
Abstract
Background
Compared to methicillin-resistant Staphylococcus aureus (MRSA), characteristics of nasal carriage and community-onset infection methicillin-susceptible S. aureus (MSSA) are less well known. No characteristics of MSSA in Taiwan have been reported previously.
Methods
We analyzed 100 nasal carriage and 34 community-onset infection MSSA isolates by pulsed-field gel electrophoresis (PFGE), spa typing, multi-locus sequence typing, agr typing, virulence gene detection, growth rate measurement, and antimicrobial susceptibility.
Results
In PFGE analysis, most (68%) infection isolates could be grouped in one major cluster using a 70% similarity cutoff. In contrast, only 17% of nasal carriage isolates belonged to this cluster. A similar classification was obtained using Based Upon Repeat Pattern analysis of spa types. The MSSA infection isolates cluster was closely related to the virulent clones of clonal complex 1 (CC1), which includes strains MW2 (USA400) and MSSA476. ST188 of CC1 was the predominant clone detected for community-onset MSSA infections. The only common ST type for MSSA and MRSA in Taiwan was ST59, the community-associated MRSA clone. It is likely, therefore, that MRSA originated from MSSA clones through SCCmec transfer. Compared to nasal carriage isolates, infection isolates less frequently possessed egc, tst and hlg genes, were more commonly susceptible to erythromycin (91% vs. 54%), and had shorter mean doubling times (38 min vs. 55 min).
Conclusions
The clonal lineages of MSSA nasal carriage and infection isolates differed in our sample of Taiwan isolates. Most community-onset MSSA infections resulted from relatively few clonal lineages. Nasal carriage isolates more frequently possessed the egc, tst and hlg genes, were more resistant to erythromycin, and grew more slowly.
Similar content being viewed by others
Background
Staphylococcus aureus can asymptomatically colonize people in the community and in the healthcare setting. S. aureus is also responsible for a wide spectrum of illnesses, ranging from superficial infection of the skin and soft tissue to life-threatening septicemia, osteomyelitis, endocarditis, and toxic shock syndrome [1]. A few clonal lineages have also been observed among epidemic methicillin-resistant S. aureus (MRSA) isolates [2]. Although successful lineages of epidemic MRSA clones may have an adaptive advantage because of antibiotic resistance, virulence, and gene regulation, the exact mechanism by which these clones successfully circulate in community and healthcare facilities has not been fully elucidated. A previous study found that most MRSA clones arose from successful epidemic methicillin-susceptible S. aureus (MSSA) clones [2]. It would be interesting to have further studies to investigate if successful MSSA clones could be used to predict prevalent strains of MSSA or MRSA infection.
It has been shown that nasal carriage of S. aureus is a major risk factor for subsequent development of community-associated and nosocomial infections [3, 4]. Although the relationship between bacterial virulence determinants and invasive disease has been explored by comparing nasal carriage and disease isolates [5], most studied isolates were obtained from the healthcare environment. However, S. aureus isolates obtained from hospital patients may represent nosocomial transmission, and could therefore confound understanding of the intrinsic pathogenesis of specific S. aureus strains. The aim of the present study was to identify differences in lineages, virulence gene prevalence, growth rates and antimicrobial susceptibility between nasal carriage and community-onset infection MSSA.
Methods
Bacterial isolates
Thirty-four community-onset infection MSSA isolates were obtained from patients at two Taiwan medical centers (Tri-Service General Hospital and Kaohsiung Medical University Hospital) during a four-month collection period in 2006. A total of 100 nasal carriage MSSA isolates were available from our previous study [6]. The 34 community-onset infections were classified as community-associated infection or healthcare-associated community-onset infection according to the definition of Klevens et al. [7]. The criteria used to classify community-associated infection and healthcare-associated community-onset infection are as follows:
-
1.
Community-associated infection: No permanent indwelling catheters or medical devices that pass through the skin into the body and no medical history in the past year of hospitalization, admission to nursing home or nursing facility or hospice. Isolates from the specimens should be obtained within 48 hours after admission to hospital.
-
2.
Healthcare-associated community-onset infection: Patient with medical history of hospitalization, admission to nursing home or nursing facility or hospice in the past year. Isolates from the specimens should also be obtained within 48 hours after admission to hospital.
Pulsed-field gel electrophoresis (PFGE)
Staphylococcal genomic DNA typing was performed using PFGE by SmaI digestion, as previously described [8]. PFGE clusters were assigned to isolate clusters having 80% or higher similarity from the dendrogram based on Dice’s coefficients [8]; genetic relatedness was also assessed by visual inspection of fragment differences [9]. Analysis of isolate similarity was also performed, using a cut-off value of 70%.
spa types, multi-locus sequence typing (MLST), agrspecificity groups, and virulence genes
We used PCR to molecularly characterize isolates by spa type, multi-locus sequence typing (MLST), agr specificity group, and virulence genes. S. aureus DNA was extracted using a DNeasy Tissue Kit (Qiagen Inc., Valencia, USA), following the manufacturer’s instructions with a slight modification. An additional step was added just prior to proteinase K digestion: incubation with 5 μL of 5 mg/mL lysostaphin at 37°C for 30 min to 1 h until the cell suspension became clear.
spa typing and the Based Upon Repeat Pattern (BURP) algorithm were performed using Ridom StaphType software (version 1.5; Ridom GmbH, Würzburg, Germany). Two clustering parameters were applied to BURP clusters, spa-clonal complexes (CCs), using dialog: “exclude spa types that are shorter than 5 repeats” and “spa types are clustered if cost is less or equal 4” [10, 11]. The cost “6” parameter was also used as discussed by Strommenger et al. [12]. The cost matrix for distances of strains between spa types was also produced using the StaphType software, and the phylogenetic tree was conducted using MEGA version 5 by the neighbor-joining method [13]. The spa repeat region was amplified and sequenced using primers spa-1113f and spa-1514r, according to the manufacturer's instructions.
MLST was performed as described previously on selected isolates of major PFGE clusters and all infection isolates [14]. Sequence type (ST) was assigned based on sequence allelic profiles using the MLST database website (http://www.mlst.net) [14].
agr specificity groups were determined by multiplex PCR as described by Lina et al. [15]. Non-typable strains were determined by agrD sequencing, which encodes the autoinducing peptide (AIP) precursor [16].
Virulence toxin genes for sea-e, seg-j, sem-o, tst, eta, etb, lukE lukD, hla, hlb, hld, hlg, and hlg-2 were detected as described by Jarraud et al. [17]. Other bacterial adhesion genes, including fnbA, cna, sdrC, sdrD, sdrE, bbp, and icaA, were detected as described by Peacock et al. [5]. Sequences specific for Panton-Valentine leukocidin (PVL) genes (lukS-lukF) were detected as described previously [18].
Bacterial growth rate
A colorimetric microplate assay was used to assess doubling time of representative clonal lineage strains [19, 20]. Isolates were grown overnight in Tryptic Soy Broth (TSB), diluted 100-fold, then grown for an additional 2 h. Samples were washed twice with phosphate-buffered saline (PBS), then the pellet was re-suspended in TSB to a concentration of approximately 109 CFU/mL. Serial ten-fold dilutions were made to 108 to 104 CFU/mL with TSB. One hundred microliters of each dilution were plated in triplicate on microplates, and TSB without cells served as a control. Then 20 μL of CellTiter-Blue® reagent (Promega Corp., Madison, USA) were added to each well, and the cells were incubated at 37°C. The absorbance was recorded every 30 min until each dilution of cells reached maximum absorbance (OD at 570 nm and 600 nm as reference wavelengths). The doubling time was calculated as previously described [19].
Antibiotic susceptibility testing
Susceptibilities were determined based on minimum inhibitory concentrations obtained from broth micro-dilution following the guidelines of the Clinical and Laboratory Standards Institute [21], using Sensititre custom-designed plates (Trek Diagnostics, West Essex, England).
Clinical data and statistical analysis
The study was performed with approval from the Institutional Review Board of Kaohsiung Medical University Hospital (KMUH-IRB-990139). Data for cases with MSSA nasal carriage were obtained following written informed consent [6]. Differences in frequencies and proportions were tested using the chi-square (χ2) test with Yates' correction or Fisher's exact test, as appropriate. A P value of less than 0.05 was considered significant. To compare virulence gene profiles, factors associated with infection at P values less than 0.01 were further studied using a logistical regression model to calculate the odd ratios (ORs) and 99% confidence intervals (CIs).
Results
Genetic profiling of nasal carriage and community-onset infection isolates
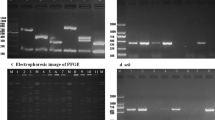
Molecular typing results, including PFGE, spa typing and MLST are presented in Figure 1. A total of 18 distinct PFGE clusters were found among 134 isolates, based on an 80% similarity cut-off value (approximately equivalent to less than or equal to six PFGE fragment differences). Thirty-nine spa types, including 7 new ones, were identified; two isolates were non-typable. Grouping by BURP, these spa types were clustered into 8 spa-CCs and 12 singletons. Four of eight spa-CCs had designated group founders: spa-CC c1:t116/t015, c2:t437/t441, c3:t338, and c4:t3400. Seven isolates with fewer than five repeats were excluded from clustering because their spa types may provide unreliable evolutionary information [11]. Although our results showed a high concordance among spa typing, MLST and PFGE, PFGE cluster C contained many spa types and STs. Four spa types (t701, t091, t213 and t160) and three STs (ST6, ST7 and ST12) were found in PFGE cluster C. These STs were distinct from one another and belonged to different clonal complexes (CCs). Because some MLST STs may be grouped in specific PFGE clusters, clonal lineages were described according to the combination of PFGE cluster and CC (PFGE cluster:CC) in the study (Figure 1).

PFGE dendrogram with molecular characterization for 100 nasal carriage and 34 infection MSSA isolates. PFGE cluster was assigned to isolates having 80% or greater similarity from the dendrograms (A similarity cut-off of 70% was also presented.). Multi-locus sequence typing (MLST), results in sequence type (ST); clonal complexes (CC); * Panton-Valentine leukocidin (PVL)-positive.
S. aureusinfections and clinical characteristics
The 34 community-onset infection MSSA isolates were classified as 24 community-associated isolates and 10 healthcare-associated community-onset isolates (Table 1). There were no significant differences between clinical manifestations caused by community-associated and healthcare-associated community-onset isolates. There were no associations between clonal lineages and disease manifestations. All STs of healthcare-associated community-onset isolates were also identified in community-associated isolates (Table 1). Therefore, all 34 MSSA infection isolates were grouped together for analysis.
Distribution of clonal lineages among nasal carriage and infection isolates
Although PFGE cluster C contained three different CCs (CC6, CC7 and CC12), isolates from this PFGE cluster were significantly more prevalent in infection than carriage isolates (32% vs. 6%, P < 0.01). An intriguing observation was that, using a 70% similarity cut-off value for cluster analysis (approximately equivalent to less than or equal to eight PFGE fragment differences) resulted in most infection isolates being grouped into one major cluster (including PFGE clusters B, C, D and E), with 68% of infection isolates and 17% of carriage isolates (P < 0.01). Carriage isolates were more diverse than infection isolates (Figure 1). The major infection isolates cluster contained several clonal lineages, including B:1, B:6, C:6, C:7, C:12, D:1 and E:97. STs of these infection isolates were ST1, ST6, ST6, ST7, ST12, ST188, and ST97, respectively. These ST isolates belonged to five CCs: CC1, CC6, CC7, CC12 and CC97. These data implicate specific MSSA clones as causing infectious diseases when clonal lineage was defined by PFGE and MLST typing. ST188 of CC1 was the predominant clone for community-onset MSSA infections. ST59 comprised 8.2% of all MSSA isolates but did not belong to the major infection isolates cluster.
BURP analysis of spa types showed a similar phenomenon to the PFGE clusters result. In an attempt to cluster more spa types in spa-CCs, we tried to adjust the cost to 6 for clustering more spa types in the BURP group; only spa-CC 267/359 could be grouped together among the major infection isolates. However, as indicated by the circle in Figure 2, the major infection PFGE cluster had closely related spa types by BURP analysis, with most infection PFGE clusters grouped together (except CC6).

Phylogenetic tree of the spa types was constructed by using MEGA version 5. The tree was produced based on the cost matrix for the distances of spa types. The cost “6” parameter for the BURP clustering was used in this figure. The circle indicates the major infection PFGE cluster. spa-CCs and the represented ST (CC) are indicated. NF (no founder) indicates clusters without group founders.
agrspecificity groups and virulence gene profiles
agr group I was the most common agr type for both nasal carriage (65%) and infection (74%) isolates, followed by agr group II for infection isolates (21%) and agr group IV for nasal carriage isolates (18%).
Among 30 virulence genes, only the LukE-LukD leukotoxin (lukE-lukD) and gamma variant hemolysin (hlg-2) genes were significantly more prevalent in infection isolates than in nasal carriage isolates (Table 2). In contrast, seven virulence genes were significantly more common in nasal carriage isolates than in infection isolates. Five of these seven virulence genes were staphylococcal enterotoxin genes (seg, sei, sem, sen, and seo), which reside at the enterotoxin gene cluster (egc) locus. Two other virulence genes found were tst and hlg, which encode toxic shock syndrome toxin 1 (TSST-1) and gamma-hemolysin, respectively. Although these were all community isolates, PVL toxin genes (lukS-lukF) were uncommonly detected in nasal carriage (7%) and infection (8.8%) isolates (P=0.727).
Bacterial growth rate
Doubling time was calculated to determine bacterial growth rates for all infection isolates and representative nasal carriage isolates (one was randomly selected from each nasal carriage clonal lineage). The mean doubling times for nasal carriage isolates were significantly higher compared to infection isolates (55.3 ± 8.1 vs. 37.9 ± 1.2 minutes, P < 0.01). Of 30 virulence genes, only egc enterotoxin genes were associated with significantly higher growth rates (P < 0.01).
Antibiotic resistance profiles
Antimicrobial resistance profiles were similar among the two groups of isolates. However, erythromycin resistance was found for 46% of nasal carriage isolates versus only 9% of infection isolates (P < 0.01) (Table 3).
Discussion
Compared to previous MRSA clone studies in Taiwan, which showed only four major infection isolate clones (ST239, ST59, ST241 and ST5) [22, 23], our study revealed more heterogeneous MSSA lineages. The major PFGE cluster of MSSA infection isolates included ST1, ST6, ST7, ST12, ST188, and ST97. However, these STs have not been reported in MRSA isolates in Taiwan [22, 23], indicating that most MSSA lineages have a different genetic background compared to MRSA lineages. Additionally, these MSSA STs differ from the most prevalent S. aureus STs worldwide [24]. Interestingly, the only ST common to MSSA and MRSA [22, 23] in Taiwan is ST59, the representative community-associated MRSA clone [25]. Our findings are consistent with those from several other countries, suggesting that MRSA probably originated from MSSA clones through SCCmec transfer [26]. These results, plus findings from a world-wide MRSA collection study, point to MRSA evolution in relatively few lineages [2], which allowed some successful clones to cause disease or spread. Although not all published studies have shown clonal differences between nasal carriage and infection isolates [27], our study revealed clonal differences in MSSA infection and nasal carriage isolates. MSSA infection isolates’ distinctive lineages and erythromycin susceptibility, as well as faster growth rates, support the clones’ uniqueness to cause community-onset MSSA infection.
Our finding that STs and clinical manifestations did not differ between community-associated and healthcare-associated community-onset isolates is consistent with Peacock et al.’s findings [5]. That team found little difference between strains associated with disease in both community and hospital settings, and suggested that bacterial factors play a role in determining invasive disease. In the present study, PFGE cluster D: CC1 (ST188) was the predominant clone in the community-onset MSSA infections. Our previous study also showed that this clone predominated among MSSA isolates from inpatients in a 2002 collection from four hospitals located in the north, middle, south, and east regions of Taiwan [16]. These results indicated that the epidemiology of MSSA infection isolates did not change much between these two periods, with no difference noted in either community or hospital settings.
Using PFGE or BURP analysis of spa types, we identified a few lineages likely to cause infection. Most of these representative CCs have been described previously as “group violations”, with the highest degree of misclassification between BURP groups and clonal lineages [12, 28]. However, these isolates may have clinical relevance because of their close relationship with severe community-associated infection strains in humans (MSSA476 and MW2 (USA400)) and ocular conjunctival infection in rabbits (UMCR1). All of these isolates belong to CC1 [29–31], and their high virulence has been shown in animal models [30, 32].
To clarify the difference between the nasal carriage isolates and infection isolates, we also compared the virulence gene profiles and growth rates among isolates. Infection isolates grew faster than carriage isolates. This difference suggests that infection isolates have better proliferation ability to outcompete other clones in the host. Additionally, the LukE-LukD leukotoxin (lukE-lukD) and gamma variant hemolysin (hlg-2) genes were more prevalent in infection isolates. Although seven virulence genes (egc enterotoxin genes, tst and hlg) were more prevalent in nasal carriage isolates, only isolates with egc enterotoxin genes were associated with slower growth rates (P < 0.01). It is possible that egc enterotoxin genes pose a fitness burden to the strains, resulting in slowed growth rates. The egc enterotoxin genes have been observed to have low-level production and weak virulence effects [33–35]. Our results reveal an epidemiologic relationship between egc enterotoxin genes and growth rate; however, the cause-effect relationship cannot be established in this study. It has been reported that virulence gene profiles are strongly linked with S. aureus clonal lineages [17]. Our nasal carriage isolates have uncommon STs, with most possessing the egc enterotoxin genes. Previous studies also showed such an epidemiological phenomenon in nasal carriage isolates [5, 36]. However, the role of the egc enterotoxin genes in nasal carriage isolates is not yet understood. The reason why our nasal carriage isolates have higher erythromycin resistance rates compared to community-onset infection isolates is also unclear.
Although the PVL genes have been considered a marker for community-acquired MRSA [37], we did not find that PVL presence was associated with community-onset MSSA infections. Goering et al. also failed to find this PVL association in a recent study [24].
agr, a global virulence gene regulator of S. aureus, is strongly linked with clonal lineages and some disease syndromes [17]. agr group I isolates were prevalent in our study, as in a previous study [38]. A similar distribution of agr group I was observed among nasal carriage (65%) and infection isolates (74%). agr types and PVL toxin genes were not useful in predicting infection MSSA isolates in the community setting.
Though only 34 infection isolates were analyzed, these isolates originated from two medical centers over a four-month period. The low MSSA isolate numbers result from a high rate of methicillin resistance among S. aureus in Taiwan during the study period [6, 22]. Also, only community-onset MSSA isolates were collected for the present study.
Conclusions
Our study presents genetic differences between nasal carriage and infection MSSA isolates from a community setting in Taiwan. The major MSSA infection isolates collected were closely related to the virulent clonal complex CC1. In addition to the difference in clonal lineages between nasal carriage and infection isolates, nasal carriage isolates more frequently possessed egc, tst and hlg genes, were more resistant to erythromycin, and had slower growth rates.
References
Lowy FD: Staphylococcus aureus infections. N Engl J Med. 1998, 339 (8): 520-532. 10.1056/NEJM199808203390806.
Enright MC, Robinson DA, Randle G, Feil EJ, Grundmann H, Spratt BG: The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA). Proc Natl Acad Sci USA. 2002, 99 (11): 7687-7692. 10.1073/pnas.122108599.
von Eiff C, Becker K, Machka K, Stammer H, Peters G: Nasal carriage as a source of Staphylococcus aureus bacteremia. N Engl J Med. 2001, 344 (1): 11-16. 10.1056/NEJM200101043440102.
Wertheim HF, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA, Nouwen JL: The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis. 2005, 5 (12): 751-762. 10.1016/S1473-3099(05)70295-4.
Peacock SJ, Moore CE, Justice A, Kantzanou M, Story L, Mackie K, O'Neill G, Day NP: Virulent combinations of adhesin and toxin genes in natural populations of Staphylococcus aureus. Infect Immun. 2002, 70 (9): 4987-4996. 10.1128/IAI.70.9.4987-4996.2002.
Lu PL, Chin LC, Peng CF, Chiang YH, Chen TP, Ma L, Siu LK: Risk factors and molecular analysis of community methicillin-resistant Staphylococcus aureus carriage. J Clin Microbiol. 2005, 43 (1): 132-139. 10.1128/JCM.43.1.132-139.2005.
Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, et al: Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA. 2007, 298 (15): 1763-1771. 10.1001/jama.298.15.1763.
McDougal LK, Steward CD, Killgore GE, Chaitram JM, McAllister SK, Tenover FC: Pulsed-field gel electrophoresis typing of oxacillin-resistant Staphylococcus aureus isolates from the United States: establishing a national database. J Clin Microbiol. 2003, 41 (11): 5113-5120. 10.1128/JCM.41.11.5113-5120.2003.
Tenover FC, Arbeit RD, Goering RV, Mickelsen PA, Murray BE, Persing DH, Swaminathan B: Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. J Clin Microbiol. 1995, 33 (9): 2233-2239.
Harmsen D, Claus H, Witte W, Rothganger J, Claus H, Turnwald D, Vogel U: Typing of methicillin-resistant Staphylococcus aureus in a university hospital setting by using novel software for spa repeat determination and database management. J Clin Microbiol. 2003, 41 (12): 5442-5448. 10.1128/JCM.41.12.5442-5448.2003.
Mellmann A, Weniger T, Berssenbrugge C, Rothganger J, Sammeth M, Stoye J, Harmsen D: Based Upon Repeat Pattern (BURP): an algorithm to characterize the long-term evolution of Staphylococcus aureus populations based on spa polymorphisms. BMC Microbiol. 2007, 7 (1): 98-10.1186/1471-2180-7-98.
Strommenger B, Braulke C, Heuck D, Schmidt C, Pasemann B, Nubel U, Witte W: spa typing of Staphylococcus aureus as a frontline tool in epidemiological typing. J Clin Microbiol. 2008, 46 (2): 574-581. 10.1128/JCM.01599-07.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011, 28 (10): 2731-2739. 10.1093/molbev/msr121.
Enright MC, Day NP, Davies CE, Peacock SJ, Spratt BG: Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J Clin Microbiol. 2000, 38 (3): 1008-1015.
Lina G, Boutite F, Tristan A, Bes M, Etienne J, Vandenesch F: Bacterial competition for human nasal cavity colonization: role of Staphylococcal agr alleles. Appl Environ Microbiol. 2003, 69 (1): 18-23. 10.1128/AEM.69.1.18-23.2003.
Chen FJ, Hiramatsu K, Huang IW, Wang CH, Lauderdale TL: Panton-Valentine leukocidin (PVL)-positive methicillin-susceptible and resistant Staphylococcus aureus in Taiwan: identification of oxacillin-susceptible mecA-positive methicillin-resistant S. aureus. Diagn Microbiol Infect Dis. 2009, 65 (4): 351-357. 10.1016/j.diagmicrobio.2009.07.024.
Jarraud S, Mougel C, Thioulouse J, Lina G, Meugnier H, Forey F, Nesme X, Etienne J, Vandenesch F: Relationships between Staphylococcus aureus genetic background, virulence factors, agr groups (alleles), and human disease. Infect Immun. 2002, 70 (2): 631-641. 10.1128/IAI.70.2.631-641.2002.
Ma XX, Galiana A, Pedreira W, Mowszowicz M, Christophersen I, Machiavello S, Lope L, Benaderet S, Buela F, Vincentino W, et al: Community-acquired methicillin-resistant Staphylococcus aureus, Uruguay. Emerg Infect Dis. 2005, 11 (6): 973-976.
Kuda T, Shimizu K, Yano T: Comparison of rapid and simple colorimetric microplate assays as an index of bacterial count. Food Control. 2004, 15 (6): 421-425. 10.1016/S0956-7135(03)00116-6.
Shiloh MU, Ruan J, Nathan C: Evaluation of bacterial survival and phagocyte function with a fluorescence-based microplate assay. Infect Immun. 1997, 65 (8): 3193-3198.
Clinical and Laboratory Standards Institute: Performance standards for antimicrobial susceptibility testing; 20th informational supplement. 2010, Wayne, PA, USA: CLSI document M100-S20, CLSI
Chen FJ, Lauderdale TL, Huang IW, Lo HJ, Lai JF, Wang HY, Shiau YR, Chen PC, Ito T, Hiramatsu K: Methicillin-resistant Staphylococcus aureus in Taiwan. Emerg Infect Dis. 2005, 11 (11): 1761-1763. 10.3201/eid1111.050367.
Wang WY, Chiueh TS, Sun JR, Tsao SM, Lu JJ: Molecular typing and phenotype characterization of methicillin-resistant Staphylococcus aureus isolates from blood in Taiwan. PLoS One. 2012, 7 (1): e30394-10.1371/journal.pone.0030394.
Goering RV, Shawar RM, Scangarella NE, O'Hara FP, Amrine-Madsen H, West JM, Dalessandro M, Becker JA, Walsh SL, Miller LA, et al: Molecular epidemiology of methicillin-resistant and methicillin-susceptible Staphylococcus aureus isolates from global clinical trials. J Clin Microbiol. 2008, 46 (9): 2842-2847. 10.1128/JCM.00521-08.
Huang TW, Chen FJ, Miu WC, Liao TL, Lin AC, Huang IW, Wu KM, Tsai SF, Chen YT, Lauderdale TL: Complete genome sequence of Staphylococcus aureus M013, a pvl-positive, ST59-SCCmec type V strain isolated in Taiwan. J Bacteriol. 2012, 194 (5): 1256-1257. 10.1128/JB.06666-11.
Deurenberg RH, Stobberingh EE: The evolution of Staphylococcus aureus. Infect Genet Evolution. 2008, 8 (6): 747-763. 10.1016/j.meegid.2008.07.007.
Feil EJ, Cooper JE, Grundmann H, Robinson DA, Enright MC, Berendt T, Peacock SJ, Smith JM, Murphy M, Spratt BG, et al: How clonal is Staphylococcus aureus?. J Bacteriol. 2003, 185 (11): 3307-3316. 10.1128/JB.185.11.3307-3316.2003.
Hallin M, Deplano A, Denis O, De Mendonca R, De Ryck R, Struelens MJ: Validation of pulsed-field gel electrophoresis and spa typing for long-term, nationwide epidemiological surveillance studies of Staphylococcus aureus infections. J Clin Microbiol. 2007, 45 (1): 127-133. 10.1128/JCM.01866-06.
Baba T, Takeuchi F, Kuroda M, Yuzawa H, Aoki K, Oguchi A, Nagai Y, Iwama N, Asano K, Naimi T, et al: Genome and virulence determinants of high virulence community-acquired MRSA. Lancet. 2002, 359 (9320): 1819-1827. 10.1016/S0140-6736(02)08713-5.
Balzli CL, Bartell J, Dajcs JJ, McCormick CC, Caballero AR, Stroman D, O'Callaghan RJ: A highly virulent Staphylococcus aureus: rabbit anterior chamber infection, characterization, and genetic analysis. Invest Ophthalmol Vis Sci. 2010, 51 (10): 5114-5120. 10.1167/iovs.10-5179.
Holden MTG, Feil EJ, Lindsay JA, Peacock SJ, Day NPJ, Enright MC, Foster TJ, Moore CE, Hurst L, Atkin R, et al: Complete genomes of two clinical Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug resistance. Proc Natl Acad Sci USA. 2004, 101 (26): 9786-9791. 10.1073/pnas.0402521101.
Voyich JM, Otto M, Mathema B, Braughton KR, Whitney AR, Welty D, Long RD, Dorward DW, Gardner DJ, Lina G, et al: Is Panton‐Valentine leukocidin the major virulence determinant in community‐associated methicillin‐resistant Staphylococcus aureus disease?. J Infect Dis. 2006, 194 (12): 1761-1770. 10.1086/509506.
Ferry T, Thomas D, Genestier A-L, Bes M, Lina G, Vandenesch F, Etienne J: Comparative prevalence of superantigen genes in Staphylococcus aureus isolates causing sepsis with and without septic shock. Clin Infect Dis. 2005, 41 (6): 771-777. 10.1086/432798.
Grumann D, Scharf SS, Holtfreter S, Kohler C, Steil L, Engelmann S, Hecker M, Völker U, Bröker BM: Immune cell activation by enterotoxin gene cluster (egc)-encoded and non-egc superantigens from Staphylococcus aureus. J Immunol. 2008, 181 (7): 5054-5061.
Omoe K, Ishikawa M, Shimoda Y, Hu D-L, Ueda S, Shinagawa K: Detection of seg, seh, and sei genes in Staphylococcus aureus isolates and determination of the enterotoxin productivities of S. aureus isolates harboring seg, seh, or sei genes. J Clin Microbiol. 2002, 40 (3): 857-862. 10.1128/JCM.40.3.857-862.2002.
van Belkum A, Melles DC, Snijders SV, van Leeuwen WB, Wertheim HFL, Nouwen JL, Verbrugh HA, Etienne J: Clonal distribution and differential occurrence of the enterotoxin gene cluster, egc, in carriage- versus bacteremia-associated isolates of Staphylococcus aureus. J Clin Microbiol. 2006, 44 (4): 1555-1557. 10.1128/JCM.44.4.1555-1557.2006.
Vandenesch F, Naimi T, Enright MC, Lina G, Nimmo GR, Heffernan H, Liassine N, Bes M, Greenland T, Reverdy ME, et al: Community-acquired methicillin-resistant Staphylococcus aureus carrying Panton-Valentine leukocidin genes: worldwide emergence. Emerg Infect Dis. 2003, 9 (8): 978-984. 10.3201/eid0908.030089.
van Leeuwen W, van Nieuwenhuizen W, Gijzen C, Verbrugh H, van Belkum A: Population studies of methicillin-resistant and -sensitive Staphylococcus aureus strains reveal a lack of variability in the agrD gene, encoding a staphylococcal autoinducer peptide. J Bacteriol. 2000, 182 (20): 5721-5729. 10.1128/JB.182.20.5721-5729.2000.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2334/12/343/prepub
Acknowledgements
The authors thank Ms. Wen-Jin Chiang and the Clinical Microbiology Division, Department of Laboratory Medicine, Kaohsiung Medical University Hospital for collection of bacterial isolates. The authors thank Ms. Li-Yun Hsieh for performing the statistical analysis.
Financial disclosure
This project was supported by intramural grants from the National Health Research Institutes (ID-099-SP-09) and Kaohsiung Municipal Hsaio-Kang Hospital, Kaohsiung Medical University. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that no competing interests exist.
Authors’ contributions
FJC and PLL designed the study. JCL provided advice regarding clinical aspects of the study. FJC and CHW performed laboratory work. PLL, LKS, and FJC prepared the manuscript. All authors read and approved the final version of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Chen, FJ., Siu, LK.K., Lin, JC. et al. Molecular typing and characterization of nasal carriage and community-onset infection methicillin-susceptible Staphylococcus aureusisolates in two Taiwan medical centers. BMC Infect Dis 12, 343 (2012). https://doi.org/10.1186/1471-2334-12-343
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2334-12-343