
Abstract
Background
Incarvillea sinensis is widely distributed from Southwest China to Northeast China and in the Russian Far East. The distribution of this species was thought to be influenced by the uplift of the Qinghai-Tibet Plateau and Quaternary glaciation. To reveal the imprints of geological events on the spatial genetic structure of Incarvillea sinensis, we examined two cpDNA segments ( trnH- psbA and trnS- trnfM) in 705 individuals from 47 localities.
Results
A total of 16 haplotypes was identified, and significant genetic differentiation was revealed (GST =0.843, NST = 0.975, P < 0.05). The survey detected two highly divergent cpDNA lineages connected by a deep gap with allopatric distributions: the southern lineage with higher genetic diversity and differentiation in the eastern Qinghai-Tibet Plateau, and the northern lineage in the region outside the Qinghai-Tibet Plateau. The divergence between these two lineages was estimated at 4.4 MYA. A correlation between the genetic and the geographic distances indicates that genetic drift was more influential than gene flow in the northern clade with lower diversity and divergence. However, a scenario of regional equilibrium between gene flow and drift was shown for the southern clade. The feature of spatial distribution of the genetic diversity of the southern lineage possibly indicated that allopatric fragmentation was dominant in the collections from the eastern Qinghai-Tibet Plateau.
Conclusions
The results revealed that the uplift of the Qinghai-Tibet Plateau likely resulted in the significant divergence between the lineage in the eastern Qinghai-Tibet Plateau and the other one outside this area. The diverse niches in the eastern Qinghai-Tibet Plateau created a wide spectrum of habitats to accumulate and accommodate new mutations. The features of genetic diversity of populations outside the eastern Qinghai-Tibet Plateau seemed to reveal the imprints of extinction during the Glacial and the interglacial and postglacial recolonization. Our study is a typical case of the significance of the uplift of the Qinghai-Tibet Plateau and the Quaternary Glacial in spatial genetic structure of eastern Asian plants, and sheds new light on the evolution of biodiversity in the Qinghai-Tibet Plateau at the intraspecies level.
Similar content being viewed by others

Background
Patterns of genetic and geographical structure in natural populations have been strongly influenced not only by intrinsic factors, such as life histories and ecological traits, but also by extrinsic factors including habitats and historical events [1–4]. Population geneticists have been interested in illustrating the factors determining genetic structure over the long term [5, 6]. During the past two decades, the phylogeographic histories of a broad spectrum of animal and plant species have been reconstructed, and phylogeographical analysis has proven to be a powerful method in searching for congruent geographical patterns of genetic variation and determining the roles that historical events have played in shaping the present spatial genetic structure of a species [3, 7–20]. The elevation of the Qinghai-Tibetan Plateau and Quaternary glaciation were key geological events that significantly affected Asian topography, atmospheric circulation, and even global climate. These events influenced floristic distributions, and therefore it is important for biologists to seek out responses of organisms to the uplift of the Qinghai-Tibetan Plateau and the climatic fluctuations during glacial periods to better understand current species distributions [3, 19, 21–25]. Until very recently, numerous phylogeographical studies have outlined Quaternary evolutionary histories of European and Northern American floras (e.g., [3, 16, 26–29]), but relatively few investigations have focused on population divergence and phylogeography of plants in the Qinghai-Tibet Plateau [22–25, 30–33]. In this study, we concentrated on elucidating the spatial genetic structure of an attractive herb, Incarvillea sinensis Lam., which is widely distributed from the southwest China through to the northeast China and the Russian Far East (Figure 1), whose biogeographic history was proposed to be closely related to the uplift of the Qinghai-Tibet Plateau and to Quaternary climate oscillations [34].

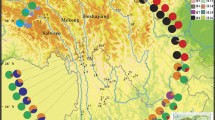
The geographical distribution of Incarvillea sinensis and 16 haplotypes (H1-H16) found among 705 individuals. QTP, NEQTP and SEQTP are the abbreviations of the Qinghai-Tibet Plateau, the northeastern Qinghai-Tibet Plateau and the southeastern Qinghai-Tibet Plateau respectively, as described in the materials and methods.
Incarvillea sinensis is the type species of Incarvillea Juss. (Bignoniaceae) including 16 species, and it also is the type of the subgenus Incarvillea including two species. Compared with other members of the genus centered in the Himalaya-Hengduan Mountains, this species has a broad range and altitudinal distribution (Figure 1). It is very rich in morphological variation, such as flower color, habit, and leaf characteristics, and its broad spectrum of morphological variants bewildered taxonomists in their attempts to propose intraspecific subdivisions; therefore, two classifications were established due to the discretionary priorities of morphological characteristics [35, 36]. Based on habit (annual or perennial), Grierson [35] subdivided I. sinsensis into two subspecies: I. sinensis subsp. sinensis and subsp. variabilis. The former is an annual herb with simple roots, while the latter is a perennial herb with woody, branched roots and often bears the remains of the previous year’s stems. In another system described in Flora Reipublicae Popularis Sinicae [36], two variants were recognized, I. sinensis var. sinensis and var. przewalskii. The former is characterized by red flowers and the latter by yellow flowers. Both intraspecific classifications seemed reasonable given that straightforward characters (flower color or habit) were used to distinguish intraspecific taxa, but the annual populations with red flowers were confusing owing to the disjunct distribution between the northern region and the southernmost Lancangjiang Valley (Figure 1). The distribution was split due to the occurrence of perennial groups with yellow and red flowers in the middle of the entire species distribution.
After investigating all of the specimens of Incarvillea sinensis in CDBI, E, K, KUN, PE, and SZ, and exploring its distribution in China, we found that its variation in morphology was regional and that all populations can be classified into three types (Figure 1, Table 1). Type 1 is an annual with red flowers, distributed from the eastern Gansu Province to the Northeast China and the Russian Far East, with one population in the Lancangjiang Valley. Type 2 is a perennial with yellow flowers and is found in the western Gansu Province and the eastern Qinghai Province. Type 3 is a perennial with red flowers found in the southeastern Qinghai-Tibet Plateau, mainly in Sichuan Province. The morphological variations within I. sinensis can be correlated with their geographic distribution, while the eastern Qinghai-Tibet Plateau (QTP) contain all three morphological types, and appears to be the center of diversity for this species. We suspected that the spatial pattern of morphological characteristics likely dropped a hint to the potential divergence in genetics.
A study on the biogeography of Incarvillea suggests that its evolution may be related to the uplift of the Qinghai-Tibet Plateau. In addition, the continuous distribution of the subgenus Incarvillea, from the eastern Qinghai-Tibet Plateau to the Russian Far East, could partially be the result of the Quaternary expansion since part of its range was glaciated [34]. Incarvillea sinensis occupies almost the full range of the subgenus except in Mongolia. Considering the morphological variations correlative to geography, we proposed that the uplift of the Qinghai-Tibetan Plateau potentially resulted in the genetic variations between populations within and outside of the Qinghai-Tibetan Plateau. Furthermore, the Pleistocene glaciation possibly induced the extinction of some northern populations and the modern distribution likely resulted from the postglacial and interglacial expansion. In order to test the hypothesis, we examined the genetic diversity of populations within and outside the Qinghai-Tibet Plateau and identified potential genetic lineages within this species.
Since the colonization of new habitats occurs through seeds, and chloroplast DNA is generally maternally inherited in angiosperms (meaning that the plastid genome is moved only by seeds) [13], chloroplast DNA (cpDNA) markers provide information on past changes in species ranges that are unaffected by subsequent pollen movements [37]. We investigated patterns of cpDNA diversity in Incarvillea sinensis using phylogeographical methods that can indicate how historical events such as range fragmentation, range expansion, and long distance dispersal, as well as current levels of gene flow, have influenced present-day distributions. The goals of this study are to provide new insights into the population genetic structure and responses of I. sinensis to geological events and reveal the imprints of geological events on the spatial pattern of the genetic diversity.
Results
Sequence variation and haplotype distribution
A matrix of 1,396 characters was obtained from concatenated alignments of trnH- psbA (377 sites) and trnS- trnfM (1,019 sites) sequences from 705 individuals representing 47 collections, in which 38 polymorphic sites were confirmed by sequencing in both directions. Sequences with polymorphisms between each other were deposited in GenBank (Accession No. JQ858492-JQ858511). Of these polymorphisms, five were indels (insertions and deletions). Two indels were single nucleotides and the other three were six bp, five bp and seven bp long respectively. In subsequent analysis, indels with length more than one base were considered as a single mutation. All five indels were coded as binary characters, 1 s and 0 s, so that the matrix was shortened to 1381 characters in length: 372 sites from trnH- psbA and 1,009 sites from trnS- trnfM. The adjusted matrix included 23 variable sites, five of which were due to the presence of indels, and all of variable sites were informative and involved in all analysis. All of the sequences from the 47 localities contained 16 unique haplotypes when indels were included (Table 2). Haplotypes and their frequency from each locality are shown in Table 1 and their spatial pattern is portrayed in Figure 1.
Genealogical relationships among haplotypes of Incarvillea sinensis
In our phylogenetic analysis, a data set including haplotype and outgroup sequences was used to reconstruct the relationships among the haplotypes. This matrix was 1,447 sites in length, and contains 38 informative sites. The consensus tree resulting from Bayesian analysis is presented in Figure 2. The ingroup sequences clustered into two major clades, the southern and northern clades, concordant with geography (Figures 1, 2). The northern clade accommodated six haplotypes (H1-H6) from the northern distribution, and 10 haplotypes (H7-H16) from the southern distribution were nested in the southern clade. Moreover, a subclade was formed within the southern clade to accommodate six haplotypes (H11-H16) from the southeastern Qinghai-Tibet Plateau, situated in the southernmost range of I. sinensis.

Phylogram among cpDNA ( trn S- trnf M and trn H- psb A regions) haplotypes of Incarvillea sinenesis resulting from Bayesian analysis. Numbers above the branches are posterior probabilities of nodes. Posterior probabilities of Bayesian run without assuming the molecular clock was enforced. RA: red flowers, annual; RP: red flowers, perennial; YP: yellow flowers, perennial; SEQTP: the southeastern Qinghai-Tibet Plateau; NEQTP: the northeastern Qinghai-Tibet Plateau.
Compared with the phylogenetic tree, the haplotype network with 95% connection constructed using the TCS program was better resolved in genealogical relationships among the haplotypes, but the network showed a loop indication ambiguous connections involving haplotypes H1, H5, H7, and H11. Following the rules of Crandall & Templeton [38] in combination with the genetic distances between haplotypes and the phylogenetic tree, the loop was resolved and the network was shown in Figure 3. Two lineages from the southern and northern regions were connected by 11 mutations. The northern lineage contained six haplotypes (H1-H6) from the northern region (that is, the northern clade in the phylogenetic tree), and the southern lineage contained 10 haplotypes (H7-H16) from the eastern Qinghai-Tibet Plateau (that is, the southern clade in the phylogenetic tree).

The parsimonious network of cpDNA Haplotypes of Incarvillea sinensis inferred using the TCS program. Every haplotype is identified by a number and each branch represents a mutational step with intermediate missing haplotypes represented by small and empty dots. The numbers in the parentheses are the frequencies of the haplotypes.
The net averaged population genetic distance per site between two regional lineages calculated using Arlequin was 0.00879, and the divergence of these two major clades within this species was dated at 4.396 MYA.
Population genetic parameters and phylogeographical structure
The total gene diversity hT was 0.677 and the average within-population gene diversity hS was 0.106 at the species level. The indices of population structure GST and NST were 0.843 and 0.975, respectively. The permutation test showed that NST was significantly larger than GST (P < 0.05). The GST and NST at the regional level implicated that significant structures existed in the SEQPT, NEQTP and their combined regions, but not in the northern region. Genetic parameters at different levels are listed in Table 3.
The analysis of molecular variance (AMOVA) of the data from the two cpDNA fragments indicates the presence of a strong differentiation in genetics among the lineages and regions (Table 4). When populations were grouped by three geographical regions, a statistically significant structure was detected by AMOVA. The comparison among the three regions shows that 93.94% (P < 0.001) of the genetic variations occurred between these regions. Only 4.7% (P < 0.001) of the variance was distributed within each of these regions, and less variance (1.36%, P < 0.001) can be attributed to the variations within the populations. When populations were grouped by two lineages, once again, the results show a similar pattern. Once the populations were grouped by subspecies and variants on taxonomy, the genetic variations between the groups decreased, and the remaining variations from the other two sources increased accordingly. The least amount of variance was contributed by the variations within populations, and this value is less than 1.5%.
Correlation between genetic and geographic distances
The Mantel test quantified the correlations between the two distance matrices, allowing for the determination of the relationships between the genetic and the geographical distances. The results of this test did not show a significant effect of isolation by distance at the species level (r = 0.001, P = 0.48, Figure 4A). The distributions of haplotype showed that only six haplotypes were detected in 31 populations from the northern region, and 20 of them possessed the same haplotype (H1), so we performed this test once again after replacing the Fsts with pairwise Nei’s genetic distances. The positive correlation between genetic difference and geographic distance (isolation by distance) was encountered in the species (r = 0.083, P = 0.034, Figure 4B). Furthermore we found significantly positive and monotonic relationships across all of the pairwise FST measures and spatial distances separating the populations of regions in SEQPT, NEQTP, and their combination (Figure 4C, D, and E), whose pattern of scatterplots were consistent with Case I proposed by Hutchison & Templeton [39]. This pattern describes the scenario of regional equilibrium between gene flow and drift (i.e., isolation by distance). In contrast, the null hypothesis of the regional equilibrium was rejected in populations from the northern region, and the scatterplot approximated case Ш of Hutchison & Templeton [39], indicating that drift is more influential than gene flow(r = 0.06, P = 0.249, Figure 4E).

Scatterplots representing relationships between genetic distance ( F ST ) and geographic distance (Km) at species and regional levels. A: species level, r = 0.001, P = 0.480; B: species level, r = 0.083, p = 0.034; C: the southeastern Qinghai-Tibet Plateau, r = 0.512, P = 0.006; D: the northeastern Qinghai-Tibet Plateau, r = 0.486, P = 0.025; E: the Qinghai-Tibet Plateau, r = 0.473, P < 0.001; F: the northern region, r = 0.06, P = 0.249; F: N region except the recently isolated and small populations, r = 0.29, P = 0.011.
Discussion
Conflict between the phylogeny and intraspecific classifications previously proposed
We found neither of the intraspecific subdivisions of the two subspecies and the two variants was supported by phylogenetic analysis, although the species was proven to be monophyletic by the cpDNA trnH- psbA sequence (not shown). Because the haplotype H15 was detected in the DQ population, the annual DQ population with red flowers phylogenetically clustered together with all of the other perennial populations with red flowers from the southeastern Qinghai-Tibet Plateau, not together with the annual populations with red flowers from the northern region (Table 1, Figure 1 & 2). According to the system described by Wang et al. [36], the DQ population should be classified into I. sinensis var. sinensis, while the other populations from the southeastern Qinghai-Tibet Plateau should be classified into I. sinensis var. przewalskii. Likewise, the perennial GW population (haplotype H9) with red flowers was most closely related to the perennial populations with yellow flowers from the northeastern Qinghai-Tibet Plateau (Table 1, Figure 1 & 2). Based on the Grierson’s system [35], the former should be classified into I. sinensis subsp. sinensis, while the latter should be I. sinensis subsp. variabilis.
Our study provides evidence in support of parallel evolution of morphological characteristics in allopatric populations (Figure 2). Character convergence within this species apparently resulted from adaptation to similar niches. From the genetic data, we found no evidence warranting the suggestion of monophyly for any of the recognized taxa. We therefore propose to view this species as a species complex that should not be subdivided into intraspecific units in taxonomy.
Genetic diversity and structure at species and regional levels
The mean within-population genetic diversity is low compared with the total diversity at both species and regional levels. The AMOVA results also showed that most of the genetic diversity was attributed to the among-population effects. The global indices of population differentiation were very high, up to 0.843 and 0.975 for GST and NST, respectively. When NST is significantly higher than GST, this usually indicates the occurrence of a phylogeographical structure [40]. Through random permutations, a significant difference between these two measures was detected within this species. On the other hand, 16 haplotypes were clustered into two lineages or three groups harmonious with geography (Figure 2 & 3), and none of them was shared among the regions (Figure 1). These results reveal that the cpDNA variation of this species is highly structured.
The southern clade consists of four haplotypes from the NEQTP and six haplotypes from the SEQTP. Two predominant haplotypes (H7 & H8) are widely distributed in NEQTP, except in Wenxian and Diebu in Gansu Province, and the remaining eight haplotypes are relatively rare and endemic to one valley or locality (Figure 1, Table 1). The indices of genetic differentiation show that geographic structure exists both in the lineage and in the subdivided regions (SEQTP and NEQTP regions, Table 3). The northern clade is distributed in the northern region where six haplotypes (H1-H6) are scattered erratically, and no geographic structure was detected (NST < GST). We note that both the differentiation index (GST) and the diversity index (hT) of the sorthern clade is higher than those of the southern clade. The degree of subdivision of the cpDNA diversity (GST) reflects the dispersal ability of the species and the effect of long-term range fragmentation, while the low GST value is indicative of high levels of gene flow through seeds [13]. Once capsules of Incarvillea sinensis split, the seeds are ejected and the seed wings contribute to seed dispersal by wind. In the southern region accommodating the southern clade, orogenesis resulted in range fragmentation, and populations of I. sinensis were distributed in valleys, except for those populations with haplotypes H7 and H8 (Figure 1). The connections between these populations were severed by towering ridges and peaks. As a result, efficient seed dispersal was reduced and the gene flow through seed dispersal was confined within the valleys, or between the neighboring populations.
In contrast, the analogous limiting factors of seed movement did not exist in the northern region, and we could not identify the existence of a significant geographic structure in the northern clade (GST > NST). Distantly related haplotypes were more often found in the same populations, as case 3 proposed by Pons & Petit [40], which indicates that the relative geographic distribution of the haplotypes may have nothing to do with their genetic distances, in this case old lineages have had ample time to become geographically redistributed since they first appeared as products of mutation. Based on the comparison of the indices of genetic diversity and differentiation between two lineages, we consider the topographical barriers in gene flow through seed dispersal to be the most likely factors leading to the higher divergence in the southern clade. The diverse niches in the eastern Qinghai-Tibet Plateau created a wide spectrum of habitats to accumulate and accommodate new mutations.
Distinctive evolutionary histories between lineages
The two major cpDNA lineages were revealed by means of reconstructing the phylogenetic tree and the haplotype network, which can be recognized by a connection of 11 mutations. The deep gap between these two clades indicates the early origin of the corresponding lineages in the evolution of the species, and the long-standing separation thereafter. We dated the divergence between the two lineages at approximately 4.4 MYA, which corresponds to the early Pliocene. The uplift of the Himalaya-Hengduan massifs was a complicated geological event, and the chronology of this process is still under debate [41–45]. It is believed that the plateau underwent its most recent dramatic uplift after the early Pliocene [43, 44]. The two lineages of Incarvillea sinensis most likely originated because of decreased effective gene flow due to topographic isolation after orogenesis and long term adaptation to the distinct habitats (if their non-overlapping distributions and unshared haplotypes are taken into consideration). The long evolutionary history allowed this species to accumulate not only mutations relative to diversity, but also high differentiation between regions.
For the northern lineage, the long history should be enough for localized gene flow to interact with drift to produce a pattern of isolation by distance across the region (i.e., regional equilibrium), but the correlation estimation of pairwise genetic and geographical distances show that drift was much more influential than gene flow on the distribution of genetic variability (Figure 4E). Populations were not at equilibrium, either because conditions required or the populations themselves have not existed long enough for regional pattern of isolation-by-distance to have been achieved [39, 46]. The northern lineage was composed of six haplotypes detected in 31 collections from the northern region. The basal and predominant haplotypes (H1) was distributed randomly and the other five haplotypes were concentrated on the haplotype H1 in the network (Figure 3). The starlike shape of the network of the six haplotypes reflected the extremely low level of the sequence divergence and the low frequency of rare haplotypes. The lack of regional equilibrium, the lower diversity and starlike shape network of the northern lineage likely indicated that possible extinction during the glacial and the interglacial and postglacial recolonization.
On the contrary, within the southern clade, including all haplotypes from the eastern Qinghai-Tibet Plateau, a significant association between the genetic variability and the geographical distribution was revealed by the Mantel test. The Mantel tests also showed that there was a significant effect of isolation by distance, which means that a regional equilibrium was reached and that more closely situated populations tended to communicate and be more genetically similar to one another [47]. Obviously, gene flow was more easily accomplished on the alpine platform than in fragmented landscapes, such as the southeastern Qinghai-Tibet Plateau. We conclude that the populations from the southeastern Qinghai-Tibet Plateau suffered severe isolation owing to orogenesis, which significantly reduced seed dispersal, so that gene flow through the seeds was confined between neighboring populations within areas without strict topographical barriers. Allopatric fragmentation became a theme throughout the evolutionary history of the southern lineage. Significant ridges and peaks of the local landscape formed permanent spatial barrier that intensified and fixed genetic isolations.
Conclusions
We detected two lineages connected by a deep gap within the species, and significant difference in genetic diversity between them. The significant genetic divergence concordant with geography and the features of genetic diversity seemed to reveal the imprints of geological events on the plant evolution. We preferred the significance of the uplift of the Qinghai-Tibet Plateau and the Quaternary Glacial in the distribution of the genetic diversity and differentiation of Incarvillea sinensis, as mentioned by Chen et al. [34].
The Qinghai-Tibet Plateau and its adjacent areas have been listed as one of the world’s biodiversity hotspots [48, 49]. Two processes may contribute to the formation of biodiversity centers: the elevation of local speciation rates (the center of origin hypothesis) and a greater accumulation of species formed elsewhere (the center of accumulation hypothesis). Available studies in this area have focused mainly on traditional taxonomy, botanical inventory, and phylogeny-based species radiation [50–53], and as evidence mounts, it becomes increasingly clear that both the origin and the accumulation hypotheses have acted in concert to form this biodiversity hotspot [51, 54–59]. Only a few plant species in this region, however, have been phylogeographically studied (e.g. [22–25, 30, 32, 33]). In the absence of phylogeographical investigations on a multitude of plants, it is very difficult to uncover the common microevolutionary patterns of the extant organisms within this area. The pattern of genetic variation within the widely distributed Incarvillea sinensis populations, however, offers clues to the mechanisms of the evolutionary of the diversity of plants in the Qinghai-Tibet Plateau. Our present study not only shows that the spatial genetic structure within this species likely is the result of tectonic events and long term adaptation to distinct habits, but also supports an evidence on the higher diversity in the Qinghai-Tibet Plateau influenced by origins.
Methods
Sample collections
We conducted extensive fieldwork following herbarium specimen records of Incarvillea sinensis from CDBI, E, K, KUN, PE, and SZ. A number of populations, in some locations, became extinct due to habitat destruction. Numerous specimens were collected in Henan, Shandong, Inner Mongolia, Jilin, Liaoning, and Heilongjiang provinces during the 1920s–1950s. Subsequently, fewer collections have been recorded; and no new specimens were recorded since 1985. We were able to obtain only eight samples in these areas, seven of which (i.e., CH, MD, QQ, SZ, XT, XY, and YH, see Table 1) were isolated and small in size. In this study, we sampled 705 individuals from 47 populations distributed in China (Figure 1, Table 1) covering the entire range of the species, except for the Russian Far East, where only a few collections were recorded. Leaves were dried using silica-gel. A modified CTAB extraction protocol was used to extract DNA from the dried leaf tissue [60].
Chloroplast DNA amplification and sequencing
Two regions of cpDNA, trnH- psbA and trnS- trnfM were amplified and sequenced. The mixture for polymerase chain reactions consisted of a total volume of 25 μL containing 2.5 μL of MgCl2-free buffer, 1.5 μL of MgCl2 solution (25 mM), 0.5 μL of dNTPs (2.5 mM each), 1.25 μL of each primer (10 μM), 1 U Taq polymerase, 1 μL of template ( c. 50–100 ng double-stranded DNA), and 17.25 μL of sterile water.
The trnH- psbA segment was amplified using the primers trnH [61] and psbA [62], and trnH was used for sequencing. The PCRs consisted of an initial denaturation at 94°C for 3 min, followed by 30 cycles of 30 s at 94°C (denaturation), 30 s at 52°C (annealing), and 30 s at 72°C (extension), and concluding with a final extension period of 7 min at 72°C.
The trnS- trnfM fragment was amplified using the trnS and trnfM primers [63] and both primers were used in sequencing. The amplification parameters for this segment were 94°C, 3 min; 30 cycles (94°C, 30 s; 52°C, 30 s; 72°C, 1 min); 72°C, 7 min.
The PCR products were purified with DNA-purification kits (Shenggong, Shanghai). The trnH- psbA and trnS- trnfM PCR products were sequenced in both directions using the ABI BigDye version 3.1 terminator cycle sequencing chemistry, with sequences read by an ABI 3730 Genetic Analyser (Applied Biosystems).
Phylogenetic analysis
Sequences were aligned using ClustalX (version 1.81, [64]), and indels were coded as substitutions following Caicedo & Schaal [65] using a '1' for present and '0' for missing. Haplotypes were identified using DnaSP (version 3.0, [66]). We performed phylogenetic analysis on haplotypes of Incarvillea sinensis using MrBayes V3.1.2 [67]. Based on the phylogenetic relationships among species of the genus, the outgroup Incarvillea arguta was used to root the phylogenetic trees, which is similar to I. sinensis on morphology such as the flower, fruit and habit. We ran 500,000 Markov chain Monte Carlo (MCMC) generations for the Bayesian analysis, with a sampling frequency of 1 in 100 generations. The consensus tree was constructed from trees saved after reaching the stationarity (the density plot took a normal shape after ca. 6000 generations). Tree-building methods might not always be the most appropriate way to represent genealogical relationships among haplotypes within a species, owing to the possibly shallow genetic divergence [68]; thus, a haplotype network was built using the TCS program [69] with the statistical parsimony method described by Templeton et al. [70] and Crandall et al. [71]. Ambiguities in the haplotype network were resolved following the recommendations of Crandall & Templeton [38].
Genetic diversity and divergence
The genetic structure was evaluated by the analysis of molecular variance [72] using ARLEQUIN 3.11 version [73], partitioning the genetic diversity within populations, among populations within groups, and among groups. Significance was assessed after 1,023 permutations. Populations were grouped by clades in phylogenetic trees, intraspecific taxonomic categories (subspecies and variants based on two taxonomic systems), and spatial regions. The geographic regions used for the AMOVA were: (1) the southeastern Qinghai-Tibet Plateau (SEQTP), including the southeast Tibet and the Sichuan Province; (2) the northeastern Qinghai-Tibet Plateau (NEQTP), including the Qinghai Province and the western Gansu Province; and (3) the northern area, including all of the remaining distributions outside the Qinghai-Tibet Plateau (Figure 1). The SEQTP and NEQTP regions composed the southern region.
We calculated within-population diversity (hS), total diversity (hT), and the level of population differentiation (GST and NST) at the species and regional levels. The occurrence of a significant phylogeographical structure was inferred by testing whether GST (the index that only considers haplotype identities) and NST (the index that takes into account a measure of haplotype divergence) were significantly different using 1000 permutations in PERMUT [40, 74].
Traditional FST and GST estimates can not distinguish different genetic structures with similar FST values because the FST is a compound product of gene flow and drift. In response, Hutchison & Templeton [39] proposed a method to evaluate the relative historical influences of gene flow and drift on regional population structure by constructing regional scatterplots of FST on geographical distances and calculating the correlation coefficients describing the relationship between them. Following this method, the FST values between pairwise populations were calculated using ARLEQUIN. Scatterplots of the FST on geographical distances were constructed, and correlation coefficients were calculated along with the significance of correlation by performing GenAlEx6 [75], in which the genetic distance matrices (pairwise FST) were compared with the matrix of geographical distance by means of a simple Mantel test to detect isolation by distance and to evaluate the relative influences of gene flow and drift on the regional population structure. We used 999 random permutations to test for the Mantel statistic significance.
Dating the divergence between the major groups
The net averaged population genetic distance, DA = diXY - (diX + diY)/2 [76], accounts for the polymorphism within populations, and is thus proportional to the time since the divergence of two reciprocally monophyletic groups, assuming homogeneity of the mutation rates across the lineage [77]. To date approximately the divergence of high-level clusters reconstructed using phylogenetic and statistic parsimony analysis, we estimated the net pairwise divergence per site (dA) using ARLEQUIN, and then calculated the divergence time following the formula T = dA/2 μ [78]. In seed plants, the approximate evolutionary rate of noncoding spacer or introns of cpDNA was estimated at 1.01 × 10-9 substitutions per site per year [79], which can be used to estimate divergence time for taxa without fossil records (e.g., [24, 80]; etc.). We also used this rate as the proximate evolutionary rate of the trnH- psbA and trns- trnfM fragments owing to the lack of any fossils for this group to use as calibration points.
References
Ge XJ, Chiang YC, Chou CH, Chiang TY: Nested clade analysis of Dunnia sinensis (Rubiaceae), a monotypic genus from China based on organelle DNA sequences. Conserv Genet. 2002, 3: 351-362. 10.1023/A:1020579617678.
Hamrick JL, Godt MJW, Sherman-Broyles SL: Factors influencing levels of genetic diversity in woody plant species. New Forest. 1992, 6: 95-124. 10.1007/BF00120641.
Hewitt GM: Genetic consequences of climatic oscillations in the Quaternary. Philos T Roy Soc B. 2004, 359: 183-195. 10.1098/rstb.2003.1388.
Newton AC, Allnutt AR, Gillies ACM, Lowe AJ, Ennos RA: Molecular phylogeography, intraspecific variation and the conservation of tree species. Trends Ecol Evol. 1999, 14: 140-145. 10.1016/S0169-5347(98)01555-9.
Avise JC: Phylogeography: The History and Formation of Species. Cambridge,Massachusetts: Harvard University Press; 2000.
Schaal BA, Hayworth DA, Olsen KM, Rauscher JT, Smith W: Phylogeography studies in plants: problems and prospects. Mol Ecol. 1998, 7: 465-474. 10.1046/j.1365-294x.1998.00318.x.
Aoki K, Suzuki T, Hsu T-W, Murakami N: Phylogeography of the component species of broad-leaved evergreen forests in Japan, based on chloroplast DNA. J Plant Res. 2004, 117: 77-94. 10.1007/s10265-003-0132-4.
Arbogast BS, Kenagy GJ: Comparative phylogeography as an integrative approach to historical biogeography. J Biogeogr. 2001, 28: 819-825.
Avise JC: Molecular population structure and the biogeographic history of a regional fauna: a case history with lessons for conservation biology. Oikos. 1992, 63: 62-76. 10.2307/3545516.
Brochmann C, Gabrielsen TM, Nordal I, Landvik JY, Elven R: Glacial survival or tabula rasa? The history of North Atlantic biota revisited. Taxon. 2003, 52: 417-450. 10.2307/3647444.
Fujii N, Ueda K, Watano Y, Shimizu T: Intraspecific sequence variation of chloroplast DNA in Pedicularis chamissonis Steven (Scrophulariaceae) and geographic structuring of the Japanese ‘alpine’ plants. J Plant Res. 1997, 110: 195-207. 10.1007/BF02509308.
Huang SF, Hwang SY, Wang JC, Lin TP: Phylogeography of Trochodendron aralioides (Trochodendraceae) in Taiwan and its adjacent areas. J Biogeog. 2004, 31: 1251-1259. 10.1111/j.1365-2699.2004.01082.x.
Petit RJ, Aguinagalde I, De Beaulieu J-L, Bittkau C, Brewer S, Cheddadi R, Ennos R, Fineschi S, Griver D, Lascoux M, Mohanty A, Müller-Starck G, Demesure-Musch B, Palmé A, Martin JP, Rendell S, Vendramin GG: Glacial refugia: hotspots but not melting pots of genetic diversity. Science. 2003, 300: 1563-1565. 10.1126/science.1083264.
Seo A, Watanabe M, Hotta M, Murakami N: Geographical patterns of allozyme variation in Angelica japonica (Umbelliferae) and Farfugium japonicum (Compositae) on the Ryukyu Islands, Japan. Acta Phytotax Geobot. 2004, 55: 29-44.
Soltis DE, Gitzendanner MA, Strenge DD, Soltis PS: Chloroplast DNA intraspecific phylogeography of plants from the Pacific Northwest of North America. Plant Syst Evol. 1997, 206: 353-373. 10.1007/BF00987957.
Soltis DE, Morris AB, McLachlan JS, Manos PS, Soltis PS: Comparative phylogeography of unglaciated eastern North America. Mol Ecol. 2006, 15: 4261-4293. 10.1111/j.1365-294X.2006.03061.x.
Stehlik I: Resistance or emigration? Response of alpine plants to the ice ages. Taxon. 2003, 52: 499-510. 10.2307/3647448.
Stewart JR, Lister AM: Cryptic northern refugia and the origins of the modern biota. Trends Ecol Evol. 2001, 16: 608-613. 10.1016/S0169-5347(01)02338-2.
Taberlet P, Fumagalli L, Wust-Saucy AG, Cosson JF: Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol. 1998, 7: 453-464. 10.1046/j.1365-294x.1998.00289.x.
Trewick SA, Wallis GP, Morgan-Richards M: Phylogeographical pattern correlates with Pliocene mountain building in the alpine scree weta (Orthoptera, Anostostomatidae). Mol Ecol. 2000, 9: 657-666. 10.1046/j.1365-294x.2000.00905.x.
Su B, Wang YX, Lan H, Wang W, Zhang Y: Phylogenetic study of complete cytochrome b genes in musk deer (genus Moschus) using museum samples. Mol Phylogenet Evol. 1999, 12: 241-249. 10.1006/mpev.1999.0616.
Wang F, Gong X, Hu C, Hao G: Phylogeography of an alpine species Primula secundiflora inferred from the chloroplast DNA sequence variation. J Syst Evol. 2008, 46: 13-22.
Yang FS, Li YF, Ding X, Wang XQ: Extensive population expansion of Pedicularis longiflora (Orobanchaceae) on the Qinghai-Tibetan Plateau and its correlation with the Quaternary climate change. Mol Ecol. 2008, 17: 5135-5145. 10.1111/j.1365-294X.2008.03976.x.
Yuan QJ, Zhang ZY, Peng H, Ge S: Chloroplast phylogeography of Dipentodon (Dipentodontaceae) in southwest China and northern Vietnam. Mol Ecol. 2008, 17: 1054-1065. 10.1111/j.1365-294X.2007.03628.x.
Zhang Q, Chiang TY, George M, Liu JQ, Abbott RJ: Phylogeography of the Qinghai-Tibetan Plateau endemic Juniperus przewalskii (Cupressaceae) inferred from chloroplast DNA sequence variation. Mol Ecol. 2005, 14: 3513-3524. 10.1111/j.1365-294X.2005.02677.x.
Abbott RJ, Brochmann C: History and evolution of the arctic flora: in the footsteps of Eric Hultén. Mol Ecol. 2003, 12: 299-313. 10.1046/j.1365-294X.2003.01731.x.
Abbott RJ, Smith LC, Milne RI, Crawford RMM, Wolff K, Balfour J: Molecular analysis of plant migration and refugia in the Arctic. Science. 2000, 289: 1343-1346. 10.1126/science.289.5483.1343.
Anderson LL, Hu FS, Nelson DM, Petit RJ, Paige KN: Ice-age endurance: DNA evidence of a white spruce refugium in Alaska. Proc Natl Acad Sci USA. 2006, 103: 12447-12450. 10.1073/pnas.0605310103.
Petit RJ, Hampe A, Cheddadi R: Climate changes and tree phylogeography in the Mediterranean. Taxon. 2005, 54: 877-885. 10.2307/25065474.
Gao LM, Möller M, Zhang XM, Hollingsworth ML, Liu J, Mill RR, Gibby M, Li DZ: High variation and strong phylogeographic pattern among cpDNA haplotypes in Taxus wallichiana (Taxaceae) in China and North Vietnam. Mol Ecol. 2007, 16: 4684-4698. 10.1111/j.1365-294X.2007.03537.x.
Ge XJ, Zhang LB, Yuan YM, Hao G, Chiang TY: Strong genetic differentiation of the East-Himalayan Megacodon stylophorus (Gentianaceae) detected by inter-simple sequence repeats (ISSR). Biodivers Conserv. 2005, 14: 849-861. 10.1007/s10531-004-0655-6.
Meng L, Yang R, Abbott RJ, Miehe G, Hu TH, Liu JQ: Mitochondrial and chloroplast phylogeography of Picea crassifolia Kom. (Pinaceae) in the Qinghai-Tibetan Plateau and adjacent highlands. Mol Ecol. 2007, 16: 4128-4137. 10.1111/j.1365-294X.2007.03459.x.
Song BH, Wang XQ, Wang XR, Ding KY, Hong DY: Cytoplasmic composition in Pinus densata and population establishment of the diploid hybrid pine. Mol Ecol. 2003, 12: 2995-3001. 10.1046/j.1365-294X.2003.01962.x.
Chen S, Guan K, Zhou Z, Olmstead R, Cronk Q: Molecular phylogeny of Incarvillea (Bignoniaceae) based on ITS and trnL-F sequences. Am J Bot. 2005, 92: 625-633. 10.3732/ajb.92.4.625.
Grierson AJC: A revision of the genus Incarvillea. Notes Roy Bot Gard Edinb. 1961, 23: 303-354.
Wang WT, Pan KY, Zhang ZY, Li ZY, Tao DD, Yin WQ: Incarvillea Juss. InFlora Reipublicae Popularis Sinicae, Volume 69. Edited by Wang WT. Beijing,China: Science Press; 1990:34–49.
Petit RJ, Kremer A, Wagner DB: Finite island model for organelle and nuclear genes in plants. Heredity. 1993, 71: 630-641. 10.1038/hdy.1993.188.
Crandall KA, Templeton AR: Empirical tests of some predictions from coalescent theory with applications to intraspecific phylogeny reconstruction. Genetics. 1993, 134: 959-969.
Hutchison DW, Templeton AR: Correlation of pairwise genetic and geographic distance measures: inferring the relative influences of gene flow and drift on the distribution of genetic variability. Evolution. 1999, 53: 1898-1914. 10.2307/2640449.
Pons O, Petit RJ: Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics. 1996, 144: 1237-1245.
Coleman M, Hodges K: Evidence for Tibetan plateau uplift before 14 Myr ago from a new minimum age for eat-west extension. Nature. 1995, 374: 49-52. 10.1038/374049a0.
Guo ZT, Ruddiman WF, Hao QZ, Wu HB, Qiao YS, Zhu RX, Peng SZ, Wei JJ, Yuan BY, Liu TS: Onset of Asian desertification by 22 Myr ago inferred from loess deposits in China. Nature. 2002, 416: 159-163. 10.1038/416159a.
Li JJ, Fang XM, Pan BT, Zhao ZJ, Song YG: Qinghai-Tibetan Plateau violent uplift and influence upon circumjacent environment in Late Cenozoic. Quaternary Sci (in Chinese). 2001, 21: 381-390.
Shi YF, Li JJ, Li BY, Yao TD, Wang SM, Li SJ, Cui ZJ, Wang FB, Pan BT, Fang XM, Zhang QS: Uplift of the Qinghai-Xizang (Tibetan) Plateau and East Asia environmental change during late Cenozoic. Acta Geograph Sin (in Chinese). 1999, 54: 10-21.
Spicer RA, Harris NBW, Widdowson M, Herman AB, Guo SX, Valdes PJ, Wolfe JA, Kelley SP: Constant elevation of southern Tibet over the past 15 million years. Nature. 2003, 421: 622-624. 10.1038/nature01356.
McCauley DE: Genetic consequences of extinction and recolonization in fragmented habitats. Biotic interactions and global change. Edited by: Kareiva PM, Kingsolver JG, Huey RB. 1993, Sinauer, Sunderland, MA, 217-233.
Wright S: Isolation by distance. Genetics. 1943, 28: 114-138.
Myers N, Mittermeier R, Mittermeier C, Fonseca G, Kent J: Biodiversity hotspots for conversation priorities. Nature. 2000, 403: 853-858. 10.1038/35002501.
Conservation International: Biodiversity-hotspots. 2006, http://www.biodiversityhotspots.org/xp/Hotspots/hotspots_by_region/Pages/default.aspx.
Guo X, He S, Zhang Y: Phylogeny and biogeography of Chinese sisorid catfishes re-examined using mitochondrial cytochrome b and 16 S rRNA gene sequences. Mol Phylogenet Evol. 2005, 35: 344-362. 10.1016/j.ympev.2004.12.015.
Liu JQ, Wang YJ, Wang AL, Hideaki O, Abbott RJ: Radiation and diversification within the Ligularia-Cremanthodium-Parasenecio complex (Asteraceae) triggered by uplift of the Qinghai-Tibetan Plateau. Mol Phylogenet Evol. 2006, 38: 31-49. 10.1016/j.ympev.2005.09.010.
Luo J, Yang D, Suzuki H, Wang YG, Chen WJ, Campbell KL, Zhang YP: Molecular phylogeny and biogeography of Oriental voles: genus Eothenomys (Muridae, Mammalia). Mol Phylogenet Evol. 2004, 33: 349-362. 10.1016/j.ympev.2004.06.005.
Sun HL, Zheng D: Formation, Evolution and Development of Qinghai-Xizang (Tibetan) Plateau. Guangzhou, China: Guangdong Science & Technology Press; 1998.
Peng Z, Ho SYW, Zhang Y, He S: Uplift of the Tibetan plateau: Evidence from divergence times of glyptosternoid catfishes. Mol Phylogenet Evol. 2006, 39: 568-572. 10.1016/j.ympev.2005.10.016.
Qiao CY, Ran JH, Wang XQ: Phylogeny and biogeography of Cedrus (Pinaceae) inferred from sequences of seven paternal chloroplast and maternal mitochondrial DNA regions. Ann Bot. 2007, 100: 573-580. 10.1093/aob/mcm134.
Ran JH, Wei XX, Wang XQ: Molecular phylogeny and biogeography of Picea (Pinaceae): implications for phylogeographical studies using cytoplasmic haplotypes. Mol Phylogenet Evol. 2006, 41: 405-419. 10.1016/j.ympev.2006.05.039.
Wei XX, Wang XQ: Recolonization and radiation in Larix (Pinaceae): evidence from nuclear ribosomal DNA paralogues. Mol Ecol. 2004, 13: 3115-3123. 10.1111/j.1365-294X.2004.02299.x.
Wu CY: Vegetation of China. Beijing: Science Press; 1980.
Yang FS, Wang XQ, Hong DY: Unexpected high divergence in nrDNA ITS and extensive parallelism in floral morphology of Pedicularis (Orobanchaceae). Plant Syst Evol. 2003, 240: 91-105. 10.1007/s00606-003-0005-2.
Doyle JJ, Doyle JL: A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987, 19: 11-15.
Tate JA, Simpson BB: Paraphyly of Tarasa (Malvaceae) and diverse origins of the polyploid species. Syst Bot. 2003, 28: 723-737.
Sang T, Crawford DJ, Stuessy TF: Chloroplast DNA phylogeny, reticulate evolution, and biogeography of Paeonia (Paeoniaceae). Am J Bot. 1997, 84: 1120-1136. 10.2307/2446155.
Demesure B, Sodzi N, Petit RJ: A set of universal primers for amplification of polymorphic non-coding regions of mitochondrial and chloroplast DNA in plants. Mol Ecol. 1995, 4: 129-131. 10.1111/j.1365-294X.1995.tb00201.x.
Thompson JD, Gibson TJ, PLewniak F, Jeanmougin F, Higgins DG: The CLUSTALX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 24: 4876-4882.
Caicedo AL, Schaal BA: Population structure and phylogeography of Solanum pimpinellifolium inferred from a nuclear gene. Mol Ecol. 2004, 13: 1871-1882. 10.1111/j.1365-294X.2004.02191.x.
Rozas J, Rozas R: DnaSP version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics. 1999, 15: 174-175. 10.1093/bioinformatics/15.2.174.
Huelsenbeck JP, Ronquist F: Mrbayes: Bayesian inference of phylogenetic trees. Bioinformatics. 2001, 17: 754-755. 10.1093/bioinformatics/17.8.754.
Posada D, Crandall KA: Intraspecific gene genealogies: trees grafting into networks. Trends Ecol Evol. 2001, 16: 37-45. 10.1016/S0169-5347(00)02026-7.
Clement M, Posada D, Crandall KA: TCS: A computer program to estimate gene genealogies. Mol Ecol. 2000, 9: 1657-1660. 10.1046/j.1365-294x.2000.01020.x. (Available from URL: http://darwin.uvigo.es/software/tcs.html)
Templeton AR, Crandall KA, Sing CF: A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics. 1992, 132: 619-633.
Crandall KA, Templeton AR, Sing CF: Intraspecific cladogram estimation:problems and solutions. In Models in Phylogeny Reconstruction. Edited byScotland RW, Siebert DJ, Williams DM. Oxford: Clarendon; 1994:273–297.
Excoffier L, Smouse PE, Quattro JM: Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992, 131: 479-491.
Schneider S, Roessli D, Excoffier L: ARLEQUIN Version 2.000: A Software for Population Genetics Data analysis. 1999, http://lgb.unige.ch/arlequin/.
Burban C, Petit RJ, Carcreff E, Jactel H: Rangewide variation of the maritime pine bast scale Matsucoccus feytaudi Duc. (Homoptera: Matsucoccidae) in relation to the genetic structure of its host. Mol Ecol. 1999, 8: 1593-1602. 10.1046/j.1365-294x.1999.00739.x.
Peakall R, Smouse PE: GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes. 2006, 6: 288-295. 10.1111/j.1471-8286.2005.01155.x.
Nei M, Li WH: Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci USA. 1979, 76: 5269-5273. 10.1073/pnas.76.10.5269.
Edwards SV, Beerli P: Perspective: gene divergence, population divergence, and the variance in coalescence time in phylogeographic studies. Evolution. 2000, 54: 1839-1854.
Nei M, Li WH: Mathematical model for studying genetic variation interms of restriction endonucleases. Proc Natl Acad Sci USA 1979, 76:5269–5273.
Graur D, Li WH: Fundamentals of Molecular Evolution.Sunderland, Massachusetts:Sinauer & Associates;1999.
Chiang YC, Hung KH, Schaal BA, Ge XJ, Hsu TW, Chiang TY: Contrasting phylogeographical patterns between mainland and island taxa of the Pinus luchuensis complex. Mol Ecol. 2006, 15: 765-779. 10.1111/j.1365-294X.2005.02833.x.
Acknowledgements
The authors thank Drs. Xun Gong, Yong Yang, Jianying Xiang, Fujuan Zhang, Bo Tian, Wentao Yu and Lianming Gao for help with this project. This project was jointly funded by the National Natural Science Foundations of China (30770137, 31070198 and 41030212), the Natural Science Foundation of Yunnan Province (2007C087M) and the West Light Foundation of The Chinese Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors’ contributions
Shaotian Chen carried out field work, the molecular genetic studies, the statistical analysis, and drafted the manuscript. Yaowu Xing and Tao Su participated in sample collections and sequencing. Zhekun Zhou designed the study. David L. Dilcher and Douglas E. Soltis polished the manuscript, and David L. Dilcher helped to conceive of the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Chen, S., Xing, Y., Su, T. et al. Phylogeographic analysis reveals significant spatial genetic structure of Incarvillea sinensisas a product of mountain building. BMC Plant Biol 12, 58 (2012). https://doi.org/10.1186/1471-2229-12-58
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2229-12-58