducts, whereas glomerula and proximal tubules were always non-reactive with either cDNA probes or polyclonal antiserum.
Valore and coworkers [17] observed interindividual variability of quantitative HBD-1 protein levels between 10 to 100 μg/l in urine from eight different donors. This led the authors to speculate about altenative forms of physiologic regulation of HBD-1 synthesis by non-classical immunostimulants such as estrogen and progesterone which were reported to modulate the concentration of antimicrobial lactoferrin and lysozyme [22, 23]. In agreement with this hypothesis Valore and coworkers noted the highest HBD-1 concentrations in urine from pregnant women and generally lower levels of HBD-1 in men than in women. When exploring this observation in vitro, we could not detect any differences in HBD-1 gene expression after stimulating CAL-54 and CAKI-1 cell lines with increasing concentrations of estrone, β-estradiol, estriol, progesterone, chorionic gonadotropin, or lactogen alone or in combination over variable lengths of time (data not shown).
Hiratsuka et al. [19] saw an average 3.1-fold increase in urinary HBD-1 concentration from 8 patients with pyelonephritis as compared to controls by radioimmunoassay, which support our real-time RT-PCR results of slightly inducible HBD-1 expression in vitro (Fig. 3a,3b,3c). However, when investigating HBD-1 gene expression in tissue samples by real-time RT-PCR our results where inconclusive with some pyelonephritic samples revealing a higher relative HBD-1 transcript level and others not compared to normal renal tissue samples (data not shown).
In the immunohistochemical experiments of our study we were also not able to detect obvious interindividual differences in respect to the quantity and intensity of HBD-1 reactivity. HBD-2 became known as the first human defensin that is expressed by epithelial cells after contact with microorganisms or proinflammatory cytokines [24] but at present no systematic investigation of HBD-2 expression in renal tissue has been performed so far. Current studies revealed no basal rates of HBD-2 gene and protein-expression in normal human kidney, salivary gland, small intestine or liver [5], except one study by Bals and co-workers reported HBD-2 mRNA expression in kidney extracts by dot blot hybridization using a commercially available filter [9].
As a novel finding in our study, induced gene- and protein expression of HBD-2 was detected in 14 out of 15 chronically infected kidneys in contrast to negative expression in normal non-infected renal tissue from tumor bearing kidneys. Similar qualitative HBD-2 gene expression was found in infected kidneys with either Escherichia coli, Klebsiella or Pseudomonas species as the causative pathogen for chronic bacterial kidney infection. These results were supported by cell culture studies that found upregulated HBD-2 gene expression in kidney-tumor derived cell lines CAL-54 and CAKI-1 after incubation with various proinflammatory stimuli such as TNFα, IL-1β, and higher LPS concentrations from Escherichia coli.
Our cell culture studies revealed inducible HBD-2 gene expression by TNFα, IL-1β and higher LPS concentrations from Escherichia coli. These results parallel previous findings of upregulated HBD-2 mRNA expression in cultured keratinocytes from human skin [5] airway epithelium [10, 11]. and oral mucosa [8] by proinflammatory cytokines [7]. In contrast to kidney-tumor derived cell lines CAL-54 and CAKI-1 the immortalized embryonal kidney cell line HEK-293 could not be activated by LPS or cytokines in our experiments (data not shown) which has also been previously noted by Brightbill et al. [25]
Conclusion
Similar to other epithelial systems of the human body epithelium, nephrons of human kidney have the capacity to produce a variety of different gene encoded antimicrobial proteins. These proteins may mount an immediate first line response against microbial pathogens before cellular and humoral defense mechanisms become effective at a later time. Although HBD-1 gene expression has been considered constitutive only, our in vitro results further support the observation of others that HBD-1 may also underlie regulation by proinflammatory cytokines or other unidentified factors to a noteworthy extent.
Further prospects for studies on human beta defensin expression and regulation in the uro-genital context include investigation of urothelium of the urinary bladder, prostatic glands and epithelia from female and male reproductive tracts under normal and inflammatory conditions as well as defining the role of the recently published HBD-3 [12, 15]. and HBD-4 [16] within the urogenital tract.
References
Schröder JM: Epithelial antimicrobial peptides: innate local host response elements. Cell Mol Life Sci. 1999, 56: 32-46. 10.1007/s000180050004.
Ganz T, Lehrer RI: Antimicrobial peptides of vertebrates. Curr Opin Immunol. 1998, 10: 41-44. 10.1016/S0952-7915(98)80029-0.
Schröder JM: Epithelial peptide antibiotics. Biochem Pharmacol. 1999, 57: 121-134. 10.1016/S0006-2952(98)00226-3.
Bensch KW, Raida M, Magert HJ, Schulz-Knappe P, Forssmann WG: hBD-1: a novel beta-defensin from human plasma. FEBS Lett. 1995, 368: 331-335. 10.1016/0014-5793(95)00687-5.
Harder J, Bartels J, Christophers E, Schröder JM: A peptide antibiotic from human skin. Nature. 1997, 387: 861-10.1038/43088.
Cole AM, Dewan P, Ganz T: Innate antimicrobial activity of nasal secretions. Infect Immun. 1999, 67: 3267-3275.
Weinberg A, Krisanaprakornkit S, Dale BA: Epithelial antimicrobial peptides: review and significance for oral applications. Crit Rev Oral Biol Med. 1998, 9: 399-414.
Krisanaprakornkit S, Kimball JR, Weinberg A, Darveau RP, Bainbridge BW, Dale BA: Inducible expression of human beta-defensin 2 by fusobacterium nucleatum in oral epithelial cells: multiple signaling pathways and role of commensal bacteria in innate immunity and the epithelial barrier. Infect Immun. 2000, 68: 2907-2915. 10.1128/IAI.68.5.2907-2915.2000.
Bals R, Wang X, Wu Z, Freeman T, Bafna V, Zasloff M, Wilson JM: Human beta-defensin 2 is a salt-sensitive peptide antibiotic expressed in human lung. J Clin Invest. 1998, 102: 874-880.
Singh PK, Jia HP, Wiles K, Hesselberth J, Liu L, Conway BA, Greenberg EP, Valore EV, Welsh MJ, Ganz T, et al: Production of beta-defensins by human airway epithelia. Proc Natl Acad Sci U S A. 1998, 95: 14961-14966. 10.1073/pnas.95.25.14961.
Harder J, Meyer-Hoffert U, Teran LM, Schwichtenberg L, Bartels J, Maune S, Schröder JM: Mucoid Pseudomonas aeruginosa, TNF-alpha, and IL-1beta, but not IL-6, induce human beta-defensin-2 in respiratory epithelia. Am J Respir Cell Mol Biol. 2000, 22: 714-721.
Garcia JR, Jaumann F, Schulz S, Krause A, Rodriguez-Jimenez J, Forssmann U, Adermann K, Kluver E, Vogelmeier C, Becker D, et al: Identification of a novel, multifunctional beta-defensin (human beta-defensin 3) with specific antimicrobial activity. Its interaction with plasma membranes of Xenopus oocytes and the induction of macrophage chemoattraction. Cell Tissue Res. 2001, 306: 257-264. 10.1007/s004410100433.
Haynes RJ, Tighe PJ, Dua HS: Antimicrobial defensin peptides of the human ocular surface. Br J Ophthalmol. 1999, 83: 737-741.
O'Neil DA, Porter EM, Elewaut D, Anderson GM, Eckmann L, Ganz T, Kagnoff MF: Expression and regulation of the human beta-defensins hBD-1 and hBD-2 in intestinal epithelium. J Immunol. 1999, 163: 6718-6724.
Harder J, Bartels J, Christophers E, Schroder JM: Isolation and characterization of human beta-defensin-3, a novel human inducible peptide antibiotic. J Biol Chem. 2001, 276: 5707-5713. 10.1074/jbc.M008557200.
Garcia JR, Krause A, Schulz S, Rodriguez-Jimenez FJ, Kluver E, Adermann K, Forssmann U, Frimpong-Boateng A, Bals R, Forssmann WG: Human beta-defensin 4: a novel inducible peptide with a specific salt-sensitive spectrum of antimicrobial activity. FASEB J. 2001, 15: 1819-1821.
Valore EV, Park CH, Quayle AJ, Wiles KR, McCray PB, Ganz T: Human beta-defensin-1: an antimicrobial peptide of urogenital tissues. J Clin Invest. 1998, 101: 1633-1642.
Schnapp D, Reid CJ, Harris A: Localization of expression of human beta defensin-1 in the pancreas and kidney. J Pathol. 1998, 186: 99-103. 10.1002/(SICI)1096-9896(199809)186:1<99::AID-PATH133>3.0.CO;2-#.
Hiratsuka T, Nakazato M, Ihi T, Minematsu T, Chino N, Nakanishi T, Shimizu A, Kangawa K, Matsukura S: Structural analysis of human beta-defensin-1 and its significance in urinary tract infection. Nephron. 2000, 85: 34-40. 10.1159/000045627.
Zucht HD, Grabowsky J, Schrader M, Liepke C, Jurgens M, Schulz-Knappe P, Forssmann WG: Human beta-defensin-1: A urinary peptide present in variant molecular forms and its putative functional implication. Eur J Med Res. 1998, 3: 315-323.
Chomczynski P, Sacchi N: Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987, 162: 156-159. 10.1006/abio.1987.9999.
Cohen MS, Britigan BE, French M, Bean K: Preliminary observations on lactoferrin secretion in human vaginal mucus: variation during the menstrual cycle, evidence of hormonal regulation, and implications for infection with Neisseria gonorrhoeae. Am J Obstet Gynecol. 1987, 157: 1122-1125.
Newbold RR, Teng CT, Beckman WC, Jefferson WN, Hanson RB, Miller JV, McLachlan JA: Fluctuations of lactoferrin protein and messenger ribonucleic acid in the reproductive tract of the mouse during the estrous cycle. Biol Reprod. 1992, 47: 903-915.
Schröder JM, Harder J: Human beta-defensin-2. Int J Biochem Cell Biol. 1999, 31: 645-651. 10.1016/S1357-2725(99)00013-8.
Brightbill HD, Libraty DH, Krutzik SR, Yang RB, Belisle JT, Bleharski JR, Maitland M, Norgard MV, Plevy SE, Smale ST, et al: Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science. 1999, 285: 732-736. 10.1126/science.285.5428.732.
Paulsen FP, Pufe T, Schaudig U, Held-Feindt J, Lehmann J, Schroder JM, Tillman BN: Detection of natural peptide antibiotics in human nasolacrimal ducts. Invest Ophthalmol Vis Sci. 2001, 42: 2157-63.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2334/2/20/prepub
Acknowledgements
We gratefully acknowledge the gift of polyclonal HBD-1 and HBD-2 antibodies from Tom Ganz, Erika Valore and Lide Liu. We are indebted to Jens Schröder for insightful discussions and Jutta Lüttges for assistance in analysing immunostaining results. We thank Jan Eggers, Holger Meyhoff, Olivera Batic, and Helga Angeli for their excellent technical assistance. This research was supported by the Gunther Voges Stiftung, Homburg/Saar, and a program grant (LE 1213/1-1) from the "Deutsche Forschungsgemeinschaft" (German Research Foundation).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
None declared.
Author's contributions
JL performed tissue asservation, supervised and coordinated all lab methods, junior head of the laboratory, applicant for funding, drafted the manuscript. MR tissue asservation, senior head of the laboratory, applicant for funding. JH performed real-time RT-PCR, provided RNA from stimulated nasal epithelium as HBD-2 positive RT-PCR control. MK advisor for immunohistochemical staining methods, analysed immunohistochemical staining results and confirmed histopathologic diagnosis of tissue samples. UK advisor for immunohistochemical staining methods, analysed immunohistochemical staining results and confirmed histopathologic diagnosis of tissue samples. JH performed in vitro stimulation of cell lines, cell line RT-PCR. KH performed RNA extraction from tissue samples, tissue RNA RT-PCR. UR performed sectioning of paraffin embedded tissues and immunohistochemical staining. MG performed tissue asservation. TL applicant for funding, interim Director of the Department of Urology, Kiel, Germany. KW-J interim Director of the Department of Urology, Kiel, Germany. MS Director of the Department of Urology, Homburg/Saar, Germany and former Director of the Department of Urology, Kiel, Germany. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Lehmann, J., Retz, M., Harder, J. et al. Expression of human beta-defensins 1 and 2 in kidneys with chronic bacterial infection. BMC Infect Dis 2, 20 (2002). https://doi.org/10.1186/1471-2334-2-20
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2334-2-20