
Abstract
Background
The immunogenicity, reactogenicity, and safety of the 10-valent pneumococcal non-typeable Haemophilus influenzae protein D conjugate vaccine (PHiD-CV) co-administered with routine childhood vaccines were evaluated among infants from Singapore and Malaysia, where PHiD-CV has been licensed.
Methods
In the primary vaccination phase, 298 infants from Singapore and 168 infants from Malaysia were randomised to receive the Phase III Clinical (Clin) or the Commercial (Com) lot of PHiD-CV at 2, 3, and 5 months of age. In the booster vaccination phase, 238 toddlers from Singapore received one dose of the PHiD-CV Commercial lot at 18–21 months of age. Immune responses to pneumococcal polysaccharides were measured using 22F-inhibition enzyme-linked immunosorbent assay (ELISA) and functional opsonophagocytic activity (OPA) assay and to protein D, using ELISA.
Results
Immune responses induced by primary vaccination with the PHiD-CV Commercial lot were non-inferior to the Phase III Clinical lot in terms of adjusted antibody geometric mean concentration (GMC) ratios for each vaccine pneumococcal serotype and protein D. For each vaccine pneumococcal serotype, ≥93.6% and ≥88.5% of infants from Malaysia and Singapore had post-primary vaccination antibody concentrations ≥0.2 μg/mL and OPA titres ≥8, in the Clin and Com groups, respectively. For each vaccine pneumococcal serotype, ≥60.8% and ≥98.2% of toddlers from Singapore had pre- and post-booster antibody concentrations ≥0.2 μg/mL, in the Clin and Com groups, respectively. All children, except one, had measurable anti-protein D antibodies and the primary and booster doses of the co-administered vaccines were immunogenic. The incidence of each grade 3 solicited symptom was ≤11.1% in both study phases. No serious adverse events considered causally related to vaccination were reported throughout the study.
Conclusions
PHiD-CV given as three-dose primary vaccination to infants in Singapore and Malaysia and booster vaccination to toddlers in Singapore was shown to be immunogenic with a clinically acceptable-safety profile.
This study has been registered at http://www.clinicaltrials.gov NCT00808444 and NCT01119625.
Similar content being viewed by others


Background
Streptococcus pneumoniae is responsible for invasive diseases, which cause significant morbidity and mortality worldwide [1]. The incidence of invasive pneumococcal disease (IPD) is especially high in Asia, where children younger than 5 years old are the most severely affected [2–5]. In Singapore, the incidence of IPD reached 15.2 per 100,000 children <5 years of age in 2008–2010; the most common serotypes were serotypes 6B, 19A, 14, and 23F [6]. In Malaysia, there is limited information on the incidence of IPD, although a previous study suggested that the incidence of pneumococcal meningitis reached 8.6 per 100,000 children <5 years of age in 2004–2006 [7]. The most common serotypes in Malaysia in 2008–2009 were serotypes 19F, 6B, 19A, and 14 [6, 8]. In both countries, emergence of antimicrobial resistant S. pneumoniae isolates is a major health concern [3, 8–14].
Prevention of pneumococcal infections through vaccination remains the best strategy to reduce the incidence of IPD. A 10-valent pneumococcal non-typeable Haemophilus influenzae (NTHi) protein D conjugate vaccine (PHiD-CV; Synflorix™, GlaxoSmithKline Vaccines), which contains serotypes 1, 4, 5, 6B, 7F, 9V, 14, 18C, 19F, and 23F, was licensed in Singapore in March 2010 and in Malaysia in September 2009. PHiD-CV has been shown to be immunogenic and well-tolerated when co-administered with commonly used paediatric vaccines in infants and toddlers [15–22]. Since eight of the ten pneumococcal serotypes included in PHiD-CV are conjugated to NTHi protein D, this vaccine has the potential to provide additional protection against NTHi diseases [23].
This study compared the immunogenicity, reactogenicity, and safety of three-dose primary vaccination with a Phase III Clinical lot or a Commercial lot of PHiD-CV co-administered with routine childhood vaccines in infants from Singapore and Malaysia. The immunogenicity and safety of a booster dose of the PHiD-CV Commercial lot were also evaluated in toddlers from Singapore.
Methods
Study design
This Phase III study comprised a primary vaccination phase conducted in three centres in Singapore and two centres in Malaysia between January 2009 and November 2009, and a booster vaccination phase conducted in the three Singaporean centres between July 2010 and February 2011. The booster vaccination phase did not encompass Malaysia since PHiD-CV obtained registration in Malaysia in September 2009 and booster vaccination could thus be offered outside clinical trial settings.
In the primary vaccination phase, infants were randomised (1:1) to two parallel groups to receive three doses of the PHiD-CV Phase III Clinical (Clin group) or Commercial lot (Com group) at 2, 3, and 5 months of age. PHiD-CV was co-administered with diphtheria, tetanus, acellular pertussis-hepatitis B virus-inactivated poliovirus, and H. influenzae type b vaccine (DTPa-HBV-IPV/Hib) in Malaysia at 2, 3, and 5 months of age and in Singapore at 2 and 5 months of age and with DTPa-IPV/Hib in Singapore at 3 months of age. All infants received two doses of a human rotavirus (HRV) vaccine at 2 and 3 months of age. The primary vaccination phase was double-blinded.
In the booster vaccination phase, all toddlers from Singapore received a booster dose of the PHiD-CV Commercial lot co-administered with DTPa-IPV/Hib at 18–21 months of age. Thus, the booster vaccination phase was conducted in an open-label manner.
The study was conducted in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki. The protocol and associated documents were reviewed and approved by the Medical Research & Ethics Committee of the Ministry of Health in Malaysia and the Medical Ethics Committee of University Malaya Medical Centre and the Domain-Specific Review Board of the National Healthcare Group in Singapore. Written informed consent was obtained before enrolment from the parents or legally acceptable representatives of each child. This study has been registered at http://www.clinicaltrials.gov NCT00808444 and NCT01119625. A protocol summary is available at http://www.gsk-clinicalstudyregister.com (GSK study IDs 111654 and 113266).
Study objectives
The primary objectives were to demonstrate the comparability of the immune response induced by three-dose primary vaccination with the Commercial lot versus the Phase III Clinical lots of PHiD-CV in infants from Malaysia and Singapore, and to assess the persistence of the antibodies induced by both PHiD-CV lots up to the booster vaccination in toddlers from Singapore. Secondary objectives included the evaluation of the immunogenicity, safety, and reactogenicity of PHiD-CV and the co-administered vaccines after primary and booster vaccinations.
Study participants
Eligible participants were healthy infants from Malaysia and Singapore aged 6–12 weeks at the time of the first vaccination, who were born after a gestation period of between 36 and 42 weeks. For the booster vaccination phase, eligible participants were healthy toddlers aged 18–21 months at the time of the booster vaccination, who had received three PHiD-CV doses in the primary vaccination phase in Singapore.
Children were excluded from participation if they were concurrently participating in another clinical study; were immunosuppressed; had used investigational products within 30 days pre-vaccination; had previously received blood products; had previous vaccination against or history of diphtheria, tetanus, pertussis, poliomyelitis, hepatitis B, Hib, or pneumococcal disease; or had allergic disease or reactions likely to be exacerbated by the vaccines, history of neurological disorders or seizures, major congenital defects, or a serious chronic illness.
Vaccines
The Commercial and Phase III Clinical lots of PHiD-CV had the same composition, which has been previously described [21]. They were administered intramuscularly in the right thigh in infants and the right deltoid or thigh in toddlers. All co-administered vaccines were manufactured by GlaxoSmithKline Vaccines. DTPa-IPV/Hib (Infanrix™-IPV/Hib) and DTPa-HBV-IPV/Hib (Infanrix™ hexa) were administered intramuscularly in the left thigh in infants and the left deltoid or thigh in toddlers. The live attenuated HRV vaccine (Rotarix™) was administered orally.
Immunogenicity assessment
Blood samples were collected one month post-primary vaccination and before and one month after the booster vaccination. Antibody concentrations against vaccine pneumococcal serotypes and cross-reactive serotypes were measured by 22F-inhibition enzyme-linked immunosorbent assay (ELISA) as described previously (cut-off: 0.05 μg/mL) [24, 25]. Percentages of children with antibody concentrations ≥0.2 μg/mL were determined. Antibody concentrations of 0.2 μg/mL measured by 22F-inhibition ELISA are equivalent to antibody concentrations of 0.35 μg/mL measured by the World Health Organization reference ELISA without 22F-inhibition, which is the threshold used for comparisons of immune responses induced by different pneumococcal conjugate vaccines [25, 26]. In the primary vaccination phase, opsonophagocytic activity (OPA) was measured by a pneumococcal killing assay using a HL 60 cell line (cut-off titre: 8) [27, 28]. Antibodies against NTHi protein D were measured by an in-house ELISA using a recombinant non-lipidated form of protein D as coating material. Anti-protein D antibodies, which are bound to protein D antigens adsorbed on polystyrene plates, are detected using an anti-human-IgG peroxidase labelled antibody followed by the addition of tetramethylbenzidine substrate. Anti-protein D antibody concentrations were expressed in ELISA units per mL (EL.U/mL), and the assay cut-off was 100 EL.U/mL.
Infants were sub-randomised (1:1) to two subsets for the analysis of immune responses to co-administered vaccines. Immune responses were measured by ELISA [29–34], except for the three poliovirus types, which were measured by a virus microneutralisation test [35]. All laboratory analyses were performed at GlaxoSmithKline (Rixensart, Belgium) or in laboratories designated by GlaxoSmithKline Vaccines. Upon discovery of an anti-hepatitis B assay specificity issue, a risk was identified that children whose post-vaccination titres were initially measured between 10 and 100 mIU/mL might have been informed of an incorrect seroprotection status. Retesting was performed as an urgent precaution using Immulite™, a commercial assay used for routine testing at Ghent University Hospital, Belgium.
Safety and reactogenicity assessment
Solicited local (injection sites pain, redness, and swelling) and general (irritability, drowsiness, fever [rectal/axillary temperature ≥38.0°C/37.5°C], and loss of appetite) symptoms and antipyretic use were recorded during a four-day post-vaccination period. In the primary vaccination phase, vomiting and diarrhoea were also recorded for four days post-vaccination. Unsolicited adverse events (AEs) were recorded for 31 days post-vaccination. The intensity of each symptom was graded on a three-grade scale. Grade 3 was reported for pain if infants/toddlers cried when the limbs were moved or if the limbs were spontaneously painful; redness and swelling, if diameters were >30 mm; fever, if rectal/axillary temperature was >40.0°C/39.5°C; loss of appetite, if infants/toddlers did not eat at all; irritability, if infants/toddlers cried and could not be comforted; diarrhoea, if infants had ≥6 looser than normal stools/day; vomiting, if infants had ≥3 episodes/day; and all other AEs, if they prevented normal activity.
Serious adverse events (SAEs) were recorded throughout the study and were defined as events that were life-threatening, required hospitalisation or prolongation of hospitalisation, or resulted in disability, incapacity, or death. As per protocol, all solicited local symptoms were considered causally related to vaccination. Causality of all other AEs was assessed by investigators.
Statistical analyses
Safety analyses were performed on the primary and booster total vaccinated cohorts. Immunogenicity analyses were performed on the primary and booster according-to-protocol (ATP) immunogenicity cohorts and on the ATP persistence cohort, which included all infants/toddlers who met all eligibility criteria, complied with protocol-defined procedures, and for whom antibody assay results were available.
Antibody geometric mean concentrations (GMCs), OPA geometric mean titres (GMTs), and percentages of infants/toddlers with concentrations or titres above pre-specified cut-offs/thresholds were calculated with 95% confidence intervals (CIs). GMCs and GMTs were calculated by taking the anti-log of the mean of the log antibody concentration/titre transformations. Antibody concentrations/titres below assay cut-offs were given an arbitrary value of half the cut-off.
Non-inferiority of the Commercial versus the Phase III Clinical lot was demonstrated if the upper limit (UL) of the two-sided 95% CI (calculated using an ANOVA model [pooled variance] adjusting for multi-country effect) on adjusted GMC ratios (Clin over Com group) for antibodies against vaccine pneumococcal serotypes and protein D was below 2.
In the primary vaccination phase, the target sample size was 460 enrolled infants to obtain ≥400 evaluable infants. When comparing both PHiD-CV lots, 200 evaluable infants per group would provide at least 98% power under equal mean or 85% power in case of 1.2-fold decrease in GMCs to show non-inferiority of the Commercial lot compared to the Phase III Clinical lot with respect to adjusted antibody GMC ratios for vaccine pneumococcal serotypes and protein D. In the booster vaccination phase, the target sample size was 298 enrolled toddlers, taking into account the actual enrolment in the primary vaccination phase in Singapore.
Incidences of solicited and unsolicited AEs were calculated with exact 95% CIs. SAEs and withdrawals due to AEs were described in detail. Non-overlapping two-sided 95% CIs were used to highlight potential group differences, which should be interpreted with caution.
Statistical analyses were performed using Statistical Analysis System (SAS® software, SAS Institute Inc., Cary, NC, United States) version 9.2 and SDD (i.e. SAS Drug and Development) web portal version 3.5.
Results
Study population
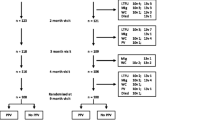
A total of 466 infants were enrolled and 464 infants completed the primary vaccination phase (Figure 1). 238 toddlers from Singapore were included in the booster vaccination phase and 231 toddlers completed the study. The demographic characteristics of the infants included in the primary ATP immunogenicity cohort were comparable in both groups and were consistent with those of the toddlers from Singapore included in the booster ATP immunogenicity cohort (Table 1).

Trial profile. Clin = group of infants from Malaysia and Singapore who received the Phase III Clinical lot of PHiD-CV in the primary vaccination phase. Com = group of infants from Malaysia and Singapore who received the Commercial lot of PHiD-CV in the primary vaccination phase. ClinCom = group of toddlers from Singapore primed with the Phase III Clinical lot of PHiD-CV who received the Commercial lot of PHiD-CV in the booster vaccination phase. ComCom = group of toddlers from Singapore primed with the Commercial lot of PHiD-CV who received the Commercial lot of PHiD-CV in the booster vaccination phase.
Immunogenicity
Pneumococcal vaccine antigens
In the primary vaccination phase, immune responses induced by the PHiD-CV Commercial lot were shown to be non-inferior to those induced by the Phase III Clinical lot (Figure 2; Additional file 1: Table S1).

Adjusted antibody GMC ratios between the Clin and the Com groups for the 10 vaccine pneumococcal serotypes and protein D at one month post-primary vaccination (primary ATP immunogenicity cohort). Clin = group of infants from Malaysia and Singapore who received the Phase III Clinical lot of PHiD-CV in the primary vaccination phase. Com = group of infants from Malaysia and Singapore who received the Commercial lot of PHiD-CV in the primary vaccination phase. Adjusted antibody GMC ratio = ratio of the geometric mean concentration at one month post-primary vaccination adjusted for country (pooled variance; Clin over Com group). PD = protein D. 95% CI = 95% confidence intervals (represented by the error bars). *Immunological non-inferiority was demonstrated if the upper limit of the 95% confidence interval of the adjusted antibody GMC ratio (Clin over Com) was below 2.0.
One month post-primary vaccination, for each vaccine pneumococcal serotype, ≥96.3% and ≥93.6% of infants had antibody concentrations ≥0.2 μg/mL in the Clin and Com groups, respectively (Table 2). Antibody GMCs for serotypes 4 and 5 seemed higher in the Clin group. For each vaccine pneumococcal serotype, percentages of infants with OPA titres ≥8 were ≥95.6% in the Clin group, except for serotype 1, and ≥96.6% in the Com group, except for serotypes 1 and 6B (Table 3). OPA GMTs for serotype 18C seemed higher in the Clin group.
For each vaccine pneumococcal serotype, ≥71.8% and ≥60.8% of toddlers at pre-booster vaccination, and ≥99.1% and ≥98.2% of toddlers one month post-booster vaccination, had antibody concentrations ≥0.2 μg/mL in the ClinCom and ComCom groups, respectively (Table 4).
One month post-primary vaccination, for cross-reactive serotypes 6A and 19A, ≥60.6% and ≥54.6% of infants had antibody concentrations ≥0.2 μg/mL (Table 2), and ≥85.5% and ≥37.7% of infants had OPA titres ≥8, in the ClinCom and ComCom groups, respectively (Table 3). One month post-booster vaccination, for each cross-reactive serotype, ≥93.4% of toddlers reached antibody concentrations ≥0.2 μg/mL (Table 4).One month post-primary vaccination, ≥99.5% of infants had measurable anti-protein D antibodies and higher anti-protein D adjusted antibody GMCs were observed in the Clin group (Figure 2). The anti-protein D antibody seropositivity rates were 100% and 98.3% of toddlers at pre-booster vaccination, and 100% and 99.1% of toddlers at one month post-booster vaccination in the ClinCom and ComCom groups, respectively (data not shown).
Co-administered vaccine antigens
One month post-primary and post-booster vaccination, all children were seroprotected against diphtheria, tetanus and poliovirus types 1, 2 and 3, and all infants were seropositive for antibodies against the three pertussis antigens and polyribosylribitol phosphate from Hib. All infants except one were seroprotected against hepatitis B surface antigen (HBs) at one month post-primary vaccination. Three children had anti-HBs antibody concentrations which were initially measured between 10 and 100 mIU/mL. Out of these 3 children, upon retesting using Immulite™, one was found to be seroprotected while the other 2 had anti-HBs antibody concentrations <10 mIU/mL. This was further followed up by investigators and one child was subsequently re-vaccinated. Efforts to reach the other child for re-vaccination were unsuccessful. Three months after the second dose of the HRV vaccine, ≥81.9% of infants were seropositive for anti-HRV antibodies (data not shown).
Safety
In the primary vaccination phase, pain and diarrhoea were the most common overall per dose Grade 3 solicited local and general symptoms, respectively (Figure 3a). Diarrhoea was the most common Grade 3 solicited general symptom considered causally related to vaccination (following 15/698 and 22/698 doses in Clin and Com groups, respectively). Antipyretic use was reported after 296/698 and 276/699 doses, and antipyretics were given prophylactically after 45/698 and 41/699 doses, in the Clin and Com groups, respectively. During 31 days post-vaccination, 113/698 and 119/699 doses were followed by at least one unsolicited AE in the Clin and Com groups, respectively. The most frequently reported unsolicited AEs were upper respiratory tract infections (35/698 doses) and cough (10/698 doses) in the Clin group, and upper respiratory tract infections (42/699 doses) and pyrexia (7/699 doses) in the Com group. No Grade 3 unsolicited AEs were considered causally related to vaccination. SAEs were reported in 18 and 7 infants in the Clin and Com groups, respectively; none were considered causally related to vaccination and none were fatal.In the booster vaccination phase, the most common Grade 3 solicited local and general symptoms were pain and irritability, respectively (Figure 3b). Grade 3 solicited general symptoms considered causally related to vaccination were reported in ≤4.3% of toddlers, including Grade 3 fever in one toddler (0.9%) in each group. Antipyretic use was reported in 50/118 and 66/120 toddlers, and prophylactic antipyretics use in 7/118 and 4/120 toddlers, in the ClinCom and ComCom groups, respectively. Unsolicited AEs were reported in 18/118 and 25/120 toddlers in the ClinCom and ComCom groups, respectively. The most frequently reported unsolicited AEs were rhinorrhea (7/118 toddlers) and cough (5/118 toddlers) in the ClinCom group, and upper respiratory tract infections (6/120 toddlers) and rhinorrhea and pyrexia (each in 5/120 toddlers) in the ComCom group. One Grade 3 unsolicited AE considered causally related to vaccination (urticaria) was reported in the ComCom group. Four toddlers in the ComCom group reported SAEs; none were considered causally related to vaccination and none were fatal.

Solicited symptoms following (a) primary vaccination (overall/dose; primary TVC) and (b) booster dose (booster TVC). TVC = total vaccinated cohort. Clin = group of infants from Malaysia and Singapore who received the Phase III Clinical lot of PHiD-CV in the primary vaccination phase. Com = group of infants from Malaysia and Singapore who received the Commercial lot of PHiD-CV in the primary vaccination phase. ClinCom = group of toddlers from Singapore primed with the Phase III Clinical lot of PHiD-CV who received the Commercial lot of PHiD-CV in the booster vaccination phase. ComCom = group of toddlers from Singapore primed with the Commercial lot of PHiD-CV who received the Commercial lot of PHiD-CV in the booster vaccination phase. Error bars represent 95% confidence intervals. Solicited symptoms are recorded during 4-days post-vaccination.
Discussion
Three primary doses of PHiD-CV given to 2-, 3-, and 5-month-old infants from Malaysia and Singapore were shown to be immunogenic with a clinically acceptable safety profile. Non-inferiority of the immunogenicity of the PHiD-CV Commercial versus the Phase III Clinical lot was demonstrated for each vaccine pneumococcal serotype and protein D, although antibody GMCs for serotypes 4 and 5 and protein D, and OPA GMTs for serotype 18C, seemed higher in infants who received the Phase III Clinical lot. Nonetheless, as percentages of infants reaching threshold antibody concentrations and OPA titres for vaccine antigens were high and comparable in both groups, the observed differences in immunogenicity may be of limited clinical relevance. For each vaccine pneumococcal serotype, post-primary vaccination antibody GMCs seemed in line with those previously measured in Korea and Taiwan [16, 19], and higher than in Europe [15, 18, 21]. Although it remains unclear why the magnitude of immune responses to pneumococcal conjugate vaccines varies in different populations, plausible explanations include genetic factors, early exposure to S. pneumoniae, or nasopharyngeal carriage of pneumococcal serotypes [36]. Pre-vaccination antibody concentration, which is influenced by waning maternal antibodies and increasing adaptive immunity due to early exposure to S. pneumoniae, in Asian children was evaluated in a previous study conducted in Taiwan, where for each vaccine pneumococcal serotype the percentage of children with pre-vaccination antibody concentrations ≥0.2 μg/ml ranged from 11.5% to 42.5%, except serotype 14 (61%) [19]. Vaccine efficacy and effectiveness of pneumococcal conjugate vaccines have been demonstrated in various countries, hence clinical relevance of population differences remains unknown [37–42].
For each vaccine pneumococcal serotype, antibody GMCs calculated before the booster dose administration were lower than those measured one month post-primary vaccination, but ≥60.8% of toddlers from Singapore had pre-booster antibody concentrations ≥0.2 μg/mL. The booster dose of the PHiD-CV Commercial lot induced a robust immune response; virtually all toddlers reached antibody concentrations ≥0.2 μg/mL one month post-booster vaccination and antibody concentrations were higher than those measured post-primary vaccination, which suggested that primary vaccination of infants with PHiD-CV induced immunological memory [15, 16, 18, 21].
Primary vaccination with either vaccine lot and booster vaccination with the PHiD-CV Commercial lot induced immune responses against cross-reactive serotypes 6A and 19A. Some functional OPA responses for cross-reactive serotype 19A were observed following primary PHiD-CV vaccination, in-line with previous studies [15, 16, 18, 21]. However, the level of protection against serotype 19A conferred by the immune response induced by PHiD-CV should be further determined [43, 44].
Both PHiD-CV lots induced antibodies against the NTHi protein D carrier, which could potentially provide protection against disease caused by NTHi. Although a clear correlation between efficacy and anti-protein D antibody concentrations has not been established, efficacy trials with the predecessor 11-valent NTHi protein D-conjugated vaccine and PHiD-CV have suggested that the protein D carrier contributed to the induction of protection against acute otitis media due to NTHi [45, 46]. The co-administered vaccines given simultaneously with primary or booster doses of PHiD-CV were also immunogenic. This is consistent with other studies and suggests that no clinically relevant interference occurred between PHiD-CV and HRV or DTPa-based vaccines [17, 19].
No SAEs considered causally related to vaccination and no fatal SAEs were reported throughout the study. The reactogenicity and safety profiles of the Commercial and the Phase III Clinical lots of PHiD-CV were comparable and clinically acceptable, in line with previous studies [20].
The primary vaccination phase was powered to demonstrate the primary objectives. Other comparisons should be considered cautiously, since there was no adjustment for multiple comparisons of the various endpoints. The clinical relevance of the observed differences remains unknown, especially for antigens with no correlate of protection. The booster vaccination phase was also limited by its open design.
Conclusions
This study showed that different lots of PHiD-CV were immunogenic with a clinically acceptable safety profile when given as 3-dose primary vaccination in 2-, 3- and 5-month-old infants from Malaysia and Singapore. A booster dose of the PHiD-CV Commercial lot induced a robust immune response in 18–21 month-old toddlers from Singapore. Both PHiD-CV lots were administered with other paediatric vaccines without compromising their immune response.
Synflorix, Infanrix-IPV/Hib, and Infanrix hexa are trademarks of the GlaxoSmithKline group of companies.
Abbreviations
- AE:
-
Adverse event
- ANOVA:
-
Analysis of variance
- ATP:
-
According to protocol
- CI:
-
Confidence interval
- DTPa-HBV-IPV/Hib:
-
Diphtheria, tetanus, acellular pertussis-hepatitis B virus-inactivated poliovirus, and H. influenzae type b vaccine
- ELISA:
-
Enzyme-linked immunosorbent assay
- GMC:
-
Geometric mean concentration
- GMT:
-
Geometric mean titre
- HRV:
-
Human rotavirus
- IPD:
-
Invasive pneumococcal disease
- OPA:
-
Opsonophagocytic activity
- PHiD-CV:
-
Pneumococcal non-typeable Haemophilus influenzae (NTHi) protein D conjugate vaccine
- SAE:
-
Serious adverse event.
References
O'Brien KL, Wolfson LJ, Watt JP, Henkle E, Deloria-Knoll M, McCall N, Lee E, Mulholland K, Levine OS, Cherian T: Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet. 2009, 374: 893-902. 10.1016/S0140-6736(09)61204-6.
Scott JA: The preventable burden of pneumococcal disease in the developing world. Vaccine. 2007, 25: 2398-2405. 10.1016/j.vaccine.2006.09.008.
Bravo LC: Overview of the disease burden of invasive pneumococcal disease in Asia. Vaccine. 2009, 27: 7282-7291. 10.1016/j.vaccine.2009.04.046.
Lin TY, Shah NK, Brooks D, Garcia CS: Summary of invasive pneumococcal disease burden among children in the Asia-Pacific region. Vaccine. 2010, 28: 7589-7605. 10.1016/j.vaccine.2010.07.053.
Jauneikaite E, Jefferies JM, Hibberd ML, Clarke SC: Prevalence of Streptococcus pneumoniae serotypes causing invasive and non-invasive disease in South East Asia: a review. Vaccine. 2012, 30: 3503-3514. 10.1016/j.vaccine.2012.03.066.
Thoon KC, Chong CY, Tee NW: Early impact of pneumococcal conjugate vaccine on invasive pneumococcal disease in Singapore children, 2005 through 2010. Int J Infect Dis. 2012, 16: e209-e215.
Suhaimi M, Asmiati AH, Soo TL: Presented at: 5th World Congress of the World Society for Pediatric Infectious Diseases (WSPID). Pneumococcal meningitis in children aged 2–60 months at Queen Elizabeth (QE) Hospital, Sabah, Malaysia: before and after the introduction of Hemophilus influenza type b vaccination. 2007, Bangkok, Thailand
Yasin RM, Zin NM, Hussin A, Nawi SH, Hanapiah SM, Wahab ZA, Raj G, Shafie N, Peng NP, Chu KK, Aziz MN, Maning N, Mohamad JS, Benjamin A, Salleh MA, Zahari SS, Francis A, Ahmad N, Karunakaran R: Current trend of pneumococcal serotypes distribution and antibiotic susceptibility pattern in Malaysian hospitals. Vaccine. 2011, 29: 5688-5693. 10.1016/j.vaccine.2011.06.004.
Soh SW, Poh CL, Lin RV: Serotype distribution and antimicrobial resistance of Streptococcus pneumoniae isolates from pediatric patients in Singapore. Antimicrob Agents Chemother. 2000, 44: 2193-2196. 10.1128/AAC.44.8.2193-2196.2000.
Song JH, Jung SI, Ko KS, Kim NY, Son JS, Chang HH, Ki HK, Oh WS, Suh JY, Peck KR, Lee NY, Yang Y, Lu Q, Chongthaleong A, Chiu CH, Lalitha MK, Perera J, Yee TT, Kumarasinghe G, Jamal F, Kamarulzaman A, Parasakthi N, Van PH, Carlos C, So T, Ng TK, Shibl A: High prevalence of antimicrobial resistance among clinical Streptococcus pneumoniae isolates in Asia (an ANSORP study). Antimicrob Agents Chemother. 2004, 48: 2101-2107. 10.1128/AAC.48.6.2101-2107.2004.
Chong CY, Koh-Cheng T, Yee-Hui M, Nancy TW: Invasive pneumococcal disease in Singapore children. Vaccine. 2008, 26: 3427-3431. 10.1016/j.vaccine.2008.04.035.
Vasoo S, Singh K, Hsu LY, Chiew YF, Chow C, Lin RT, Tambyah PA: Increasing antibiotic resistance in Streptococcus pneumoniae colonizing children attending day-care centres in Singapore. Respirology. 2011, 16: 1241-1248. 10.1111/j.1440-1843.2011.02036.x.
Le CF, Palanisamy NK, Mohd Yusof MY, Sekaran SD: Capsular serotype and antibiotic resistance of Streptococcus pneumoniae isolates in Malaysia. PLoS One. 2011, 6: e19547-10.1371/journal.pone.0019547.
Nathan JJ, Taib NM, Desa MN, Masri SN, Yasin R, Jamal F, Sagineedu SR, Karunanidhi A: Prevalence of macrolide resistance and in vitro activities of six antimicrobial agents against clinical isolates of Streptococcus pneumoniae from a multi-center surveillance in Malaysia. Med J Malaysia. 2013, 68: 119-124.
Bermal N, Szenborn L, Chrobot A, Alberto E, Lommel P, Gatchalian S, Dieussaert I, Schuerman L: The 10-valent pneumococcal non-typeable Haemophilus influenzae protein D conjugate vaccine (PHiD-CV) coadministered with DTPw-HBV/Hib and poliovirus vaccines: assessment of immunogenicity. Pediatr Infect Dis J. 2009, 28: S89-S96. 10.1097/INF.0b013e318199f901.
Kim CH, Kim JS, Cha SH, Kim KN, Kim JD, Lee KY, Kim HM, Kim JH, Hyuk S, Hong JY, Park SE, Kim YK, Kim NH, Fanic A, Borys D, Ruiz-Guinazu J, Moreira M, Schuerman L, Kim KH: Response to primary and booster vaccination with 10-valent pneumococcal nontypeable Haemophilus influenzae protein D conjugate vaccine in Korean infants. Pediatr Infect Dis J. 2011, 30: e235-e243. 10.1097/INF.0b013e31822a8541.
Knuf M, Szenborn L, Moro M, Petit C, Bermal N, Bernard L, Dieussaert I, Schuerman L: Immunogenicity of routinely used childhood vaccines when coadministered with the 10-valent pneumococcal non-typeable Haemophilus influenzae protein D conjugate vaccine (PHiD-CV). Pediatr Infect Dis J. 2009, 28: S97-S108. 10.1097/INF.0b013e318199f61b.
Wysocki J, Tejedor JC, Grunert D, Konior R, Garcia-Sicilia J, Knuf M, Bernard L, Dieussaert I, Schuerman L: Immunogenicity of the 10-valent pneumococcal non-typeable Haemophilus influenzae protein D conjugate vaccine (PHiD-CV) when coadministered with different Neisseria meningitidis serogroup C conjugate vaccines. Pediatr Infect Dis J. 2009, 28: S77-S88. 10.1097/INF.0b013e318199f609.
Lin TY, Lu CY, Chang LY, Chiu CH, Huang YC, Bock HL, Tang H, François N, Moreira M, Schuerman L, Huang LM: Immunogenicity and safety of 10-valent pneumococcal non-typeable Haemophilus influenzae protein D-conjugate vaccine (PHiD-CV) co-administered with routine childhood vaccines in Taiwan. J Formos Med Assoc. 2012, 111: 495-503. 10.1016/j.jfma.2011.07.014.
Chevallier B, Vesikari T, Brzostek J, Knuf M, Bermal N, Aristegui J, Borys D, Cleerbout J, Lommel P, Schuerman L: Safety and reactogenicity of the 10-valent pneumococcal non-typeable Haemophilus influenzae protein D conjugate vaccine (PHiD-CV) when coadministered with routine childhood vaccines. Pediatr Infect Dis J. 2009, 28: S109-S118. 10.1097/INF.0b013e318199f62d.
Vesikari T, Wysocki J, Chevallier B, Karvonen A, Czajka H, Arsène JP, Lommel P, Dieussaert I, Schuerman L: Immunogenicity of the 10-valent pneumococcal non-typeable Haemophilus influenzae protein D conjugate vaccine (PHiD-CV) compared to the licensed 7vCRM vaccine. Pediatr Infect Dis J. 2009, 28: S66-S76. 10.1097/INF.0b013e318199f8ef.
Lalwani S, Chatterjee S, Chhatwal J, Verghese VP, Mehta S, Shafi F, Borys D, Moreira M, Schuerman L: Immunogenicity, safety, and reactogenicity of the 10-valent pneumococcal non-typeable Hemophilus influenzae protein D conjugate vaccine (PHiD-CV) when co-administered with the DTPw-HBV/Hib vaccine in Indian infants: a single-blind, randomized, controlled study. Hum Vaccin Immunother. 2012, 8: 612-622. 10.4161/hv.19287.
Bakaletz LO, Kennedy BJ, Novotny LA, Duquesne G, Cohen J, Lobet Y: Protection against development of otitis media induced by nontypeable Haemophilus influenzae by both active and passive immunization in a chinchilla model of virus-bacterium superinfection. Infect Immun. 1999, 67: 2746-2762.
Concepcion NF, Frasch CE: Pneumococcal type 22F polysaccharide absorption improves the specificity of a pneumococcal-polysaccharide enzyme-linked immunosorbent assay. Clin Diagn Lab Immunol. 2001, 8: 266-272.
Henckaerts I, Goldblatt D, Ashton L, Poolman J: Critical differences between pneumococcal polysaccharide enzyme-linked immunosorbent assays with and without 22F inhibition at low antibody concentrations in pediatric sera. Clin Vaccine Immunol. 2006, 13: 356-360. 10.1128/CVI.13.3.356-360.2006.
Poolman JT, Frasch CE, Kayhty H, Lestrate P, Madhi SA, Henckaerts I: Evaluation of pneumococcal polysaccharide immunoassays using a 22F adsorption step with serum samples from infants vaccinated with conjugate vaccines. Clin Vaccine Immunol. 2010, 17: 134-142. 10.1128/CVI.00289-09.
Romero-Steiner S, Frasch CE, Carlone G, Fleck RA, Goldblatt D, Nahm MH: Use of opsonophagocytosis for serological evaluation of pneumococcal vaccines. Clin Vaccine Immunol. 2006, 13: 165-169. 10.1128/CVI.13.2.165-169.2006.
Henckaerts I, Durant N, De Grave D, Schuerman L, Poolman J: Validation of a routine opsonophagocytosis assay to predict invasive pneumococcal disease efficacy of conjugate vaccine in children. Vaccine. 2007, 25: 2518-2527. 10.1016/j.vaccine.2006.09.029.
Granstrom M, Thoren M, Blennow M, Tiru M, Sato Y: Acellular pertussis vaccine in adults: adverse reactions and immune response. Eur J Clin Microbiol. 1987, 6: 18-21. 10.1007/BF02097184.
Camargo ME, Silveira L, Furuta JA, Oliveira EP, Germek OA: Immunoenzymatic assay of anti-diphtheric toxin antibodies in human serum. J Clin Microbiol. 1984, 20: 772-774.
Melville-Smith ME, Seagroatt VA, Watkins JT: A comparison of enzyme-linked immunosorbent assay (ELISA) with the toxin neutralization test in mice as a method for the estimation of tetanus antitoxin in human sera. J Biol Stand. 1983, 11: 137-144. 10.1016/S0092-1157(83)80038-9.
Karpinski KF, Hayward S, Tryphonas H: Statistical considerations in the quantitation of serum immunoglobulin levels using the enzyme-linked immunosorbent assay (ELISA). J Immunol Methods. 1987, 103: 189-194. 10.1016/0022-1759(87)90289-4.
World Health Organization: Progress in the control of viral hepatitis: memorandum from a WHO meeting. Bull World Health Organ. 1988, 66: 443-455.
Hepatitis B virus: a comprehensive strategy for eliminating transmission in the United States through universal childhood vaccination. Recommendations of the Immunization Practices Advisory Committee (ACIP). MMWR Recomm Rep. 1991, 40: 1-25.
World Health Organization: Standard Procedures for Determining Immunity to Poliovirus using the Microneutralization Test (WHO/EPI/GEN 93.9). 1993
Puumalainen T, Dagan R, Wuorimaa T, Zeta-Capeding R, Lucero M, Oligren J, Kayhty H, Nohynek H: Greater antibody responses to an eleven valent mixed carrier diphtheria- or tetanus-conjugated pneumococcal vaccine in Filipino than in Finnish or Israeli infants. Pediatr Infect Dis J. 2003, 22: 141-149.
Black SB, Shinefield HR, Ling S, Hansen J, Fireman B, Spring D, Noyes J, Lewis E, Ray P, Lee J, Hackell J: Effectiveness of heptavalent pneumococcal conjugate vaccine in children younger than five years of age for prevention of pneumonia. Pediatr Infect Dis J. 2002, 21: 810-815. 10.1097/00006454-200209000-00005.
Cutts FT, Zaman SM, Enwere G, Jaffar S, Levine OS, Okolo JB, Oluwalana C, Vaughan A, Obaro SK, Leach A, McAdam KP, Biney E, Saaka M, Onwuchekwa U, Yallop F, Pierce NF, Greenwood BM, Adegbola RA: Efficacy of nine-valent pneumococcal conjugate vaccine against pneumonia and invasive pneumococcal disease in The Gambia: randomised, double-blind, placebo-controlled trial. Lancet. 2005, 365: 1139-1146. 10.1016/S0140-6736(05)71876-6.
Klugman KP, Madhi SA, Huebner RE, Kohberger R, Mbelle N, Pierce N: A trial of a 9-valent pneumococcal conjugate vaccine in children with and those without HIV infection. N Engl J Med. 2003, 349: 1341-1348. 10.1056/NEJMoa035060.
Lucero MG, Nohynek H, Williams G, Tallo V, Simoes EA, Lupisan S, Sanvictores D, Forsyth S, Puumalainen T, Ugpo J, Lechago M, de Campo M, Abucejo-Ladesma E, Sombrero L, Nissinen A, Soininen A, Ruutu P, Riley I, Makela HP: Efficacy of an 11-valent pneumococcal conjugate vaccine against radiologically confirmed pneumonia among children less than 2 years of age in the Philippines: a randomized, double-blind, placebo-controlled trial. Pediatr Infect Dis J. 2009, 28: 455-462. 10.1097/INF.0b013e31819637af.
Whitney CG, Pilishvili T, Farley MM, Schaffner W, Craig AS, Lynfield R, Nyquist AC, Gershman KA, Vazquez M, Bennett NM, Reingold A, Thomas A, Glode MP, Zell ER, Jorgensen JH, Beall B, Schuchat A: Effectiveness of seven-valent pneumococcal conjugate vaccine against invasive pneumococcal disease: a matched case–control study. Lancet. 2006, 368: 1495-1502. 10.1016/S0140-6736(06)69637-2.
Palmu AA, Jokinen J, Borys D, Nieminen H, Ruokokoski E, Siira L, Puumalainen T, Lommel P, Hezareh M, Moreira M, Schuerman L, Kilpi TM: Effectiveness of the ten-valent pneumococcal Haemophilus influenzae protein D conjugate vaccine (PHiD-CV10) against invasive pneumococcal disease: a cluster randomised trial. Lancet. 2013, 381: 214-222. 10.1016/S0140-6736(12)61854-6.
Deceuninck G, De Wals P: Effectiveness of three pneumococcal conjugate vaccines (PCVS) to prevent invasive pneumococcal disease (IPD) in Quebec, Canada [abstract]. Pneumonia. 2014, 3: 163-
Domingues CMAS, Verani JR, Montenegro Renoiner EI, Brandileone MCC, Flannery B, De Oliveira LH, Santos JB, De Moraes JC: Effectiveness of ten-valent pneumococcal conjugate vaccine against invasive pneumococcal disease in Brazil: a matched case–control study. Lancet Respir Med. 2014, 2: 464-471. 10.1016/S2213-2600(14)70060-8.
Prymula R, Peeters P, Chrobok V, Kriz P, Novakova E, Kaliskova E, Kohl I, Lommel P, Poolman J, Prieels J-P, Schuerman L: Pneumococcal capsular polysaccharides conjugated to protein D for prevention of acute otitis media caused by both Streptococcus pneumoniae and non-typable Haemophilus influenzae: a randomised double-blind efficacy study. Lancet. 2006, 367: 740-748. 10.1016/S0140-6736(06)68304-9.
Tregnaghi MW, Saez Llorens X, Lopez P, Abate H, Smith E, Pósleman A, Calvo A, Wong D, Cortes Barbosa C, Ceballos A, Tregnaghi M, Sierra A, Rodriguez M, Troitiño M, Carabajal C, Falaschi A, Leandro A, Castrejon MM, Lepetic A, Lommel P, Hausdorff WP, Borys D, Guiñazú JR, Ortega Barría E, Yarzabal JP, Schuerman L, on behalf of the COMPAS Group: Efficacy of pneumococcal nontypable Haemophilus influenzae protein D conjugate vaccine (PHiD-CV) in young Latin American children: A double-blind randomized controlled trial. PLoS Med. 2014, 11: e1001657-10.1371/journal.pmed.1001657.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2334/14/530/prepub
Acknowledgements
The authors thank the children and their parents/legally acceptable representatives who participated in the study, the study nurses, and other staff members without whom this study would not have been possible. The authors also thank Dr Tan Kim Kiat for his contribution to the study, Josephine Tan Liu, Natalie Tan Woon Hui, and Thoon Koh Cheng for their involvement in recruitment and follow-ups of patients. We are grateful to all teams of GlaxoSmithKline Vaccines for their contribution to this study, especially Aurelie Fanic for statistical input to protocol and primary phase analysis, Sudheer Ravula and Nancy François for performing statistical analyses for booster phase and quality check, and Liliana Manciu, Marta Moreira and Tineke Ryckaert for their contribution to the study. The authors also thank Aneta Skwarek-Maruszewska and Bart van Heertum (XPE Pharma & Science, Belgium c/o GlaxoSmithKline Vaccines) for publication management and Claire Verbelen (XPE Pharma & Science, Belgium) for drafting the manuscript.
Role of the funding source
GlaxoSmithKline Biologicals SA was the funding source and was involved in all stages of the study conduct and analysis. GlaxoSmithKline Biologicals SA also took responsibility for all costs associated with the development and publishing of the present manuscript.
Previous publications
The results of this study were presented in part at the 32nd Annual Congress of the Malaysian Paediatric Association (MPA), October 15–17, 2010 and at the 14th Asia Pacific Congress of Pediatrics (APCP), September 8–12, 2012.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
CYC declared having received grants from GlaxoSmithKline Biologicals SA for research coordination, clinics, and vaccines. MTK and FSL declared having received grants for carrying out the reported study and other studies and travel fees for presenting the study results at conferences. PCC received payment for giving talks on vaccination to doctors and nurses and for travelling to a conference. CYC received payment for training workshop lectures and for travelling to a conference. MTK received honorarium as speaker at meetings organised by the Malaysian Paediatric Association. KKT received reimbursement for travelling expenses to attend investigators meeting for trial initiation. YWSY had no competing interest. DB, FS, YLT were employed by GlaxoSmithKline Vaccines during the study period. KS and MH worked as consultant for GlaxoSmithKline Vaccines. DB declared stock options ownership in GlaxoSmithKline Vaccines.
Authors’ contributions
Study design was done by FSL, MTK, KKT, YLT, KS and DB. Recruitment of centres and/or investigators was done by FSL, PCC, MTK, YLT, CYC, whereas provision of subjects by FSL, PCC, MTK, KKT, YLT, YWSY and CYC. Performing or supervising the analysis was done by PCC, FS, YLT, MH, DB and interpretation of results by FS, KKT, MH, YLT, KS and DB. Supervision of the study/research group was done by PCC, MTK, KT, YLT, KS, YWSY, DB and CYC. Acquisition of funding by KKT, YLT and DB. All authors read and approved the final manuscript.
Poh Chong Chan, Chia Yin Chong contributed equally to this work.
Electronic supplementary material
12879_2014_3854_MOESM1_ESM.docx
Additional file 1: Between groups adjusted antibody GMC ratios post-primary vaccination (primary ATP immunogenicity cohort).(DOCX 17 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Lim, F.S., Koh, M.T., Tan, K.K. et al. A randomised trial to evaluate the immunogenicity, reactogenicity, and safety of the 10-valent pneumococcal non-typeable Haemophilus influenzaeprotein D conjugate vaccine (PHiD-CV) co-administered with routine childhood vaccines in Singapore and Malaysia. BMC Infect Dis 14, 530 (2014). https://doi.org/10.1186/1471-2334-14-530
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2334-14-530