
Abstract
Background
The most common causes of cholestatic jaundice are biliary atresia and idiopathic neonatal hepatitis (INH). Specific disorders underlying INH, such as various infectious and metabolic causes, including neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) especially, in East Asian populations are increasingly being identified. Since most NICCD infants recovered from liver disease by 1 year of age, they often are misdiagnosed with INH, leading to difficulty in determining the true prevalence of NICCD. Mutation(s) of human SLC25A13 gene encoding a mitochondrial aspartate/glutamate carrier isoform 2 (AGC2), can lead to AGC2 deficiency, resulting in NICCD and an adult-onset fatal disease namely citrullinemia type II (CTLN2). To study the prevalence of NICCD and SLC25A13 mutations in Thai infants, and to compare manifestations of NICCD and non-NICCD, infants with idiopathic cholestatic jaundice or INH were enrolled. Clinical and biochemical data were reviewed. Urine organic acid and plasma amino acids profiles were analyzed. PCR-sequencing of all 18 exons of SLC25A13 and gap PCR for the mutations IVS16ins3kb and Ex16+74_IVS17-32del516 were performed. mRNA were analyzed in selected cases with possible splicing error.
Results
Five out of 39 (12.8%) unrelated infants enrolled in the study were found to have NICCD, of which three had homozygous 851del4 (GTATdel) and two compound heterozygous 851del4/IVS16ins3kb and 851del4/1638ins23, respectively. Two missense mutations (p.M1? and p.R605Q) of unknown functional significance were identified. At the initial presentation, NICCD patients had higher levels of alkaline phosphatase (ALP) and alpha-fetoprotein (AFP) and lower level of alanine aminotransferase (ALT) than those in non-NICCD patients (p< 0.05). NICCD patients showed higher citrulline level and threonine/serine ratio than non-NICCD infants (p< 0.05). Fatty liver was found in 2 NICCD patients. Jaundice resolved in all NICCD and in 87.5% of non-NICCD infants at the median age of 9.5 and 4.0 months, respectively.
Conclusion
NICCD should be considered in infants with idiopathic cholestasis. The preliminary estimated prevalence of NICCD was calculated to be 1/48,228 with carrier rate of 1/110 among Thai infants. However, this number may be underestimated and required further analysis with mutation screening in larger control population to establish the true prevalence of NICCD and AGC2 deficiency.
Similar content being viewed by others


Background
Cholestatic jaundice affects approximately 1 in every 2,500 infants world-wide [1, 2]. The most common causes are biliary atresia and idiopathic neonatal hepatitis (INH). An increase in specific disorders underlying INH are being identified, which include various infectious and metabolic causes such as tyrosinemia, alpha-1 antitrypsin deficiency, and galactosemia [3]. INH could be caused by unidentified metabolic disorders.
Deficiency of AGC2, liver-type mitochondrial aspartate-glutamate carrier (AGC) [4], is an autosomal recessive disorder caused by mutations of the SLC25A13 gene (7q21.3) [5]. It results in two distinct phenotypes: adult-onset type II citrullinemia (CTLN2; OMIM 603471), and neonatal intrahepatic cholestasis caused by AGC2 deficiency (NICCD; OMIM 605814) [6]. CTLN2 is characterized by recurrent episodes of altered mental status, hyperammonemia and coma which can occur at any age but usually in adulthood [5, 7, 8]. NICCD patients present in the first few months of life with milder symptoms characterized by intrahepatic cholestasis, diffuse fatty liver, parenchymal cellular infiltration associated with hepatic fibrosis, hypoalbuminemia, coagulopathy, liver dysfunction with or without hypoglycemia, galactosuria, multiple aminoacidemia including elevated citrulline, arginine, threonine, methionine, phenylalanine, and tyrosine concentrations [8–12].
NICCD was first described by Ohura et al. in 2001 [13]. Increasing numbers of NICCD patients have been reported with the majority from Japanese and East Asian populations, including Taiwanese [14, 15], Korean [16] and Chinese [12, 17]. A handful of patients with NICCD have been identified in Arabic, Pakistani, Caucasian descendants, suggesting a panethnic disease [16, 18–20]. Since most NICCD infants recovered from liver disease by 1 year of age, they often are misdiagnosed with INH, leading to difficulty in determining the true prevalence of NICCD [10]. Because clinical manifestations and biochemical findings are nonspecific for NICCD, DNA analysis or Western blot analysis of AGC2 protein in lymphocytes is the most reliable diagnostic tool [21]. The objectives were to study the prevalence of NICCD in Thai infants with idiopathic cholestasis, mutation spectrum of SLC25A13 in Thai NICCD, and comparison of clinical manifestations and blood chemistry between NICCD and non-NICCD infants.
Results
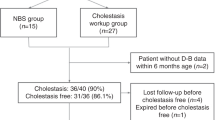
Thirty-nine unrelated infants with idiopathic cholestasis or INH participated in the study, 18 from cohort-A (12 males and 6 females) and 21 from cohort-B (17 males and 4 females), making a male/female ratio 3:1. Median age at onset of the jaundice was 1 month (range 0.5-5.0) in both cohorts. The median age at enrollment was 60.5 months (range 12.0-131.0) in cohort-A, and 3.0 months (range 1.0-16.0) in cohort-B. At the time of enrollment, in cohort-A, jaundice was resolved in all but one patient, whereas all patients in cohort-B presented with jaundice. Five male patients (1 from cohort-A and 4 from cohort-B) were confirmed to have NICCD.
Mutation data
Genotypes in the NICCD cases were homozygous 851del4 or GTATdel (mutation [I]) in Patients 1, 2, and 5, 851del4/IVS16ins3kb (or mutation [XIX]) in Patient 3 (Figure 1A), and 851del4/c.1638-1660dup (or mutation [III]: 1638ins23) in Patient 4. There were two suspected NICCD cases with genotype p.M1?/wt in Patient 6 and R605Q/wt in Patient 7 (both from cohort-A). None of the patients had mutation [XX] (or Ex16+74_IVS17-32del516). Two reported single nucleotide polymorphisms (SNPs) including IVS4+6A>G (rs6957975) and L398L (rs2301629); and 2 newly identified SNPs, IVS4-52 A>G and IVS17-12C>A were discovered (Table 1).

Agraose gel electrophoresis showing IVS16ins3kb (XIX), Mutations 851del4 (I), and p.M1? variant. A: 1.5% gel revealing 3.65 kb fragment representing the IVS16ins3kb (XIX) and the 990bp fragment representing normal allele.; B: 1.5% gel showing exon 9-PCR-HpyCH4IV restriction digest. The 851del4 mutation abolishes the restriction site, whereas the normal allele was cut, yielding 179 and 122bp fragments.; C: 2.5% gel showing 109bp-product of PCR using primers E1F/E1_1MT-R. The p.M1? allele produces an EagI restriction site, yielding 89 and 20bp-fragments.
After an identification of 851del4 in Patients 1-5, we established a screening method using PCR with primer SLC25A13_E9 forward and reverse (for all primers, see Additional file 1) followed by HpyCH4IV (A′CGT) restriction digestion (Figure 1B). This test was used for simple and rapid screening of the 851del4 in the control specimens and also as a second method of confirming the deletion in the patients.
The p.M1? variant does not alter restriction site, therefore, a reverse primer E1_M1T-R was designed and used with forward primer E1-F to yield PCR product containing the mutated site and to create an artificial EagI restriction site (C′GGCCG) in the p.M1? allele (Figure 1C). This test was used to screen the 100 controls.
Sequencing of cDNA from an 851del4-homozygous patient and the p.R605Q-heterozygous individual revealed correct splicing of the exons involved (data not shown). None of the controls were found to have 851del4, IVS16ins3kb, 1638ins23, and the p.R605Q variant (creating a BsrDI restriction site). The p.M1? was present in only heterozygous state in 3 controls (Table 1).
Comparison of clinical and laboratory findings between NICCD and non-NICCD infants
The major presentations of five definite NICCD patients (Patients 1-5) were cholestatic jaundice and hepatomegaly. Median age at onset of cholestatic jaundice was 0.5 months (range 0.1-4.0) in NICCD patients and 1.0 month (range 0.5-5.0) in non-NICCD patients (Patients 8-39). Splenomegaly was initially found in 2 NICCD (40%) and 11 non-NICCD patients (34.4%). At presentation, NICCD patients had significantly higher alkaline phosphatase (ALP), lower ALT and higher alpha-fetoprotein (AFP) than in non-NICCD infants (Table 2). Coagulopathy, galactosuria and hypoglycemia were remarkably observed among NICCD patients but not in the non-NICCD infants. Serum AFP levels were not available in suspected NICCD cases. Plasma amino acid (PAA) profiles were only available from four NICCD patients (from cohort-B) and 18 non-NICCD patients which showed elevated citrulline and elevated threonine/serine ratio in NICCD patients compared to those in non-NICCD patients (p<0.05) (Table 3).
Liver histology was available in 3 NICCD and 15 non-NICCD infants. Cholestasis and fatty changes were found in two NICCD patients (Patients 3 and 4). In addition, histologic features of cirrhosis were demonstrated in Patient-4. Patient-2 had marked cellular swelling, disarray of hepatic cords, cholestasis, multinucleated giant cell transformation, and decreased interlobular bile ducts. The major histological findings in non-NICCD infants were multinucleated giant cell transformation and features compatible with INH (n=9), paucity of interlobular bile duct (n=4), fatty changes (n=1; Patient 10) and severe periportal fibrosis (n=1). The liver histologic findings of the 2 suspected NICCD patients were compatible with INH.
The management of all the patients was directed toward treating the consequence of cholestasis, consisting of non-lactose and medium chain triglycerides formula and supplementation with fat-soluble vitamins. Jaundice resolved in all NICCD patients at the median age of 6.6 months (range 5-11) and liver biochemistry became normal in 4/5 NICCD patients (except patient 4) at the median age of 9.5 months (range 5-10). Patient 4 had the most severe manifestation in the NICCD group in this study. He had jaundice started at 1 month old, developmental delay due to unrecognized hypoglycemia till the age of 8 months when he was referred to our department. After 4 months of treatment, jaundice and hypoglycemia resolved and liver chemistry became near normal. At the time of this report, he is 38 month-old with appropriate growth and psychomotor development.
Jaundice resolved in the two suspected NICCD cases, at the age of 5 and 9 months, respectively. Among the non-NICCD infants, 87.5% (28/32) had jaundice resolved at median age of 4 months (range 2-24). The remainder had progressive jaundice leading to cirrhosis; one patient underwent liver transplantation and has been well, and another patient died of complications of end-staged liver disease.
Discussion
In the present study, the 851del4 was accounting for 80% of the mutant alleles identified. We demonstrate 12.8% (5/39) prevalence of NICCD among Thai infants with idiopathic cholestatic jaundice/INH. Ko et al and Fu et al reported 6% (3/47) and 9.5% (38/400) prevalence of NICCD among Korean and Chinese infants with idiopathic cholestasis, respectively [22, 23]. By employing the above prevalence of 12.8%, 40% of Thai infantile cholestatsis being have idiopathic cholestasis [24] (and our unpublished data), and panethnic incidence of cholestatic jaundice 1/2,500 infants (no specific data for Thai infants) [1, 2], the incidence of NICCD among Thai infants is calculated to be 1 in 48,828. By reverse calculation based on Hardy-Weinberg equation, the frequency of SLC25A13 mutation-carrier among Thai infants calculated from the incidence of NICCD is predicted to be 1 in 110 (q2=1/48,828; 2pq = 2x≅1x1/220). This number is lower than the frequency of SLC25A13 mutations carrier among Chinese (1/63), Japanese (1/65), and Korean (1/108) populations, respectively [15, 20, 25], plausibly due to underestimation of the prevalence in the present study because not all infants with SLC25A13 mutation(s) on both alleles manifest NICCD symptoms and that the carrier rates described in those East Asian populations were obtained through molecular genetic screening of control population which is more reliable method to provide epidemiologic data.
If p.M1? is considered pathologic allele and its carrier rate (3/100) by population analysis is taken into account with the carrier rate of the three mutations (851del4, IVS16ins3kb, and 1638ins23) obtained from the cholestatic infants group, it makes a carrier rate of 1 in 52 (4/210), giving a new estimated incidence of NICCD 1 in 11,025. Given no available data of p.M1? in other population to compare and the absence of functional prove of the p.M1?, it may be too premature to include the carrier rate of p.M1? for estimation of the incidence of NICCD in Thai population.
We tried to compare the genotype of the most severe case (Patient 4) in the present study to those severe NICCD cases published in the literature. Only four NICCD patients have been reported to have progressive liver disease requiring liver transplantation [9, 26–28]. Genotypes of those cases were 851del4/IVS11+1G>A [9, 27], 1638ins23/S225X [26], and genotype not specified in two cases; therefore, genotype-phenotype association could not be concluded.
Among over 50 mutations of SLC25A13 described [25], the 851del4 (mutation I) and IVS11+1G>A (mutation II) are the most frequent mutations described among Japanese affected population, accounting for 70% of the mutant alleles, and 851del4 is the most frequent in Chinese [8, 20]. The other common mutations were 1638ins23, S255X, and IVS6+5G>A [20]. There is no doubt about the pathogenicity of the mutations 851del4, IVS16ins3kb, and c.1638-1660dup because these mutations have been identified multiple times in East Asian affected population although, functional data is quite limited.
The pathogenicity of the novel p.M1? variant is uncertain. With the absence of the original initiation start codon, the first AUG codon is located at nt 71-73 leading to translation of a non functional short polypeptide of 21 amino acids and possible degradation of the mutant mRNA through a nonsense-mediated mRNA decay mechanism [29]. Whether or not this particular allele has deleterious effect on AGC2 function, it remains to be elucidated. The p.R605Q mutation occurs at a conserved amino acid position of SLC25A13 (AGC2) and SLC25A12 (aralar) across various species (AGC2: cow, chicken, mouse, rat, chimpanzee, horse; aralar: human, monkey, macaque, dog, mouse); although possibly be deleterious, its pathogenicity remains to be proven.
When comparing with the non-NICCD cases, the NICCD patients had significantly higher ALP and lower ALT levels, higher citrulline concentration and threonine/serine ratio, supporting previous studies [9, 11, 30]. The AST/ALT ratio seemed higher in NICCD group, but was not statistically significant. Failure to thrive was a presenting feature in 37-50% of NICCD patients, but not found in our cohort [11, 12]. NICCD cannot be excluded on the basis of normal PAA profiles, as evidenced in Patients-3. Liver histology was not always diagnostic for NICCD. The characteristic histology of NICCD including cholestatsis and fatty change [10, 11, 31, 32] was found in two cases in this study. Bile duct paucity, an uncommon finding in NICCD which has been demonstrated in one report [31] was found in one patient (Patient 2). His clinical manifestations were moderately severe with cholestatic jaundice, coagulopathy, galactosuria and jaundice resolved at 6 months of age. Fatty liver was found in a non-NICCD patient (Patient 10) who had jaundice since 1 month and the jaundice resolved by 3 months without identified other metabolic liver diseases. Another non-NICCD case, Patient 11, had markedly high ALP and AFP (1935 and >60,500 respectively, shown in Table 2). Her jaundice started at 1 month and resolved at 6 months of age, and her liver histology was the feature of INH. Of note, Patients 10 and Patient 11 could have NICCD but with unidentifiable mutation(s).
There were limitations of the present study. Firstly, using the panethnic incidence of infantile cholestasis for the estimation of the prevalence of NICCD among Thai infants raises concern on the precision of the predicted prevalence. It would be more reliable to use prevalence of infantile cholestasis among Thai population; however, such data does not exist. Secondly, the prevalence of NICCD obtained by calculation from manifesting infants may be underestimated because AGC2 deficiency is a disease with incomplete penetrance. Thirdly, large deletion and/or insertion could be missed by PCR-direct sequencing. Fourthly, the lack of functional analysis to support the pathologic significance of novel SLC25A13 variants identified in the present study leads to inconclusive data of some patients (Patients 6 and 7). Existing system for functional analysis of AGC2 protein is complicated and difficult to conduct [33]. New method of functional analyses of AGC2 mutation is needed to provide more insight into SLC25A13 variants.
Conclusion
NICCD has been underestimated and should be considered in infants with idiopathic cholestatic jaundice, especially when associated with markedly high ALP and AFP, fatty liver and elevated plasma citrulline. It is impossible to establish the true incidence of NICCD and AGC2 deficiency among Thai infants by the method used in the present study. Molecular genetic analysis of general population and functional analysis to indicate the disease association with the variants is required to answer that question. Development of alternative and simple system for functional testing for AGC2 variants is necessary for better understanding pathogenic mechanism of AGC2 genetic variants, especially for those newly identified.
Material and methods
Patients and clinical analysis
Infants with idiopathic cholestasis or idiopathic neonatal hepatitis were recruited. Those diagnosed during January 1996 - September 2006 were assigned to cohort-A (retrospective cohort), and those diagnosed during October 2006 - February 2010 to cohort-B (prospective cohort). Idiopathic cholestasis was defined as serum total bilirubin (TB) exceeding 5 mg/dL and direct bilirubin (DB) level > 20% of TB or DB > 1 mg/dL if TB was < 5 mg/dL, and without an identifiable cause. Written informed consents were obtained from each parent, following the approval of Ramathibodi Institutional Review Board. Clinical data were reviewed. Routine investigations for infantile cholestasis included viral studies, abdominal ultrasonography, hepatobiliary scintigraphy and metabolic screening (urine reducing substance). Biliary atresia and extrahepatic obstruction were excluded in all cases. Liver biopsy was performed if the diagnosis remained inconclusive. Urine organic acids and plasma amino acid (PAA) were analyzed in the patients from cohort-A who still had jaundice at the time of enrollment, and all patients from cohort-B. Fischer and threonine/serine ratio were calculated (Fischer ratio = branched-chain amino acids valine + leucine + isoleucine/aromatic amino acids tyrosine + phenylalanine). Specific investigations for progressive familial intrahepatic cholestasis (PFIC), Alagille syndrome and specific metabolic liver diseases were performed in selected cases. Investigations for alpha-1 antitrypsin deficiency were not routinely performed since a previous study showed that this disorder is unlikely to be a cause of liver diseases among Thai children [34].
Genetic analysis
Genomic DNA (gDNA) was prepared from peripheral blood lymphocytes by phenol-chloroform extraction. PRIMER 3 was used for primer design (http://www.Frodo.wi.mit.edu/cgi-bin/primer3). GenBank reference sequences were NT_079595 and NM_014251.2 (AGC2 isoform 2). Sequencing was performed on an ABI 3100 DNA sequencer after purification with QIAquick PCR purification kits (QAIGEN®; California, USA). Long-range PCR was performed according to the manufacturer’s instructions (Long PCR Enzyme Mix; Fermentas Life Sciences, California, USA). mRNA transcripts were isolated from peripheral blood with the QIAamp RNA Blood Mini Kit (QIAGENTM), then reverse transcribed into single-stranded complementary DNA (cDNA) using Superscript III Reverse Transcriptase (Invitrogen). Primer sequences for gDNA and cDNA amplification are available as supplementary data at the journal website.
PCR-sequencing of all 18 exons of the SLC25A13 gene of genomic DNA from all the infants and of cDNA from selected cases was performed. To detect both 3kb insertion of intron 16 (mutation [XIX] or IVS16ins3kb) and 516bp deletion of exon 16 connecting intron 17 ([XX] or Ex16+74_IVS17-32del516), gap PCR was performed by using long-range PCR and primers Ex16F and Ex18-3′R, as previously described [20]. Positive controls for the insertion were kindly provided by Prof. Kobayashi from Kagoshima University. Once a mutation(s) was identified, genetic analysis of family members was performed and PCR-restriction digest with appropriate endonuclease restriction enzymes was carried out in 100 healthy controls (regular blood donors). The IVS16ins3kb was also screened in the 100 controls. cDNA analysis was performed in selected case with possible splicing error. Primer sequences and detailed molecular method are available as an Additional file 1. Nomenclature of newly identified SLC25A13 variants is assigned following the guidelines of the Human Genome Variation Society (http://www.hgvs.org/mutnomen) [35].
Infants with pathogenic SLC25A13 mutation(s) identified on both alleles were considered definite case of NICCD while those with pathogenic/unclear significant mutation identified on only one allele were considered suspected NICCD, and those known as nonpathogenic/silent or unidentifiable mutation were considered non-NICCD cases.
Statistical analysis
Data were analyzed using SPSS (version 13.0, SPSS Inc, Chicago, Illinois, USA). Mann–Whitney U test and Fisher's Exact test were used to compare continuous variables and dichotomous variables, respectively. A P-value of <0.05 was considered statistically significant.
Author’s contributions
ST, SJ, PP, collected and analyzed clinical data, and wrote/edited the manuscript. DW, PS, SJ designed laboratory work and wrote/edited the manuscript. PK and DC performed laboratory work and edited the manuscript. KK advised the study design and editing the manuscript. All authors read and approved the final manuscript.
References
Moyer V, Freese DK, Whitington PF, Olson AD, Brewer F, Colletti RB, Heyman MB: Guideline for the evaluation of cholestatic jaundice in infants: recommendations of the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2004, 39 (2): 115-128. 10.1097/00005176-200408000-00001.
Dick MC, Mowat AP: Hepatitis syndrome in infancy–an epidemiological survey with 10 year follow up. Arch Dis Child. 1985, 60 (6): 512-516. 10.1136/adc.60.6.512.
Suchy FJ: Neonatal cholestasis. Pediatr Rev. 2004, 25 (11): 388-396.
Del Arco A, Agudo M, Satrustegui J: Characterization of a second member of the subfamily of calcium-binding mitochondrial carriers expressed in human non-excitable tissues. Biochem J. 2000, 345 (Pt 3): 725-732.
Kobayashi K, Sinasac DS, Iijima M, Boright AP, Begum L, Lee JR, Yasuda T, Ikeda S, Hirano R, Terazono H, Crackower MA, Kondo I, Tsui LC, Scherer SW, Saheki T: The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein. Nat Genet. 1999, 22 (2): 159-163. 10.1038/9667.
Saheki T, Kobayashi K: Mitochondrial aspartate glutamate carrier (citrin) deficiency as the cause of adult-onset type II citrullinemia (CTLN2) and idiopathic neonatal hepatitis (NICCD). J Hum Genet. 2002, 47 (7): 333-341. 10.1007/s100380200046.
Imamura Y, Kobayashi K, Shibatou T, Aburada S, Tahara K, Kubozono O, Saheki T: Effectiveness of carbohydrate-restricted diet and arginine granules therapy for adult-onset type II citrullinemia: a case report of siblings showing homozygous SLC25A13 mutation with and without the disease. Hepatol Res. 2003, 26 (1): 68-72. 10.1016/S1386-6346(02)00331-5.
Kobayashi K, Saheki T: Citrin deficiency. in GeneReviews at GeneTests: Medical Genetics Information Resource [database online]. 1997, Copyright, University of Washington, Seattle
Ohura T, Kobayashi K, Tazawa Y, Abukawa D, Sakamoto O, Tsuchiya S, Saheki T: Clinical pictures of 75 patients with neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD). J Inherit Metab Dis. 2007, 30 (2): 139-144. 10.1007/s10545-007-0506-1.
Kimura A, Kage M, Nagata I, Mushiake S, Ohura T, Tazawa Y, Maisawa S, Tomomasa T, Abukawa D, Okano Y, Sumazaki R, Takayanagi M, Tamamori A, Yorifuji T, Yamato Y, Maeda K, Matsushita M, Matsuishi T, Tanikawa K, Kobayashi K, Saheki T: Histological findings in the livers of patients with neonatal intrahepatic cholestasis caused by citrin deficiency. Hepatol Res. 2010, 40 (3): 295-303. 10.1111/j.1872-034X.2009.00594.x.
Tazawa Y, Kobayashi K, Abukawa D, Nagata I, Maisawa S, Sumazaki R, Iizuka T, Hosoda Y, Okamoto M, Murakami J, Kaji S, Tabata A, Lu YB, Sakamoto O, Matsui A, Kanzaki S, Takada G, Saheki T, Iinuma K, Ohura T: Clinical heterogeneity of neonatal intrahepatic cholestasis caused by citrin deficiency: case reports from 16 patients. Mol Genet Metab. 2004, 83 (3): 213-219. 10.1016/j.ymgme.2004.06.018.
Song YZ, Li BX, Chen FP, Liu SR, Sheng JS, Ushikai M, Zhang CH, Zhang T, Wang ZN, Kobayashi K, Saheki T, Zheng XY: Neonatal intrahepatic cholestasis caused by citrin deficiency: clinical and laboratory investigation of 13 subjects in mainland of China. Dig Liver Dis. 2009, 41 (9): 683-689. 10.1016/j.dld.2008.11.014.
Ohura T, Kobayashi K, Tazawa Y, Nishi I, Abukawa D, Sakamoto O, Iinuma K, Saheki T: Neonatal presentation of adult-onset type II citrullinemia. Hum Genet. 2001, 108 (2): 87-90. 10.1007/s004390000448.
Yeh JN, Jeng YM, Chen HL, Ni YH, Hwu WL, Chang MH: Hepatic steatosis and neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) in Taiwanese infants. J Pediatr. 2006, 148 (5): 642-646. 10.1016/j.jpeds.2005.12.020.
Lu YB, Kobayashi K, Ushikai M, Tabata A, Iijima M, Li MX, Lei L, Kawabe K, Taura S, Yang Y, Liu TT, Chiang SH, Hsiao KJ, Lau YL, Tsui LC, Lee DH, Saheki T: Frequency and distribution in East Asia of 12 mutations identified in the SLC25A13 gene of Japanese patients with citrin deficiency. J Hum Genet. 2005, 50 (7): 338-346. 10.1007/s10038-005-0262-8.
Dimmock D, Kobayashi K, Iijima M, Tabata A, Wong LJ, Saheki T, Lee B, Scaglia F: Citrin deficiency: a novel cause of failure to thrive that responds to a high-protein, low-carbohydrate diet. Pediatrics. 2007, 119 (3): e773-e777. 10.1542/peds.2006-1950.
Song YZ, Guo L, Yang YL, Han LS, Kobayashi K, Saheki T: [Failure to thrive and dyslipidemia caused by citrin deficiency: a novel clinical phenotype]. Zhongguo Dang Dai Er Ke Za Zhi. 2009, 11 (5): 328-332.
Ben-Shalom E, Kobayashi K, Shaag A, Yasuda T, Gao HZ, Saheki T, Bachmann C, Elpeleg O: Infantile citrullinemia caused by citrin deficiency with increased dibasic amino acids. Mol Genet Metab. 2002, 77 (3): 202-208. 10.1016/S1096-7192(02)00167-1.
Dimmock D, Maranda B, Dionisi-Vici C, Wang J, Kleppe S, Fiermonte G, Bai R, Hainline B, Hamosh A, O'Brien WE, Scaglia F, Wong LJ: Citrin deficiency, a perplexing global disorder. Mol Genet Metab. 2009, 96 (1): 44-49. 10.1016/j.ymgme.2008.10.007.
Tabata A, Sheng JS, Ushikai M, Song YZ, Gao HZ, Lu YB, Okumura F, Iijima M, Mutoh K, Kishida S, Saheki T, Kobayashi K: Identification of 13 novel mutations including a retrotransposal insertion in SLC25A13 gene and frequency of 30 mutations found in patients with citrin deficiency. J Hum Genet. 2008, 53 (6): 534-545. 10.1007/s10038-008-0282-2.
Tokuhara D, Iijima M, Tamamori A, Ohura T, Takaya J, Maisawa S, Kobayashi K, Saheki T, Yamano T, Okano Y: Novel diagnostic approach to citrin deficiency: analysis of citrin protein in lymphocytes. Mol Genet Metab. 2007, 90 (1): 30-36. 10.1016/j.ymgme.2006.09.009.
Fu HY, Zhang SR, Yu H, Wang XH, Zhu QR, Wang JS: Most common SLC25A13 mutation in 400 Chinese infants with intrahepatic cholestasis. World J Gastroenterol. 2010, 16 (18): 2278-2282. 10.3748/wjg.v16.i18.2278.
Ko JS, Song JH, Park SS, Seo JK: Neonatal intrahepatic cholestasis caused by citrin deficiency in Korean infants.J Korean Med Sci. 2007, 22 (6): 952-956. 10.3346/jkms.2007.22.6.952.
Aanpreung P, Laohapansang M, Ruangtrakool R, Kimhan J: Neonatal cholestasis in Thai infants. J Med Assoc Thai. 2005, 88 (Suppl 8): S9-S15.
Kobayashi K, Ushikai M, Song Y-Z, Gao H-Z, Sheng J-S, Tabata A, Okumura F, Ikeda S, Saheki T: Overview of citrin deficiency: SLC25A13 mutations and the frequency. J Applied Clin Pediatr. 2008, 23 (20): 1553-1557.
Shigeta T, Kasahara M, Kimura T, Fukuda A, Sasaki K, Arai K, Nakagawa A, Nakagawa S, Kobayashi K, Soneda S, Kitagawa H: Liver transplantation for an infant with neonatal intrahepatic cholestasis caused by citrin deficiency using heterozygote living donor.Pediatr Transplant. 2010, 14 (7): E86-E88. 10.1111/j.1399-3046.2009.01172.x.
Tamamori A, Okano Y, Ozaki H, Fujimoto A, Kajiwara M, Fukuda K, Kobayashi K, Saheki T, Tagami Y, Yamano T: Neonatal intrahepatic cholestasis caused by citrin deficiency: severe hepatic dysfunction in an infant requiring liver transplantation. Eur J Pediatr. 2002, 161 (11): 609-613. 10.1007/s00431-002-1045-2.
Xing Y, Qiu W, Ye J, Han L, Xu S, Zhang H, Gao X, Wang Y, Gu X: Studies on the clinical manifestation and SLC25A13 gene mutation of Chinese patients with neonatal intrahepatic cholestasis caused by citrin deficiency. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2010, 27 (2): 180--185.
Maquat LE: Nonsense-mediated mRNA decay in mammals. J Cell Sci. 2005, 118 (Pt 9): 1773-1776.
Tazawa Y, Abukawa D, Sakamoto O, Nagata I, Murakami J, Iizuka T, Okamoto M, Kimura A, Kurosawa T, Iinuma K, Kobayashi K, Saheki T, Ohura T: A possible mechanism of neonatal intrahepatic cholestasis caused by citrin deficiency. Hepatol Res. 2005, 31 (3): 168-171. 10.1016/j.hepres.2005.01.001.
Takaya J, Kobayashi K, Ohashi A, Ushikai M, Tabata A, Fujimoto S, Yamato F, Saheki T, Kobayashi Y: Variant clinical courses of 2 patients with neonatal intrahepatic cholestasis who have a novel mutation of SLC25A13. Metabolism. 2005, 54 (12): 1615-1619. 10.1016/j.metabol.2005.06.009.
Tomomasa T, Kobayashi K, Kaneko H, Shimura H, Fukusato T, Tabata M, Inoue Y, Ohwada S, Kasahara M, Morishita Y, Kimura M, Saheki T, Morikawa A: Possible clinical and histologic manifestations of adult-onset type II citrullinemia in early infancy. J Pediatr. 2001, 138 (5): 741-743. 10.1067/mpd.2001.113361.
Palmieri L, Pardo B, Lasorsa FM, del Arco A, Kobayashi K, Iijima M, Runswick MJ, Walker JE, Saheki T, Satrustegui J, Palmieri F: Citrin and aralar1 are Ca(2+)-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 2001, 20 (18): 5060-5069. 10.1093/emboj/20.18.5060.
Chongsrisawat V, Jantaradsamee P, Vivatvakin B, Pongpaew P, Poovorawan Y: Alpha 1-antitrypsin phenotype of children with liver diseases in Thailand. Asian Pac J Allergy Immunol. 1998, 16 (1): 27-30.
den Dunnen JT, Antonarakis SE: Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000, 15 (1): 7-12. 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-230X/12/141/prepub
Acknowledgement
The study was supported by grant from Mahidol University directed to ST and DW, and Grant-in-Aid for Asia-Africa Science Platform Program from the Japan Society for the Promotion of Science directed to KK. DW is a recipient of Research Career Development Awards from the Faculty of Medicine Ramathibodi Hospital, Mahidol University as was PS. We thank Professors Prapon Wilairat and Amnuay Thithapandha for critical advice and revising the manuscript. Prof. Kobayashi passed away in December, 2011.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interest.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Treepongkaruna, S., Jitraruch, S., Kodcharin, P. et al. Neonatal intrahepatic cholestasis caused by citrin deficiency: prevalence and SLC25A13mutations among thai infants. BMC Gastroenterol 12, 141 (2012). https://doi.org/10.1186/1471-230X-12-141
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-230X-12-141