
Abstract
Background
Although endemic cholera causes significant morbidity and mortality each year in Nepal, lack of information about the causal bacterium often hinders cholera intervention and prevention. In 2012, diarrheal outbreaks affected three districts of Nepal with confirmed cases of mortality. This study was designed to understand the drug response patterns, source, and transmission of Vibrio cholerae associated with 2012 cholera outbreaks in Nepal.
Methods
V. cholerae (n = 28) isolated from 2012 diarrhea outbreaks {n = 22; Kathmandu (n = 12), Doti (n = 9), Bajhang (n = 1)}, and surface water (n = 6; Kathmandu) were tested for antimicrobial response. Virulence properties and DNA fingerprinting of the strains were determined by multi-locus genetic screening employing polymerase chain reaction, DNA sequencing, and pulsed-field gel electrophoresis (PFGE).
Results
All V. cholerae strains isolated from patients and surface water were confirmed to be toxigenic, belonging to serogroup O1, Ogawa serotype, biotype El Tor, and possessed classical biotype cholera toxin (CTX). Double-mismatch amplification mutation assay (DMAMA)-PCR revealed the V. cholerae strains to possess the B-7 allele of ctx subunit B. DNA sequencing of tcpA revealed a point mutation at amino acid position 64 (N → S) while the ctxAB promoter revealed four copies of the tandem heptamer repeat sequence 5'-TTTTGAT-3'. V. cholerae possessed all the ORFs of the Vibrio seventh pandemic island (VSP)-I but lacked the ORFs 498–511 of VSP-II. All strains were multidrug resistant with resistance to trimethoprim-sulfamethoxazole (SXT), nalidixic acid (NA), and streptomycin (S); all carried the SXT genetic element. DNA sequencing and deduced amino acid sequence of gyrA and parC of the NAR strains (n = 4) revealed point mutations at amino acid positions 83 (S → I), and 85 (S → L), respectively. Similar PFGE (NotI) pattern revealed the Nepalese V. cholerae to be clonal, and related closely with V. cholerae associated with cholera in Bangladesh and Haiti.
Conclusions
In 2012, diarrhea outbreaks in three districts of Nepal were due to transmission of multidrug resistant V. cholerae El Tor possessing cholera toxin (ctx) B-7 allele, which is clonal and related closely with V. cholerae associated with cholera in Bangladesh and Haiti.
Similar content being viewed by others


Background
Toxigenic Vibrio cholerae is the causative agent of cholera, an acute life-threatening diarrheal disease, which occurs in many developing countries, particularly South Asia, Africa, and Latin America [1, 2]. Based on the phenotypic and genotypic differences, V. cholerae O1 strains are classified into two biotypes: ‘classical’ and ‘El Tor’ [3]. Seven distinct pandemics of cholera have been recorded since 1817 [4]. The sixth pandemic, and presumably the earlier pandemics, were caused by the classical biotype of V. cholerae O1, which was displaced by V. cholerae O1 biotype El Tor in the 1960’s to become the causative agent of the ongoing seventh cholera pandemic [1].
Over the past few years, V. cholerae O1 biotype ET causing Asiatic cholera has shown remarkable changes in its phenotypic and genetic characteristics [5]. V. cholerae O139, also known as Bengal strain emerged in 1992 as the major cause of epidemic cholera in Bangladesh and India by displacing ET biotype strains [6]. V. cholerae O1 ET continues to prevail as the major cause of cholera worldwide. The most recent development in the evolution of global cholera has been the emergence and spread of a new variant ET, or altered ET, in Bangladesh that carries ctxB of the classical (CL) biotype (ctxB CL) [7]. Since 2001, V. cholerae O1 biotype ET strains associated with endemic cholera in Bangladesh have been altered ET, while those isolated before 2001 contained ctxB of the ET biotype [7], which is the prototype associated with the 7th cholera pandemic. According to recent reports, altered ET strains have been spreading globally [8–15] causing more severe disease [16].
Following the earthquake in Haiti in 2010, epidemic cholera broke out in the country that has since affected tens of thousands of people and led to the death of more than 4,000 [17]. Although the 7th cholera pandemic expanded from Asia reaching Africa in the 1970’s and Latin America in 1991, cholera had never been reported in Haiti after 1960 [18]. The source of the recent cholera epidemic in Haiti remains controversial but it is widely believed that it was imported by UN peacekeeping forces arriving from Nepal [17, 19].
Nepal is a cholera endemic country where cholera continues to be a major public health concern, especially among lower socio-economic groups. Although cholera causes significant morbidity and mortality each year, Nepal lacks the requisite laboratory infrastructures to conduct epidemiological and related surveillance to determine disease burden, as well as to identify the source and transmission of cholera. Both of these types of information are important for the effective management of all infectious diseases, not only cholera and diarrhea. In 2012, outbreaks of cholera were reported in three districts of Nepal with death of some of the affected individuals in remote villages in the north-western district of Doti. Cholera treatment is often hindered due to emergence of multi-drug resistance in V. cholerae[20, 21]. Compounding the problem related to selecting an effective antibiotic for cholera treatments, even less is known about drug resistance properties of V. cholerae O1 bacterium in Nepal [22–25]. In the present study, V. cholerae strains associated with the 2012 cholera outbreaks in three district of Nepal were investigated for microbiological and molecular characteristics. The major tests applied to this study included antibiotic response patterns, and multi-locus genetic screening by polymerase chain reaction, DNA fingerprinting analyses by sequencing, and pulsed-field gel electrophoresis (PFGE) of genomic DNA.
Methods
Ethical approval
Ethical approval for the present study was obtained from the Ethical Review Board of Nepal Health Research Council (NHRC) (Reg. 68/2011; 2068-12-20 approved on 2 April 2012). Informed consent was obtained for all patients enrolled in the present study, for patients under 18 years of age parental consent was obtained.
Isolation of V. cholerae
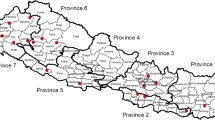
During the 2012 cholera outbreaks in three districts of Nepal (Figure 1), V. cholerae isolated from clinical [Kathmandu (n = 12), Doti (n = 9), and Bajhang (n = 1)] and natural surface water [Kathmandu (n = 6)] sources were confirmed by microbiological, serological and molecular methods. V. cholerae isolation involved enriching the samples in alkaline peptone water (APW) (pH-8.4) at 37°C for 4–6 h, followed by culturing overnight on selective bacteriological media such as taurocholate tellurite gelatin agar (TTGA). V. cholerae colonies were confirmed using a combination of biochemical and serological methods, as described previously [26]. V. cholerae isolates were examined for phenotypic test such as antimicrobial response, as well as for virulence, molecular, and phylogenetic characteristics. Reference V. cholerae O1 strains N16961 and O395 served as biotypes El Tor (ET) and Classical (CL) control, respectively.

Map of Nepal showing the cholera affected three districts: Doti, Bajhang, and Kathmandu, 2012. Cholera affected two north-western districts, Doti and Bajhang, including the capital city, Kathmandu, appear in red.
Determination of serogroup and biotype
Serogroups of the V. cholerae isolates were identified by biochemical and molecular methods and confirmed by slide agglutination using specific polyvalent antisera for V. cholerae O1 and O139, followed by screening with a monoclonal antibody specific for both serogroups [26]. Biotyping primarily involved selective phenotypic tests, including chicken erythrocyte agglutination, sensitivity to polymyxin B, and Mukherjee CL phage IV and Mukherjee ET phage V [1].
Genomic DNA preparation
Genomic DNA extraction was carried out following previously described methods [11].
PCR assays for serogroup and biotype determination
Subtypes of all strains were reconfirmed using V. cholerae species-specific ompW PCR [27]. Serogroups were reconfirmed using multiplex PCR targeted at O1- (rfbO1) and O139- (rfbO139) specific O biosynthetic genes and the cholera toxin (CTX) gene (ctxA) [28]. Biotype specific characteristics were determined using PCR assays targeted to tcpA (CL and ET) [29], rstR, a gene encoding phage transcriptional regulator [30], presence of the repeat in toxin (rtxC) [31], rstC that encodes an anti-repressor protein, and tlc, that codes for the toxin-linked cryptic plasmid [32].
Determination of ctxBgenotype
The double mismatch amplification mutation assay (DMAMA-PCR) was recently developed to discriminate the classical (ctxB genotype 1), El Tor (ctxB gneotype 3), and Haitian types (ctxB genotype 7) of ctxB alleles by focusing on nucleotide positions 58 and 203 of the ctxB gene [33]. DMAMA-PCR was performed in this study to detect the ctxB genotype using primers and conditions as described previously [33]. V. cholerae O1 strains O395 (CL), N16961 (ET), and 2010EL-1786 (Haiti variant, genotype 7) were used as control strains for DMAMA-PCR analysis.
Nucleotide sequencing and analysis of genes ctxB and tcpA
Genes ctxB and tcpA of randomly selected V. cholerae O1 strains in 2012 were sequenced following conditions as described elsewhere [34]. PCR amplification of genes ctxB and tcpA was performed in a 25 μl reaction mixture in an automated Peltier thermal cycler (PTC-200, M. J. Research). Subsequently, PCR products were purified with a Microcon centrifugal filter device (Millipore Corporation, Bedford, MA) and sequenced using an ABI PRISM Big Dye Terminator Cycle Sequencing Reaction kit (Applied Biosystems, Foster City, CA) on an ABI PRISM 310 automated sequencer (Applied Biosystems). The deduced amino acid sequences of the respective genes from all strains were aligned using CLUSTAL-W.
The promoter sequence of ctxAB classoperon
The 600 bp DNA fragments containing the entire P ctxAB of V. cholerae strains (n = 4) from Nepal were PCR-amplified using zot gene (the gene preceding the ctx AB operon)-specific forward and ctxA gene-specific reverse primers, ZtPF and CtPR respectively [35], followed by direct sequencing of the amplified products.
ORF content analysis of VSP-I and VSP-II region
The presence or absence of ORFs in the VSP-I and VSP-II cluster of V. cholerae O1 isolates was examined by PCR using primers described elsewhere [32]. V. cholerae O1 strains N16961 El Tor (ET) and O395 Classical (CL) biotypes served as controls.
Antibiotic susceptibility
Susceptibility to antibiotics was performed by disk diffusion, as described by both Bauer et al. [36] and the Clinical and Laboratory Standards Institute [37], using commercial antibiotic discs. Nine antibiotics (Oxoid, United Kingdom) were employed: erythromycin (E, 15 μg); gentamicin (CN, 10 μg); trimethoprim/sulfamethoxazole (SXT, 30 μg), tetracycline (TE, 30 μg), ampicillin (AMP, 30 μg), streptomycin (S, 10 μg), azithromycin (AZM, 15 μg), nalidixic acid (NA, 30 μg) and ciprofloxacin (CIP, 5 μg). Characterizations of the resistance or susceptibility profiles of the isolates were determined by measuring the inhibitory zone and comparing it with an interpretative chart to determine sensitivity to each antibiotic.
PCR assay for the detection of SXT element
Using PCR assays, all V. cholerae O1 strains were examined for the presence of the SXT element. The detection of SXT was performed using primers and procedures described previously [38].
Sequence analysis of gyrA and parCgenes
The DNA sequencing of the PCR amplified genes encoding DNA gyrase (gyrA) and topoisomerase IV (parC) was performed according to the previously described procedure [39].
Pulsed-field gel electrophoresis (PFGE)
Whole agarose-embedded genomic DNA from the V. cholerae isolates was prepared. PFGE was carried out using a contour-clamped homogeneous electrical field (CHEF-DRII) apparatus (Bio-Rad), according to procedures described previously [40]. Conditions for separation were as follows: 2 to 10s for 13 h, followed by 20 to 25 s for 6 h. An electrical field of 6 V/cm was applied at an included field angle of 120°. Genomic DNA of the test strains was digested by NotI restriction enzyme (Gibco-BRL, Gaithersburg, MD), and Salmonella enterica serovar Braenderup was digested using XbaI, with fragments employed as molecular size markers. Restriction fragments were separated in 1% pulsed-field-certified agarose in 0.5X TBE (Tris-borate-EDTA) buffer. Post-electrophoresis gel-treatment included gel-stained and de-stained. The DNA was visualized using a UV transilluminator, and images were digitized via a one-dimensional gel documentation system (Bio-Rad).
Image analysis
The fingerprint pattern in the gel was analyzed using a computer software package, Bionumeric (Applied Maths, Belgium). After background subtraction and gel normalization, the fingerprint patterns were typed according to banding similarity and dissimilarity, using the Dice similarity coefficient and unweighted-pair group method employing average linkage (UPGMA) clustering, as recommended by the manufacturer. The results were graphically represented as dendrograms.
Results
Phenotypic and genetic characteristics
V. cholerae O1 strains (n = 28) included in this study from 2012 diarrhea outbreaks and environmental sources in Nepal produced translucent colonies with black center on TTGA, and gave biochemical reactions typical of V. cholerae. All strains reacted positively to serogroup O1-specific antibody, but not to O139, confirming all to be V. cholerae O1. Serological results also showed that all V. cholerae O1 belonged to the Ogawa serotype (Table 1). All V. cholerae strains exhibited chicken cell agglutination (CCA), sensitivity to ET-specific phage V, and resistance to both polymyxin B and CL specific phage IV, confirming biotype El Tor (ET)-specific phenotypic traits (Table 1).
The phenotypically confirmed V. cholerae O1 biotype ET strains amplified the primers for species-specific gene ompW, and all amplified primers specific for O biosynthetic gene wbe of V. cholerae O1, but not wbf, which is specific for serogroup O139 (Table 1). All of the V. cholerae O1 strains isolated from cholera and from surface water sources in Nepal amplified the primers for the cholera-toxin gene ctxA (Table 1). In addition, all of the ctxA + V. cholerae strains amplified the primers for biotype ET-specific marker gene rtxC, confirming ET attributes. All strains had tcpA, the major virulence-associated gene of the VPI-I gene cluster, and all amplified primers for ET-specific marker gene tcpA ET, but not tcpA CL. All of the V. cholerae ET strains confirmed in this study carried the ET biotype-specific repressor gene rstR ET, confirming that they carried ET-biotype CTX prophage, and possessed rstC and tlc genes (Table 1).
CtxBtyping by DMAMA-PCR
All V. cholerae O1 strains isolated from both clinical and environmental sources (n = 28), including the O395 (CL), N16961 (ET) and 2010EL-1786 (Haiti variant, ctxB genotype 7) were analyzed using double-mismatch amplification mutation assay (DMAMA)-PCR technique to determine the CTX-B genotype. As shown in Table 1, all of the ET Biotype strains amplified the primers specific for ctxB genotype 7 irrespective of their source and place of isolation.
Sequencing of ctxB and tcpA
PCR-amplified genes ctxB (460 bp) and tcpA (675 bp) from selected V. cholerae O1 strains (n = 4; one clinical strain from each of the three districts including one environmental strain from Kathmandu) were sequenced and the N’-amino acid sequences determined using bioinformatic tools. Sequencing data revealed that all of the tested V. cholerae O1 strains contained the deduced amino acid sequence of CTXB. This is identical to that of the CL biotype CT, having histidine and threonine at positions 39 and 68, respectively, but an additional sequence variation was observed in position 20, where histidine found in CL and ET biotype CT was replaced by asparagine (H → N) (Nucleotide accession numbers are KJ596538, KJ596539, KJ596540 and KJ596541). The DNA sequence and deduced amino acids matched against the ctxB genotype 7. The ctxB sequencing data were consistent and supported the results of DMAMA-PCR.
DNA sequencing data of the amplified tcpA gene revealed the presence of a point mutation that resulted in an amino acid substitution at position 64 (N → S) of the deduced peptide (accession numbers are KJ596546, KJ596547, KJ596548 and KJ596549).
The promoter sequence of ctxAB classoperon
Analysis of the promoter sequences of the randomly selected Nepalese V. cholerae O1 strains isolated in 2012 revealed that they contained four copies of heptamer repeat sequences in the P ctxAB regions (accession numbers are KJ596554, KJ596555, KJ596556 and KJ596557) irrespective of their source of isolation. This indicates that the CT regulatory regions of these strains are like those of canonical El Tor strains and dissimilar to those of classical strains, which usually carry seven to eight such repeats [38].
VSP Islands
All Nepalese V. cholerae O1 examined in this study carried all the tested ORFs of the VSP-I genomic island region. However, all strains lacked the VSP-II genomic island ORFs VCO498, VCO502, VCO504 and VCO511 (Table 2).
Antibiotic susceptibility assay
Antibiotic susceptibility assay revealed that all of the 28 tested V. cholerae O1 strains isolated in 2012 from the three districts of Nepal, irrespective of their source and place of isolation, were multi-drug resistant with resistance to NA, SXT, and S. However, all strains were sensitive to AZM, TE, AMP, CN, E, and CIP (Table 1).
Detection of SXT element
All of the V. cholerae strains (n = 28) amplified the primer for the SXT gene, a mobile genetic element carrying multi-drug resistance gene cassettes in bacteria (Table 1).
Sequencing of gyrA and parC
Sequencing of gyrA and parC of V. cholerae O1 strains (n = 4) representing three districts of Nepal, including both clinical and environmental sources of Kathmandu, detected one point mutation in each; in gyrA serine was substituted by isoleucine at position 83 (accession numbers are KJ596542, KJ596543, KJ596544 and KJ596545), and in parC serine was substituted by leucine at position 85 (accession numbers are KJ596550, KJ596551, KJ596552 and KJ596553).
PFGE and cluster analysis
The PFGE of NotI-digested genomic DNAs of the V. cholerae O1 strains yielded 20 to 23 fragments (Figure 2) (Data available from the Dryad Digital Repository: doi:10.5061/dryad.600nd), and their molecular sizes ranged from 20.5 to 350 kb. The PFGE pattern of all the Nepalese strains irrespective of their source and place of isolation matched both with each other, and with that of the altered El Tor PFGE pattern in the number and position of the DNA fragments, suggesting genetic homogeneity .

Dendrogram showing genomic fingerprints of V . cholerae O1 strains isolated in Nepal in 2012. Genomic fingerprinting of multi-drug resistant V. cholerae O1 El Tor strains isolated from 2012 diarrhea outbreaks in three districts (Doti, Bajhang, and Katmandu) of Nepal. Dendrogram was constructed by Dice similarity coefficient and UPGMA clustering method by using pulsed-field gel electrophoresis (PFGE) images of NotI- restriction digested genomic DNA of the tested Nepalese V. cholerae strains; PFGE images of contemporary V. cholerae O1 El Tor strains from Haiti and Bangladesh, including El Tor reference strain N16961 were also included. The scale bar at the top (left) indicates similarity coefficient. The banding patterns and the similarity coefficient revealed 100% similarity for all the tested Nepalese, Haitian, and Bangladeshi V. cholerae strains included in the dendrogram, suggesting° high level of clonal relatedness (Data available from the Dryad Digital Repository: doi:10.5061/dryad.600nd).
In order to understand the clonal link between the V. cholerae O1 strains associated with cholera outbreaks in Doti, Kathmandu, and Bajhang, cluster analysis was performed by dendogram using the PFGE (NotI) images of the Nepalese V. cholerae O1 strains, together with PFGE (NotI) images of representative V. cholerae O1 strains isolated in Bangladesh (2010) and Haiti (2010), which were available in our soft database. All of the Nepalese strains were clonal as they shared the same cluster showing 100% similarity, and were related closely with representative V. cholerae O1 altered El Tor strains isolated in Bangladesh and Haiti.
Discussion
This study presents microbiological and molecular data on V. cholerae isolated from 2012 Nepal cholera and surface water sources showing the transmission of a highly clonal, multi-drug resistant V. cholerae O1, which was associated with simultaneous cholera outbreaks occurring in Kathmandu and the remote villages of the western districts of Doti and Bajhang, Nepal. The study also revealed very close clonal relationships between the Nepalese V. cholerae O1 strains with those from Bangladesh and Haiti isolated in 2010.
The conventional culture, biochemical, and serological test results confirmed that both clinical and environmental V. cholerae isolates belonged to serogroup O1 and serotype Ogawa. The microbiological test results were complemented by the molecular data obtained from PCR assays performed for the amplification of V. cholerae species-specific gene ompW[27], genes ctxA encoding subunit A of cholera toxin (CTX), and wbe encoding serogroup O1-specific antigenic polysaccharides [28]. These results confirmed that all of the V. cholerae isolates were toxigenic and belonged to serogroup O1. The phenotypic characteristics, together with the presence of biotype specific marker genes, such as rtxC, rstC, tcpA ET, rstR ET, and ctxB CL, confirmed that all of the V. cholerae O1 strains occurring in surface water and associated with the 2012 cholera outbreaks in Nepal were biotype El Tor possessing the ctxB marker gene of classical biotype. This important data identified all strains as altered El Tor, first described in Bangladesh in 2006 [7, 11].
Although treatment of cholera involves a course of effective antibiotics, together with appropriate oral or intravenous rehydration fluid(s) [41], antibiotic therapy world-wide has faced with challenges related to the rapid emergence and spread of multi-drug resistant (MDR) V. cholerae strains resistant to atibiotics, such as TE, AMP, kanamycin (KN), S, SXT, NA, E, and most recently, to CIP and norfloxacin (NOR) [17, 21, 42, 43]. In Nepal, at least three different resistance phenotypes of V. cholerae were previously reported to be in circulation [24]. A recent study reported temporal variation in drug resistance patterns of V. cholerae associated with cholera in Nepal between 2007 and 2010 [25]. In the present study, V. cholerae O1 associated with diarrheal outbreaks in Nepal, including those isolated from natural surface water samples, were MDR showing resistance towards SXT, NA, and S, suggesting that drug resistance patterns can change temporally, and spatially, and thus require continuous monitoring in order to select an effective drug of choice at any given time.
In V. cholerae, multi-drug resistance was shown to be attributed to lateral acquisition of self-transmissible genetic element designated SXT, carrying multiple antibiotic-resistance markers [44]. All V. cholerae strains tested in the present Nepalese study were MDR and likewise, all had the SXT element, presumably carrying MDR marker genes in their genome, a fact that appears to be in line with their consistent resistance towards trimethoprim-sulfamethxazole. Resistance to fluoroquinolones such as NA is mostly associated with genes encoding gyrase (gyrA and gyrB) and topoisomerase IV (parC and parE). DNA sequencing of the gyrA and parC of the Nepalese V. cholerae strains suggests a similar molecular basis for quinolone resistance, as a single point mutation in each of the genes resulted in amino acid switching from Serine to Isoleucine at 83 position, and Serine to Leucine at 85 position, respectively, as found in the quinolone resistant V. cholerae associated with cholera in Africa [20], India [39], and Haiti [45].
The polymorphism of ctxB gene-encoding cholera toxin (CT) subunit B and the corresponding amino acid substitution was first reported in the early 1990’s [34]. Subsequent investigation of the ctxB gene sequence revealed the presence of eleven distinct genotypes in different serogroups of V. cholerae[46]. Genotypes 1, 2, 3, 7, 10, and 11 were found in serogroup O1 strains, genotypes 3, 4, 5, and 6 were found in serogroup O139 strains, and genotypes 8 and 9 were found only in serogroups O27 and O37 [46]. A previous study showed the presence of two different ctxB genotypes, namely 1 and 7, among V. cholerae associated with cholera in Nepal between 2007 and 2010 [25]. In the present study, ctxB genotype 7 was confirmed for all 28 V. cholerae O1 strains isolated in 2012 from three districts of Nepal. Data presented in this study provide ample evidence of genetic switching from ctxB genotype 1 to 7 in Nepal, a phenomenon seen in 2007 and 2008 for V. cholerae causing endemic cholera in India [47] and Bangladesh [48], respectively. The global dissemination of ctxB genotype 7 has been growing with reports of the genotype in the West African countries of Nigeria and Cameroon, as well as in Haiti [17, 20].
The toxin co-regulated pilus (TCP), an essential colonization factor of V. cholerae[49] that also serves as a receptor for CTX-Φ [44], is a homopolymer of the major pilus protein, TcpA pilin [49] encoded by tcpA. The DNA sequence of tcpA differs slightly at the C-terminal domain for classical and El Tor biotype strains [3]. In the present study, DNA sequencing and the deduced amino acid sequences of TcpA of V. cholerae O1 El Tor strains associated with cholera outbreaks in Nepal (2012) differed from the El Tor reference strain N16961 due to a mutation at the amino acid position 64 (Asparagine → Serine). This change may be subtle and it is not known whether such genetic switching of the tcpA gene has any epidemiological impact on V. cholerae causing endemic cholera in Nepal. Nonetheless, a change of amino acid at position 64 of TcpA was first reported in the V. cholerae serogroup O1 biotype El Tor strain associated with cholera in Bangladesh [50]. This was followed by reports from Haiti [51], suggesting that this genotype is spreading globally.
ToxR is a global transcriptional regulator protein responsible for virulence gene expression, and toxR sequence repeats (TTTTGAT) located between CTX prophage genes zot and ctxA are essential for ToxR binding and activation of the ctxAB promoter [52]. The Nepalese V. cholerae O1 strains contained four copies of such repeats (data not shown), while five copies were found among the El Tor variant of V. cholerae associated with cholera in Haiti [17, 45]. These are less than the seven copies that were reported for the classical biotype strains of V. cholerae O395.
The VSP-I gene cluster encompasses a 16 kb region from VC0175 to VC0185, and most of the genes encode hypothetical or conserved proteins with no known function. On the other hand, the VSP-II region is a ~27 kb region that encompasses VC0490–VC0516 [29]. These two clusters are unique to the El Tor strains of the seventh pandemic. The Nepalese V. cholerae O1strains carried intact VSP-I but harbored a variant VSP-II lacking the ORFs 498–511 of the VSP-II genomic island region, which was first reported from a V. cholerae strain (CIRS101) associated with cholera in Bangladesh [53] and Haiti [45].
A recent MLVA-based study carried out with V. cholerae O1 strains associated with cholera between 2007 and 2010 showed the circulation of four different groups of altered V. cholerae O1 El Tor strains in Western Nepal including Butwal and Kathmandu [25]. The molecular basis and epidemiological significance of such genetically divergent V. cholerae O1 altered El Tor remains an interesting area to explore. However, V. cholerae O1 associated with cholera outbreaks in the three Nepalese districts proved to be highly clonal, since all of the strains had an indistinguishable PFGE (Not-I) banding pattern irrespective of their source of isolation, reflecting high genetic homogeneity in the V. cholerae population. The PFGE pattern generated by the V. cholerae O1 strains in this study also matched with the pattern reported for V. cholerae O1 associated with cholera outbreaks in Bangladesh and Haiti, suggesting that closely related multi-drug resistant strains are undergoing global dissemination.
Conclusion
In conclusion, this study presents data on the transmission of a multi-drug resistant V. cholerae showing identical PFGE pattern in three districts (Doti, Bajhang, and Kathmandu) of Nepal during the 2012 diarrhea outbreaks. Considering the changing climate and increasing global burden of cholera, and the emergence and spread of new hyper-infective variants of V. cholerae[16], regular surveillance of V. cholerae outbreaks is highly recommended for monitoring the distribution, clonal type and evolution of the pathogen, for effective cholera management in Nepal and elsewhere.
References
Kaper JB, Morris JG, Levine MM: Cholera. Clin Microbiol Rev. 1995, 8: 48-86.
Faruque SM, Albert MJ, Mekalanos JJ: Epidemiology, genetics and ecology of toxogenic Vibrio cholerae. Microbiol Mol Biol Rev. 1998, 62: 1301-1314.
Safa A, Nair GB, Kong RYC: Evolution of new variants of Vibrio cholerae O1. Trends Microbiol. 2010, 18: 46-54.
Pollitzer R, Swaroop S, Burrows W: Cholera Monogr Ser World Health Organ. 1959, 58 (43): 1001-1019.
Nair GB, Faruque SM, Bhuiyan NA, Kamruzzaman M, Siddique AK, Sack DA: New variants of Vibrio cholerae O1 biotype El Tor with attributes of the classical biotype from hospitalized patients with acute diarrhea in Bangladesh. J Clin Microbiol. 2002, 40: 3296-3299.
Albert MJ, Siddique AK, Islam MS, Faruque ASG, Ansaruz-zaman M, Faruque SM, Sack RB: Large outbreak of clinical cholera due to Vibrio cholerae non-O1 in Bangladesh. Lancet. 1993, 341: 704-
Nair GB, Qadri F, Holmgren J, Svennerholm AM, Safa A, Bhuiyan NA, Ahmad QS, Faruque SM, Faruque ASG, Takeda Y, Sack DA: Cholera due to altered El Tor strains of Vibrio cholerae O1 in Bangladesh. J Clin Microbiol. 2006, 44: 4211-4213.
Ansaruzzaman M, Bhuiyan NA, Nair GB, Sack DA, Lucas M, Deen JL, Ampuero J, Chaignat CL, The Mozambique Cholera Vaccine Demonstration Project Coordination Group: Cholera in Mozambique, variant of Vibrio cholerae. Emerg Infect Dis. 2004, 10: 2057-2059.
Safa A, Sultana J, Cam PD, Mwansa JC, Kong RYC: Vibrio cholerae O1hybrid El Tor strains, Asia and Africa. Emerg Infect Dis. 2008, 14: 987-988.
Nguyen BM, Lee JH, Cuong NT, Choi SY, Hien NT, Anh DD, Lee HR, Ansaruzzaman M, Endtz HP, Chun J, Lopez AL, Czerkinsky C, Clemens JD, Kim DW: Cholera outbreaks caused by an altered Vibrio cholerae O1 El Tor biotype strain producing classical cholera toxin B in Vietnam in 2007 to 2008. J Clin Microbiol. 2009, 47: 1568-1571.
Alam M, Nusrin S, Islam A, Bhuiyan NA, Rahim N, Delgado G, Morales R, Mendez JL, Navarro A, Gil AI, Watanabe H, Morita M, Nair GB, Cravioto A: Cholera between 1991 and 1997 in Mexico was associated with infection by Classical, El Tor, and El Tor Variants of Vibrio cholerae. J Clin Microbiol. 2010, 48: 3666-3674.
Morita M, Ohnishi M, Arakawa E, Yamamoto S, Nair GB, Matsushita S, Yokoyama K, Kai A, Seto K, Watanabe H, Izumiya H: Emergence and genetic diversity of El Tor Vibrio cholerae O1 that possess classical biotype ctxB among travel associated cases of cholera in Japan. J Med Microbiol. 2010, 59: 708-712.
Okada K, Chantaroj S, Roobthaisong A, Hamada S, Sawanpanyalert P: A cholera outbreak of the Vibrio cholerae O1 El Tor variant carrying classical ctxB in northeastern Thailand in 2007. Am J Trop Med Hyg. 2010, 82: 875-878.
Goel AK, Jain M, Kumar P, Sarguna P, Bai M, Gosh N, Gopalan N: Molecular characterization reveals involvement of altered El Tor biotype Vibrio cholerae O1 strains in cholera outbreak at Hyderabad, India. J Microbiol. 2011, 49: 280-284.
Islam MS, Mahmud ZH, Ansaruzzaman M, Faruque SM, Talukder KA, Qadri F, Alam M, Islam S, Bardhan PK, Mazumder RN, Khan AI, Ahmed S, Iqbal A, Chitsatso O, Mudzori J, Patel S, Midzi SM, Charimari L, Endtz HP, Cravioto A: Phenotypic, genotypic and antibiotic sensitivity patterns of strains isolated from the cholera epidemic in Zimbabwe. J Clin Microbiol. 2011, 49: 2325-2327.
Siddique AK, Nair GB, Alam M, Sack DA, Huq A, Nizam A, Longini IM, Qadri F, Faruque SM, Colwell RR, Ahmed S, Iqbal A, Bhuiyan NA, Sack RB: El Tor cholera with severe disease: a new threat to Asia and beyond. Epidemiol Infect. 2010, 138: 347-352.
Chin CS, Sorenson J, Harris JB, Robins WP, Charles RC, Jean-Charles RR, Bullard J, Webster DR, Kasarskis A, Peluso P, Paxinos EE, Yamaichi Y, Calderwood SB, Meklanos JJ, Schadt EE, Waldor MK: The origin of the Haitian cholera outbreak strain. N Engl J Med. 2011, 364: 33-42.
News BBC: Haiti cholera outbreak causes not clear, experts say. 2010, Oct 28 [cited 2010 Dec 3]. http://www.bbc.co.uk/news/world-latin-america-11618352
News BBC: Haiti cholera outbreak: Nepal troops not tested- 8 December 2010. http://www.bbc.co.uk/news/world-south-asia-11949181,
Quilici ML, Massenet D, Gake B, Bwalki B, Olson DM: Vibrio cholerae O1 variant with reduced susceptibility to ciprofloxacin, Western Africa. Emerg Infect Dis. 2010, 16: 804-1805.
Jain M, Goel AK, Bhattacharya P, Ghatole M, Kamboj DV: Multidrug resistant Vibrio cholerae O1 El Tor carrying classical ctxB allele involved in cholera outbreak in South Western India. Acta Trop. 2011, 117: 152-156.
Ise T, Pokharel BM, Rawa S, Shrestha RS, Dhakhwa JR: Outbreaks of cholera in Kathmandu Valley in Nepal. J Trop Pediatr. 1996, 42: 305-307.
Tamang MD, Sharma N, Makaju RK, Sarma AN, Koju R, Nepali N, Mishra SK: An outbreak of El Tor cholera in Kavre district, Nepal. Kathmandu Univ Med J. 2005, 3: 138-142.
Karki R, Bhatta DR, Malla S, Dumre SP: Cholera incidence among patients with diarrhea visiting National public health Laboratory, Nepal. Jpn J Infect Dis. 2010, 63: 185-187.
Shakya G, Kim DW, Clemens JD, Malla S, Upadhyaya BP, Dumre SP, Shrestha SD, Adhikari S, Sharma S, Rija N, Shrestha SK, Mason C, Kansakar P: Phenotypic and genetic characterization of Vibrio cholerae O1 clinical isolates collected through national antimicrobial resistance surveillance network in Nepal. World J Microbiol Biotechnol. 2012, 28: 2671-2678.
Alam M, Sultana M, Nair GB, Siddique AK, Hasan NA, Sack RB, Sack DA, Ahmed KU, Sadique A, Watanabe H, Grim CJ, Huq A, Colwell RR: Viable but nonculturable Vibrio cholerae O1 in biofilms in the aquatic environment and their role in cholera transmission. Proc Natl Acad Sci U S A. 2007, 104: 17801-17806.
Nandi B, Nandy RK, Mukhopadhyay S, Nair GB, Shimada T, Ghose AC: Rapid method for species-specific identification of Vibrio cholerae using primers targeted to the gene of outer membrane protein OmpW. J Clin Microbiol. 2000, 38: 4145-4151.
Hoshino K, Yamasaki S, Mukhopadhyay AK, Chakraborty S, Basu A, Bhattacharya SK, Nair GB, Shimada T, Takeda Y: Development and evaluation of a multiplex PCR assay for rapid detection of toxigenic Vibrio cholerae O1 and O139. FEMS Immunol Med Microbiol. 1998, 20: 201-207.
Rivera ING, Chun J, Huq A, Sack RB, Colwell RR: Genotypes associated with virulence in environmental isolates of Vibrio cholerae. Appl Environ Microbiol. 2001, 67: 2421-2429.
Mwansa JCL, Mwaba J, Lukwesa C, Bhuiyan NA, Ansaruzzaman M, Ramamurthy T, Alam M, Nair GB: Multiply antibiotic-resistant Vibrio cholerae O1 biotype El Tor strains emerge during cholera outbreaks in Zambia. Epidemiol Infect. 2007, 135: 847-853.
Chow KH, Ng TK, Yuen KY, Yam WC: Detection of RTX toxin gene in Vibrio cholerae by PCR. J Clin Microbiol. 2001, 39: 2594-2597.
O’Shea YA, Reen FJ, Quirke AM, Boyd EF: Evolutionary genetic analysis of the emergence of epidemic Vibrio cholerae isolates on the basis of comparative nucleotide sequence analysis and multilocus virulence gene profiles. J Clin Microbiol. 2004, 42: 4657-4671.
Naha A, Pazhani GP, Ganguly M, Ghosh S, Ramamurthy T, Nandy RK, Nair GB, Takeda Y, Mukhopadhyay AK: Development and evaluation of a PCR assay for tracking the emergence and dissemination of Haitian variant ctxB in Vibrio cholerae O1 strains isolated from Kolkata, India. J Clin Microbiol. 2012, 50: 1733-1736.
Olsvik O, Wahlberg J, Petterson B, Uhle M, Popovic T, Wachsmuth IK, Fields P: Use of automated sequencing of polymerase chain reaction generated amplicons to identify three types of CholeraToxin subunit B in Vibrio cholerae 01 strains. J Clin Microbiol. 1993, 31: 22-25.
Halder K, Das B, Nair GB, Bhadra RK: Molecular evidence favoring step-wise evolution of Mozambique V. cholerae O1 El Tor hybrid strain. Microbiology. 2010, 156: 99-107.
Bauer AW, Kirby WM, Sheris JC, Turck M: Antibiotics susceptibility testing by standardized single disk method. Am J Clin Pathol. 1966, 45: 493-496.
CLSI: Approved Guideline 2nd Edn, Document M45-A2. Methods for Antimicrobial Dilution and Disc Susceptibility Testing of Infrequently Isolated or Fastidious Bacteria. 2010, Wayne, PA: Clinical Laboratory Standards Institute, 1-56238-732-4
Thungapathra M, Amita, Sinha KK, Chaudhuri SR, Garg P, Ramamurthy T, Nair GB, Ghosh A: Occurrence of antibiotic resistance gene cassettes aac (6’)-Ib, dfrA5, dfrA12, and ereA2 in class I integrons in Non-O1, Non-O139 Vibrio cholerae strains in India. Antimicrob Agents Chemother. 2002, 46: 2948-2955.
Baranwal S, Dey K, Ramamurthy T, Nair GB, Kundu M: Role of active efflux in association with target gene mutations in fluoroquinolone resistance in clinical isolates of Vibrio cholerae. Antimicrob Agents Chemother. 2002, 46: 2676-2678.
Cameron DN, Khambaty FM, Wachsmuth IK, Tauxe RV, Barrett TJ: Molecular characterization of Vibrio cholerae O1 strains by pulsed-field gel electrophoresis. J Clin Microbiol. 1994, 32: 1685-1690.
Sack DA, Sack RB, Nair GB, Siddique AK: Cholera. Lancet. 2004, 363: 223-233.
Mhalu FS, Mmari PW, Ijumba J: Rapid emergence of El Tor Vibrio cholerae resistant to antimicrobial agents during first six months of fourth cholera epidemic in Tanzania. Lancet. 1979, 1 (8112): 345-347.
Glass RI, Huq I, Alim ARMA, Yunus M: Emergence of multiple antibiotic-resistant Vibrio cholerae in Bangladesh. J Infect Dis. 1980, 142: 939-942.
Waldor MK, Tschape H, Mekalanos JJ: A new type of conjugative transposon encodes resistance to sulfamethoxazole, trimethoprim, and streptomycin in Vibrio cholerae O139. J Bacteriol. 1996, 178: 4157-4165.
Hasan NA, Choi SC, Eppinger M, Clark PW, Chen A, Alam M, Haley BJ, Taviani E, Hine E, Su Q, Tallon LJ, Prosper JB, Furth K, Hoq M, Li H, Fraser-Liggett CM, Cravioto A, Huq A, Ravel J, Cebula TA, Colwell RR: Genomic diversity of, Haitian Cholera outbreak strains. Proc Natl Acad Sci U S A. 2010, 2012 (109): 2010-2017.
Marin MA, Vicente AC: Variants of Vibrio cholerae O1 El Tor from Zambia showed new genotypes of ctxB. Epidemiol Infect. 2011, 23: 1-2.
Kumar P, Jain M, Goel AK, Bhadauria S, Sharma SK, Kamboj DV, Singh L, Ramamurthy T, Nair GB: A large cholera outbreak due to a new cholera toxin variant of the Vibrio cholerae O1 El Tor biotype in Orissa, Eastern India. J Med Microbiol. 2009, 58: 234-238.
Rashed SM, Mannan SB, Johura FT, Islam MT, Sadique A, Watanabe H, Sack RB, Huq A, Colwell RR, Cravioto A, Alam M: Genetic characteristics of drug resistant V. cholerae O1 causing endemic cholera in Dhaka, 2006–2011. J Med Microbiol. 2012, 61: 1736-1745.
Taylor RK, Miller VL, Furlong DB, Mekalanos JJ: Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc Natl Acad Sci U S A. 1987, 84: 2833-2837.
Grim CJ, Hasan NA, Taviani E, Haley B, Chun J, Brettin TS, Bruce DC, Detter JC, Han CS, Chertkov O, Challacombe J, Huq A, Nair GB, Colwell RR: Genome sequence of hybrid Vibrio cholerae O1 MJ-1236, B-33, and CIRS101 and comparative genomics with V. cholerae. J Bacteriol. 2010, 192: 3524-3533.
Reimer AR, Domselaar GV, Stroika S, Walker M, Kent H, Tarr C, Talkington D, Rowe L, Olsen-Rasmussen M, Frace M, Sammons S, Dahourou GA, Boncy J, Smith AM, Mabon P, Petkau A, Graham M, Gilmour MW, Gerner-Smidt P: V. cholerae outbreak genomics task force: Comperative genomics of Vibrio cholerae from Haiti, Asia, and Africa. Emerg Infect Dis. 2011, 17: 2113-2121.
Pfau JD, Taylor RK: Genetic footprint on the ToxR-binding site in the promoter for cholera toxin. Mol Microbiol. 1996, 20: 213-222.
Taviani E, Grim CJ, Choi J, Chun J, Haley B, Hasan NA, Huq A, Colwell RR: Discovery of novel Vibrio cholerae VSP-II genomic islands using comparative genomic analysis. FEMS Microbiol Lett. 2010, 308: 130-137.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2334/14/392/prepub
Acknowledgements
This research was supported in part by the International Center for Diarrheal Disease Research, Bangladesh (icddr, b), the National Institutes of Health (NIH) Grant No. 1RO1A13912901 through agreements between the Johns Hopkins Bloomberg School of Public Health and icddr, b. This research was also supported partially by the National Institute of Infectious Diseases (NIID), Tokyo, Japan. icddr, b gratefully acknowledges its core donors: AusAID, GoB, CIDA, Sida & DFID, which provide unrestricted support to its activity and research. We are grateful to Dr. G.D. Thakur, Director of EDCD, Nepal for all the support while visiting affected sites, and for sharing preliminary data from the outbreaks in Doti and Bajhang. We thank Dr. A. Pant at Shahid Shukraraj Tropical Hospital, Kathmandu for facilitating acquisition of samples from Kathmandu outbreaks. We also thank Kantipur College of Medical Sciences for facilitating some of the laboratory work in Kathmandu, Nepal.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SMD, MA, AC, designed and HW and RBS supported the study. MA coordinated and SMD, FTJ, SM, AS, SBM, MUR, and SI carried out the study. SMD, RMR, and DK arranged the sample collection. SMD, FTJ, and MA analyzed data and prepared the manuscript. AC, HW, and RBS helped in manuscript writing and revision. All authors reviewed and approved the final version of the manuscript.
Sameer M Dixit, Fatema-Tuz Johura contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article

Cite this article
Dixit, S.M., Johura, FT., Manandhar, S. et al. Cholera outbreaks (2012) in three districts of Nepal reveal clonal transmission of multi-drug resistant Vibrio choleraeO1. BMC Infect Dis 14, 392 (2014). https://doi.org/10.1186/1471-2334-14-392
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2334-14-392