
Abstract
Background
Salicylic acid is a critical signalling component in plant defence responses. In Arabidopsis, isochorismate synthase encoded by SID2 is essential for the biosynthesis of salicylic acid in response to biotic challenges. Recently, both the calmodulin binding protein CBP60g and its closest homolog, the non-calmodulin binding SARD1, have been shown to bind to the promoter region of SID2. Loss of both CBP60g and SARD1 severely impacts the plants ability to produce SA in response to bacterial inoculation and renders the plant susceptible to infection. In an electrophoretic mobility shift assay CBP60g and SARD1 were shown to bind specifically to a 10mer oligonucleotide with the sequence GAAATTTTGG.
Results
Gene expression profiling on a custom microarray identified a set of genes, like SID2, down-regulated in cbp60g sard1 mutant plants. Co-expression analysis across a defined set of ATH1 full genome microarray experiments expanded this gene set; clustering analysis was then applied to group densely interconnected genes. A stringent threshold for co-expression identified two related calmodulin-like genes tightly associated with SID2. SID2 was found to cluster with genes whose promoter regions were significantly enriched with GAAATT motifs. Genes clustering with SID2 were found to be down-regulated in the cbp60g sard1 double mutant. Representative genes from other clusters enriched with the GAAATT motif were found to be variously down-regulated, unchanged or up-regulated in the double mutant. A previously characterised co-expression between SID2 and WRKY28 was not reproduced in this analysis but was contained within a subset of the experiments where SID2 was co-expressed with CBP60g or SARD1.
Conclusion
Putative components of the CBP60g SARD1 signalling network have been uncovered by co-expression analysis. In addition to genes whose regulation is similar to that of SID2 some are repressed by CBP60g and SARD1.
Similar content being viewed by others

Background
Two principal mechanisms, with overlapping components, exist to protect plants from infection. Pattern triggered immunity (PTI) involves the recognition of conserved, indispensible microbial structures, such as flagellin. These Microbe Associated Molecular Patterns (MAMPs) are recognised by Pattern Recognition Receptors (PRRs) such as FLS2, which recognizes flagellin, and stimulate a signalling network to elaborate an appropriate defence. In order to break free of this basal immune response adapted pathogens must produce and deliver effectors capable of disarming the plants surveillance and countermeasures. The second mechanism of protection therefore is Effector Triggered Immunity (ETI) whereby plants monitor effectors or their targets with Resistance (R) gene products. Recognition of pathogen effectors again stimulates a signalling network with many elements common to that of PTI but with typically more drastic consequences [1]. Plant hormones play critical roles in the signalling following both PTI and ETI, with salicylic acid (SA) central in mediating protection against biotrophic and hemi-biotrophic pathogens [2, 3]. Salicylic acid accumulates both locally and systemically following infection and is essential for establishment of Systemic Acquired Resistance (SAR) and the development of durable, broad spectrum resistance against normally virulent pathogens [4, 5].
For both ETI and PTI the accumulation and action of SA is dependent on several shared components. In Arabidopsis thaliana the EDS1/PAD4 node lies upstream of SA biosynthesis, as the two interacting proteins are essential for activation of the SA signalling sector [6, 7]. SID2 has been identified as a critical component in the biosynthesis of SA in response to biotic challenge; SID2 encodes an isochorismate synthase capable of catalysing the formation of the SA precursor isochorismate from chorismate [8]. Also critical for the accumulation of SA is the MATE transporter EDS5, which may be involved in the transport of a biosynthetic precursor of SA [9]. Downstream of SA biosynthesis, NPR1 is involved in the activation of SA-dependent gene expression. Suitably high SA levels and appropriate redox conditions result in NPR1 monomerisation allowing it to enter the nucleus and interact with transcription factors of the TGA family [10–12]. Recent studies have identified either NPR1 or the paralogs NPR3 and NPR4 as SA receptors [13, 14]. Wu et al. showed that the interaction of SA and NPR1 produced a conformational change allowing the NPR1 BTB/POZ domain to interact with TGA2. While Fu et al. did not observe SA binding by NPR1 they showed that NPR3 and NPR4 could act as SA-dependent adapters for the proteasomal degradation of NPR1 whose different affinity for SA subtly modulates the response to different SA concentrations.
With SID2 occupying a critical role in the transduction of defence signalling through SA, much effort has been made to understand its regulation. Positive regulators of SID2 expression have been identified, such as WRKY28 which has been shown to bind to the SID2 promoter and induce SID2 expression in transfection assays. Electrophoretic mobility shift assays (EMSA) revealed that WRKY28 bound to a modified version of the consensus W-box motif that retains the TGAC core [15]. Negative regulators of SID2 expression have also been uncovered; EIN3 has been shown to bind to the SID2 promoter and combined mutations of ein3 and its close homolog eil1 showed elevated SID2 expression, SA accumulation and increased resistance to bacterial infection [16]. Similarly, three related NAC transcription factors (ANAC019, ANAC055 and ANAC072) were found to inhibit SID2 expression, SA accumulation and resistance to bacterial infection with ANAC019 shown to bind to the SID2 promoter [17].
Two further genes involved in the regulation of SID2 are CBP60g and SARD1. CBP60g is a member of a family of calmodulin (CaM) binding proteins that was identified as being strongly induced in response to MAMPs treatment. Plants carrying cbp60g null mutations were compromised in the induction of SID2 and accumulation of SA [18]. CBP60g was shown to bind CaM in a Ca2+ dependent fashion; cbp60g transgenes with mutations in the CaM binding domain that abolished the CaM interaction were incapable of complementing the null mutant. Independently, the closest homolog of CBP60g was identified in a screen for mutants defective in systemic acquired resistance and named SARD1 [19]. SARD1, while more closely related to CBP60g than the rest of the CBP60 family, does not bind CaM [19, 20]. Both cbp60g and sard1 were impaired in SAR with the double mutant more strongly affected [19]. Lines over-expressing SARD1 accumulated more SA than wildtype plants [19] while the double knockout mutant was severely compromised in SA accumulation in response to infection [19, 20]. Both CBP60g and SARD1 were shown to bind to the promoter of SID2 and in EMSA experiments a central DNA binding domain of both proteins was found to bind to an oligomer with the sequence GAAATTTTGG selected from the SID2 promoter [19]. CBP60g and SARD1 have partially redundant function in SA signalling with both mutants affecting SA accumulation and pathogen growth but the double mutant exhibiting a greater than additive effect [19, 20]. While clearly overlapping in function there are a variety of distinctions in addition to the requirement of CaM binding: CBP60g appears to have more influence over the early events in defence signalling with SARD1 playing a more prominent role later; MAMPs triggered signalling is more greatly affected by the loss of CBP60g than the loss of SARD1 [20]. At the transcriptomic level the expression fingerprint of cbp60g more closely resembles that of sid2 than sard1 does during MAMPs responses while the trend was reversed later time-points with virulent bacterial infection [20].
While the cbp60g sard1 mutant drastically reduces SID2 expression and SA accumulation upon biotic challenge the double mutant is more susceptible to Pseudomonas syringae pv maculicola ES4326 (Pma ES4326) than sid2-2 indicating a role for CBP60g and SARD1 in SA-independent defence signalling [20]. Potential targets for CBP60g SARD1 regulation were identified using a custom microarray where 25 genes (from an array of 571 genes) including SID2 were down-regulated in the double mutant. Analysis of the promoters of these genes found a significant enrichment of a GAAATTT motif, a fragment of the oligomer used in Zhang et al.’s EMSA study. Similarly Zhang et al. noted an enrichment of AATTTT motifs in genes up-regulated in other PTI and ETI studies.
With the advent of whole genome transcriptional profiling many studies have made use of the abundance of microarray data to identify new pathway components based on their co-expression with known elements. For example, additional enzymes involved in cellulose synthesis and flavonoid biosynthesis were uncovered based on their correlation with known genes across publicly available array data [21, 22]. This type of analysis has also uncovered new regulatory elements controlling glucosinolate biosynthesis and fatty acid biosynthesis [23, 24]. In fact it was co-expression analysis that first uncovered the regulation of SID2 by WRKY28 [25]. Interestingly, the correlation between SID2 and WRKY28 was only observed when restricted to a subset of array experiments involving stress treatments. Given the enrichment of putative CBP60g SARD1 motifs within the sample of genes down-regulated in cbp60g sard1 plants we decided to use co-expression analysis to expand this subset and search for similar motif enrichment in order to identify additional potential targets for CBP60g SARD1 control and additional components of the signalling network.
Results
Expression patterns of CBP60g, SARD1, and SID2are correlated
Our previous work suggested that CBP60g plays a greater role than SARD1 during a MAMP response, while the reverse is true during the response to Pma ES4326. To test for a similar effect in the relationship of the expression levels of CBP60g and SARD1 to that of SID2, we monitored expression of these three genes following infiltration of leaves with the flagellin fragment flg22 or Pma ES4326, sampling every hour for the first 9 h following infection followed by a final sampling at 24 h. Figure 1 shows that following flg22 treatment, CBP60g induction precedes SARD1 induction, with expression of CBP60g already significantly up-regulated at 1 h, and maintaining a stronger fold-induction throughout the time course. The expression profile of CBP60g is closely mirrored by that of SID2 with the slight difference that CBP60g induction is more rapidly activated. Accordingly, the Pearson’s correlation for the similarity between the CBP60g and SID2 expression patterns was high (0.92) compared to the SARD1:SID2 correlation (0.66). A different pattern was observed following inoculation with Pma ES4326. While CBP60g is again faster to respond to infection, from 3h SARD1 up-regulation matches that of CBP60g and then exceeds it from 8h onwards. The later elevation of SARD1 transcription more closely matches that of SID2 than CBP60g. The correlation values are reversed relative to flg22 inoculation, at 0.64 for CBP60g:SID2 and 0.93 for SARD1:SID2. These results are consistent with the idea that CBP60g has a greater effect than SARD1 on SID2 expression using a MAMPs response, while the reverse is true during response to Pma ES4326.

CBP60g , SARD1 and SID2 are co-expressed in response to MAMPs treatment and Pma ES4326 infection. Wildtype Col-0 Arabidopsis plants were treated with 1 μM flg22, Pma ES4326 (OD600 = 0.01) or mock treated with water. qPCR data from two biological replicates were combined using a mixed linear model and the expression relative to Actin2 calculated. The log2 ratio of treatment relative to mock is plotted with error bars representing the standard error of the difference between means. (A) With flg22 the Pearson correlation coefficient for CBP60g:SID2 was 0.92 and 0.66 for SARD1:SID2. (B) Following Pma ES4326 inoculation the Pearson correlation coefficient values are 0.64 for CBP60g:SID2 and 0.93 for SARD1:SID2.
Selection of data sets for defining the CBP60g/SARD1regulon
We hypothesized that genes under the control of CBP60g and SARD1 could be identified by mining public gene expression data for genes co-expressed with the 45 genes whose expression was found to be suppressed in cbp60g, sard1 or cbp60g sard1 mutant plants using a small defence-related custom microarray (GEO: GSE18865 [20]). To do this, we wanted to select an appropriate set of data, as inclusion of many experiments in which defence genes are not induced is likely to increase the noise in the analysis. For example, the relationship between WRKY28 and SID2 was only observed in a subset of the ATTEDII co-expression database restricted to stress-associated profiling experiments and excluding other data [25]. We collected data from 308 expression profiling experiments conducted using the Affymetrix ATH1 Arabidopsis array, with treatments related to pathogen infection, stress responses, hormone treatment or associated mutations, and developmental series. The experiments ranged in size from six to several hundred arrays. Data from each experiment were processed identically; a conservative approach was taken to quality control with any outlying arrays being removed prior to batch normalization and summarization by RMA. In each experiment, the Spearman rank correlation coefficients for CBP60g:SID2 and SARD1:SID2 were calculated along with the associated p-value for significance of correlation. The results are shown in Figure 2. In many experiments, both correlations are similarly strong, but there are also experiments in which only CBP60g or only SARD1 is strongly correlated with SID2. In several experiments where a MAMPs elicitor is applied the correlation between CBP60g and SID2 is greater than the correlation between SARD1 and SID2. For example in the experiment marked (a) (NASCARRAYS122) seedlings were treated with flg22, the bacterial protein HrpZ, the fungal protein NPP1 or a preparation of bacterial lipopolysacharrides, the CBP60g correlation is 0.83 while the SARD1 correlation is 0.39. Similarly in (b) (Ausubel lab IMDS – Flg22 and OGs [26]) with treatments of flg22 and the plant-derived danger signal oligogalacturonides the CBP60g correlation is 0.81 while the SARD1 correlation is 0.52. However, not all MAMP treatment experiments gave rise to the same discrimination, in (c) (Ausubel lab IMDS – Chitin 8mer) treatment with fungal-derived chitin fragments produce equally strong correlations for both CBP60g and SARD1, 0.88 and 0.90 respectively. Other experiments which exhibited stronger CBP60g SID2 correlation included experiments investigating plastid function. In (d) (EBI Arrayexpress E-MEXP-2927 [27]) null mutations in the plastid biogenesis component SCO3 produce a CBP60g correlation of 0.89 while the SARD1 correlation was 0.2. The impact of excess light or application of an electron transport inhibitor in (e) (EBI Arrayexpress E-MTAB-403) also resulted in a specific CBP60g:SID2 correlation, 0.90 compared with 0.28 for SARD1. Array experiments where a stronger SARD1:SID2 correlation was observed included: (f) Phytophthora parasitica infection of roots; (g) combined mutations in ABA signalling and SA biosynthesis; (h) mutations in the exocycst component EXO70A1; (i) comparisons of potassium starvation and caesium toxicity (NASCARRAYS468; GEO GSE16913, [28]; NASCARRAYS435; NASCARRAYS105). While there are interesting trends in the specific, discriminatory associations between CBP60g or SARD1 and SID2 there are not currently sufficient datasets to construct specific co-expression networks. In the majority of experiments where CBP60g or SARD1 was strongly and significantly correlated with SID2 the homolog was also positively correlated, providing a sufficiently large dataset to form robust co-expression networks. Based on these results, we decided to use the 125 experiments, comprising 2,245 arrays, in which either CBP60g or SARD1 showed a strong and significant correlation with SID2 of at least 0.7 and a p-value of no more than 0.05.

Significant SID2:CBP60g/SARD1 correlation is observed in numerous publicly available microarray datasets. Affymetrix ATH1 array experiments were RMA normalised and the Spearman rank correlation coefficient calculated between probesets representing SID2 (262177_at) and CBP60g (246821_at) and SARD1 (260046_at) plotted. Darker points represent experiments where at least one correlation has a p-value no greater than 0.05. Highlighted experiments represent: (a-c) assorted MAMPs treatments; (d-e) plastid function; (f) oomycete infection; (g) SA and ABA signalling mutants; (h) exocyst component mutants; (i) potassium starvation.
Some clusters of genes co-expressed with CBP60g/SARD1-dependent genes have promoters enriched with GAAATT motifs
We reasoned that by clustering genes with expression patterns similar to the 45 genes that showed reduced expression in cbp60g, sard1, or cbp60g sard1 plants, we might identify additional defence genes whose expression levels are controlled by CBP60g and/or SARD1. Prior to clustering, we explored different criteria for selection of co-expressed genes. We used Spearman’s rank correlation as a measure of similarity, and determined the number of probesets obtained using different cut-off values from 0.85 to 0.70 (Table 1). For each set of genes obtained, we counted the numbers of GAAATT motifs in their promoters. Previous promoter analysis of differentially expressed genes had scored GAAATTT as the most significant sub-component of the CBP60g SARD1 binding motif [20]. However, in pilot clustering analysis and in all subsequent analyses the shortened GAAATT motif was found to have greater statistical over-representation in clusters containing SID2 (Additional file 1: Table S1). Hence we used the GAAATT motif rather than the longer GAAATTT motif to evaluate the various thresholds and clustering metrics for analysis. The 0.85 correlation cut-off was clearly too stringent, as it identified 59 probesets, only 14 more than the 45 used to begin the analysis. The 0.80 cut-off identified 128 probesets, with 2.77 motifs per gene. This seemed a reasonably stringent criterion, and we used these 128 probesets for one clustering analysis - experiment #1. When the correlation cut-off was relaxed to 0.70, 518 probesets were identified, albeit with a lower average density of motifs per gene of 2.52. However, these included many corresponding to genes with 4 or more motifs in their promoters, suggesting that the 0.80 cut-off may exclude some potential targets for CBP60g SARD1 regulation. We used this larger set for a second clustering analysis - experiment #2.
For both clustering experiments, we used DPClus [29] to identify densely interconnected nodes within the co-expression network with connections being defined as correlations of 0.8 or greater for experiment #1 and 0.7 or greater for experiment #2. The generation of clusters was constrained by a threshold for the minimum density of connections within a cluster and a parameter controlling the periphery tracking of clusters such that sparsely connected nodes would be ejected from a cluster even if the average connection density passed the threshold. These two parameters affect the clustering resolution and the capacity to distinguish sub-networks within the co-expression network. Overlapping clusters could be formed permitting one gene node to span multiple clusters while some sparsely connected genes could be excluded from all clusters. Various parameters were tested and the cluster or clusters containing SID2 evaluated. In each instance the GAAATT motif was significantly over-represented in the SID2 cluster and parameters that gave rise to the maximum statistical significance observed were chosen for the final analysis – a minimum density value of 0.75 and a CP threshold of 0.75.
Figure 3a shows the result of experiment #1. The 128 probesets formed 15 clusters, ranging in size from 3 to 16 genes, with average numbers of motifs per gene ranging from 1.4 to 4.33 motifs per gene. Figure 3b shows the result of experiment #2. The 518 probesets formed 58 clusters, ranging in size from 5 to 113 genes, with average numbers of motifs per gene ranging from 0.86 to 4.2 motifs per gene (Additional file 2: Table S2 and Additional file 3: Table S3).

Clusters of genes with GAAATT enriched promoters identified within the CBP60g / SARD1 co-expression network. 45 genes down-regulated in cbp60g, sard1 or cbp60g sard1 mutants were used to seed a co-expression network. (A) In experiment #1 a Spearman rank correlation coefficient threshold of 0.8 defined a network of 128 probesets. DPClus formed 15 overlapping clusters. (B) In experiment #2 a correlation threshold of 0.7 defined a network of 518 probesets which were organised into 61 overlapping clusters. Circles represent clusters of genes and edges represent correlation between members of different clusters. The number of genes in a cluster is proportionate to the size of the circle. The colour of the cluster reflects the average number of GAAATT motifs within the 1500 bp promoter region of cluster members, red indicates an over-representation, blue under-representation and white the genome average.
The SID2clusters are enriched for GAAATT motifs and defence genes
As SID2 has been reported to be a direct target of CBP60g and SARD1, we studied the clusters containing SID2 more closely. In Experiment #1, SID2 is in cluster 3. The internal structure of this cluster is shown in Figure 4a. It includes five genes in addition to SID2: The density of connections for the cluster is 1 and the gene most strongly co-expressed with SID2 is CML46. CML46 and CML47 are two members of a gene family encoding calmodulin-like proteins containing calcium binding EF-hand domains. They are phylogenetically closely related to a third family member not represented on the ATH1 array, CML45[30]. SARD1 is contained in this cluster (though it is only represented by the more reliable of its two probesets) alongside a putative heavy-metal transporter and AGP5. Of the five genes linked to SID2 only AGP5 was present on the custom array used to seed this analysis. As shown in Figure 4b, analysis of the GAAATT motif in the promoters of these genes by POBO shows that enrichment of this motif is highly significant (p-value < 0.0001) [31]. qPCR confirmed that expression of all cluster members was induced in response to infection with Pma ES4326, this induction was significantly suppressed in the cbp60g sard1 mutant for AGP5, At5g52760 and CML47; CML46 expression was lower in the double mutant but with a high p-value of 0.097 (Figure 4c). Intriguingly, CML45, the nearest homolog of CML46 and CML47, is up-regulated in response to Pma ES4326 and this induction is significantly enhanced in cbp60g sard1 plants (Additional file 4: Figure S1).

SID2 is tightly co-expressed with SARD1 and calmodulin-like genes with GAAATT abundant promoters. (A) The internal structure of cluster 3 from experiment #1. Edges between genes show correlation above the threshold of 0.8 with thickness proportional to the magnitude of correlation. Genes are coloured according to the number of GAAATT motifs present in the 1500 bp promoter region from white for genes with 0 motifs to red for a maximum of 7. (B) POBO analysis of GAAATT motif frequency in the 1500 bp upstream of transcription start sites. 1000 pseudoclusters of six genes were generated both from within cluster 3 and the genome background; jagged lines show the motif frequencies from which a fitted curve was derived. GAAATT motifs were found to be significantly over-represented with a p-value < 0.0001. (C) qRT-PCR measurement of gene expression 24 hpi Pma ES4326 inoculation (OD600 =0.01). Data from four or five biological replicates were merged using a mixed linear model and the mean log2 ratio to Actin2 expression plotted along with the standard error. Asterisks denote a significant differential expression between wildtype and the cbp60g sard1 mutant with p-value ≤ 0.05 from a two-tailed t-test.
In the expanded clustering of experiment #2 SID2 is also present in only one cluster – cluster 2. Figure 5a shows that the SID2 cluster from experiment #2 contains all the genes from the more stringent analysis but is much larger, comprising 31 probesets. The POBO analysis shown in Figure 5b indicates that the enrichment of the GAAATT motif remains highly significant in this cluster. SID2 and CML46 are the cluster members with the highest GAAATT motif density with 7 motifs while ARCK1, a receptor-like kinase involved in suppressing ABA responses has 6 [32]. qPCR was used to investigate the impact of the cbp60g sard1 double mutation on two of the genes with GAAATT rich promoters not previously identified as being involved in defence, ARCK1 and the phospholipase At4g38560 (Figure 5c). In addition to both SARD1 probesets and CBP60g, this cluster also includes many genes known to play important roles in plant defence because loss-of-function mutations compromise resistance. These include PAD4[33], EDS1[34], ADR1-L1[35], SOBIR1[36] and WRKY46[15]. The cluster was significantly enriched for genes with known or putative receptor kinase function and genes implicated in calcium signalling when compared to all the probesets on the array using the Mapman over-representation tool (http://mapman.mpimp-golm.mpg.de/general/ora/ora.shtml, [37]). The heavy-metal transporter gene from the experiment #1 SID2 cluster was joined by a neighbouring close homolog. Three genes in the cluster were predicted to contain ankyrin repeats.

An expanded SID2 regulon contains CBP60g , SARD1 and a variety of defence genes with GAAATT rich promoters. (A) The internal structure of cluster 2 from experiment #2. Edges between genes show correlation above the threshold of 0.7 with thickness proportional to the magnitude of correlation. Genes are coloured according to the number of GAAATT motifs present in the 1500 bp promoter region from white for genes with 0 motifs to red for a maximum of 7. (B) POBO analysis of GAAATT motif frequency in the 1500 bp upstream of transcription start sites. 1000 pseudoclusters of 30 genes were generated both from within cluster 2 and the genome background; jagged lines show the motif frequencies from which a fitted curve was derived. GAAATT motifs were found to be significantly over-represented with a p-value < 0.0001. (C) qRT-PCR measurement of gene expression 24 hpi Pma ES4326 inoculation (OD600 =0.01). Data from five biological replicates were merged using a mixed linear model and the mean log2 ratio to Actin2 expression plotted along with the standard error. Asterisks denote significant differential expression between wildtype and the cbp60g sard1 mutant with p-value ≤ 0.05 from a two-tailed t-test.
All 7 members of the cluster tested by qPCR were found to be up-regulated in response to bacterial infection in a cbp60g sard1 dependent fashion except CML46 (Figure 4c and 5c). While some cluster members may prove to be targets for CBP60g SARD1 regulation, genetic analysis and transcriptome profiling has placed other components (PAD4, EDS1) upstream of CBP60g and SARD1 in the immune signalling pathway, thus the co-expression network reveals multiple stages in the control of SA mediated defence signalling. Both EDS1 and PAD4 have previously been characterised as SA inducible and their roles in the feed-forward loop amplifying SA signals may best explain their co-expression with SID2[38, 39].
Other motifs were observed to be over-represented within this cluster in addition to GAAATT. A CCT n7 TCC dyad was over-abundant while the CCT or TCC submotifs or the dyad with any intervening length other than 7 were all under-represented (Figure 6a-c). This motif was found in the SARD1 promoter close to the transcription start site. 17 of the 23 CCT n7 TCC motifs present in the cluster fall in the proximal 750 bp of the 1500 bp promoter, a significant bias when compared with the genome distribution by Fisher’s exact test (p-value=0.005) (Additional file 5: Figure S2). No such bias was observed for GAAATT position or strand in any cluster. Various permutations of the consensus WRKY transcription factor binding site W-box and the W-like box described in [15] were also observed to be enriched in the SID2 cluster (Figure 6d-f).

The SID2 regulon is enriched with known cis-elements and novel motifs. POBO analysis of motif distribution in 1500 bp promoters from experiment #2 cluster 2. 1000 pseudoclusters of 30 genes were generated both from within cluster 2 and the genome background; jagged lines show the motif frequencies from which a fitted curve was derived. (A-C) CCTNNNNNNNTCC motifs are significantly over-represented with a p-value < 0.0001 though CCT and TCC motifs are significantly under-represented in the cluster. (D-E) Different versions of the W-box consensus sequence binding site for WRKY transcription factors and (F) the defined WRKY28 binding site are significantly over-represented in cluster 2.
Genes in other clusters enriched for GAAATT motifs are suppressed, induced or unaffected by the cbp60g sard1mutant
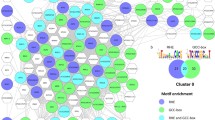
Expanding the analysis to other clusters with a significant over-representation of the GAAATT motif (Additional file 6: Table S4) uniformly identified pathogen inducible gene expression (Figures 7a and b). However, while some representative (GAAATT abundant) genes from GAAATT-rich clusters, for example cluster 17, were demonstrated to be cbp60g sard1 dependent for their full induction by qPCR (Figure 7a) or already known to be so from the custom microarray study (PBS3, AIG1) other GAAATT rich cluster members, for example CNGC13 from cluster 60, were found to be induced in a cbp60g sard1 independent fashion. Since GAAATT is a commonly occurring motif in Arabidopsis promoters small clusters when repeatedly sampled with replacement by POBO may be unduly skewed by the presence of one or two genes with a large number of motifs.

Putative targets for CBP60g / SARD1 regulation are repressed in cbp60g sard1 plants. Expression of candidate genes with promoters containing multiple GAAATT motifs from the 14 clusters, other than cluster 2, most significantly enriched for GAAATT motifs in experiment #2. (A) Candidates from clusters 1, 14, 15, 17, 21, 22, 28, 35, 36, 37, 40, 53, 58, 60. (B) Candidates from cluster 8. qRT-PCR measurement of gene expression 24 hpi Pma ES4326 inoculation (OD600 =0.01). Data from five biological replicates were merged using a mixed linear model and the mean log2 ratio to Actin2 expression plotted along with the standard error. Asterisks denote a significant differential expression between wildtype and the cbp60g sard1 mutant with p-value ≤ 0.05 from a two-tailed t-test.
One larger cluster, cluster 14 with 26 genes enriched with GAAATT motifs, was found to contain CBP60g alongside multiple vesicle trafficking components, including some shown to be important for SA homeostasis [40]. However, no cbp60g sard1 dependent expression was observed for SYP122 or SNAP33 (Figure 7a). W-boxes and another motif, AAGTC, were both observed with significant over-representation in this cluster and may better explain potential co-regulation within the cluster (Additional file 6: Table S4). Another cluster representing genes of clearly linked function was cluster 15, containing the pathogen responsive genes PR1, PR2, PR5 and PNP-A. However, while PR1 has previously been shown to require CBP60g and SARD1 for full induction, PR2 was not significantly affected.
In some instances the GAAATT rich gene selected to monitor a given cluster was found to be up-regulated in the cbp60g sard1 double mutant relative to wildtype e.g. At1g51890 and At1g64610. This phenomenon was observed in several members of cluster 12 with PBP1 having a significantly enhanced response to infection in the double mutant while At5g41750 and At2g32030 were up-regulated in the mock inoculated mutants. Intriguingly for MPK11, which encodes a MAP kinase activated during PTI [41], both a significantly suppressed pathogen response and elevated basal expression were observed in the mutant.
WRKY28:SID2co-expression occurs in a subset of the conditions included in this analysis
Prior co-expression analysis of biotic stress microarray data had uncovered the regulation of SID2 by WRKY28 and the regulation of SA biosynthesis component PBS3 by WRKY46[25]. While WRKY46 clustered with SID2, the proposed downstream target for WRKY46, PBS3 does not co-cluster with WRKY46. Similarly, WRKY28 was not found to be robustly co-expressed with SID2 in this study and was not included in the 518 genes selected for analysis in experiment #2. In the selected dataset of 2245 arrays WRKY28 ranks as the1775th most closely correlated gene with SID2 with a Spearman rank coefficient of 0.34. The modified W-box motif identified through EMSA as the binding site for WRKY28 is enriched in cluster 2 but weakly in comparison with other motifs investigated (Figure 6f). To identify conditions responsible for the previously reported WRKY28:SID2 co-expression, microarray studies with a significant WRKY28:SID2 correlation were plotted alongside CBP60g:SID2 and SARD1:SID2 correlations in a heatmap (Figure 8). The association of WRKY28 and SID2 comprised a subset of the studies with significant CBP60g or SARD1 co-expression. However, analysis of the 31 experiments comprising 386 arrays where WRKY28:SID2 expression is significant and greater than 0.7 still resulted in a stronger association between CBP60g or SARD1 and SID2 than WRKY28 and SID2 with WRKY28 only rising to become the 69th gene most closely correlated with SID2.

WRKY28:SID2 co-expression is restricted to a subset of conditions engendering CBP60g/SARD1:SID2 co-expression. Spearman rank correlation coefficient between SID2 and CBP60g, SARD1 and WRKY28 plotted for the 182 microarray experiments where at least one correlation was significant (p-value ≤ 0.05). Correlation values were sorted by a self-organising maps algorithm and subjected to complete linkage hierarchical clustering. Positive correlations are yellow while negative correlations are blue.
Discussion
The various strands of evidence pointing to the discriminatory action of the partially redundant CBP60g and SARD1 in mediating different aspects of the immune response were reinforced when surveying the microarray datasets (Figure 2). As with the flg22 timecourse (Figure 1) a closer association of CBP60g and SID2 was observed in several MAMPs treatment studies and intriguingly, several experiments unrelated to defence. Unfortunately, despite the abundance and variety of microarray experiments publicly available there are currently too few to build independent correlation networks for CBP60g and SARD1. Aoki et al. reported that a minimum of 100 arrays is required for stability in the density of gene co-expression networks [42] and the addition of several MAMPs specific experiments sampling at early time-points would be required to pass this threshold for a distinct CBP60g analysis. The presence of SARD1 and absence of CBP60g in the SID2 cluster with the more stringent correlation threshold of experiment #1 (Figure 4a) imply that the combined dataset may be weighted towards later timepoints in infection, understandably since these have higher probability of uncovering large scale differential expression in relatively costly gene expression profiling studies. CBP60g has recently been implicated in mediating responses to abiotic stress such as drought [43]. Whilst many abiotic stress studies were included few passed the threshold for CBP60g or SARD1 correlation with SID2. Closer inspection of two well-characterised studies of drought and osmotic stresses (NASCARRAYS – 139 and 141) revealed strong up-regulation of CBP60g in response to stress with only weak SID2 response and hence low correlation. A similar pattern was observed in several other experiments with an abiotic stress treatment. Preliminary analysis revealed CBP60g to be co-expressed with a subset of the genes from the 518 used in experiment #2 with SARD1 co-expressed with only one gene across this dataset (Additional file 7: Figure S3). Once more information has been uncovered concerning the role of CBP60g in mediating abiotic stress responses, revisiting such a co-expression analysis may prove fruitful.
Potentially the most exciting finding from the co-expression analysis of CBP60g and/or SARD1 and SID2 has been the two calmodulin-like genes in the core SID2 cluster from experiment #1. Their strength of correlation combined with the frequency of GAAATT motifs in both CML46 and CML47 promoters and their potential for physically interacting with CBP60g makes them ideal candidates for further investigation. While the pathogen associated induction of CML47 was dependent on CBP60g and SARD1 there was but a weak and insignificant impact on CML46. A further distinction between the two is that in the main analysis CBP60g and CML46 have a Spearman correlation co-efficient of 0.84 and 0.74 in the abiotic stress data set used in Additional file 7: Figure S3, whereas between CBP60g and CML47 the value falls from 0.76 to 0.31 indicating a potential abiotic stress specific role for CML46. The up-regulation of CML45 (unmonitored by the ATH1 microarray) in cbp60g sard1 mutants was also surprising and points to potentially complex interplay of feedback loops in controlling the expression of these putative CBP60g interactors.
Zooming out of the core SID2 cluster by relaxing the co-expression threshold in experiment #2 revealed several more putative targets for CBP60g and SARD1 regulation. Signalling components such as the kinases in this cluster provide key targets for investigating pathogen susceptibility in knock-out mutants as these may lie upstream of key defence responses. Cluster 2 already includes several genes known to confer resistance to infection. However, several of these (PAD4, EDS1, WRKY46) lie upstream of CBP60g and SARD1[7, 20] and so while their presence in this cluster provides an interesting insight into the various feedback loops that govern their co-expression they will not explain the SID2-independent defence response. Two genes affecting pathogen resistance that may be downstream of CBP60g and SARD1 are SOBIR1 and ADR1-L1. SOBIR1 is a negative regulator of defence responses [36] and so a poor candidate but the NB-LRR receptor ADR1-L1 has been implicated as a positive regulator in the establishment of PTI, ETI and basal defence against virulent pathogens [35].
Outside the clusters containing SID2, cluster 17 of experiment #2 contains the greatest density of observed cbp60g sard1 dependent genes with PBS3 and AIG1, known from the custom array study, and FMO1 and CML47 confirmed by qPCR [20]. FMO1 regulates EDS1-dependent, SID2-independent defence signalling, a process inhibited by NUDT7 the homolog of which, NUDT5, is present in this cluster [44].
Of the genes whose promoters have abundant GAAATT motifs and which cluster within GAAATT enriched regulons there are perhaps four ways to account for those whose expression are not CBP60g / SARD1 dependent. Firstly, since GAAATT is a relatively abundant sequence within the promoters of Arabidopsis genes with an average of close to two motifs per 1500 bp promoter the chance occurrence of two genes with several motifs within a small cluster of, say, five genes will easily produce false positives. It is however difficult to say how large a cluster should be to be worthy of investigation since at the higher stringency of experiment #1 clusters we know to be of interest can be relatively small. A second possibility is that, for CBP60g / SARD1 function, binding to the GAAATT motif is necessary but not sufficient and additional transcription factors and their binding sites are required for cooperative activation. The heatmap in Figure 8 implies that WRKY28 is not essential for such activation. However, the SID2 cluster and several other GAAATT rich clusters are enriched with the consensus W-box element for WRKY binding. Third, for some clusters although GAAATT enrichment may accurately imply CBP60g SARD1 binding, another transcription factor may exert dominant control over induction in response to biotic challenge. Thus the influence of CBP60g and SARD1 may only be observed when this factor is absent. Finally, there are six other members of the CBP60 family with as yet no demonstrated DNA binding potential but in some instances moderate induction in response to biotic stresses which may potentially interact with some motif similar to the one defined for CBP60g and SARD1.
The role of CBP60g and SARD1 in repressing the expression of genes with multiple GAAATT motifs and co-expression with other GAAATT enriched genes was surprising given our previous finding of a significant under-representation of GAAATTT motifs in the promoters of genes up-regulated in the cbp60g sard1 double mutant [20]. However, such complexity is not without precedence. The calmodulin-regulated transcription factor SR1 has been shown to positively regulate CBF2 but negatively regulate EDS1[45, 46]. The complexity of Ca2+ mediated control over SA-mediated defence signalling is further underlined by the observation that SR1 inhibits expression of two important positive regulators upstream of SID2, EDS1 and NDR1, but also inhibits the negative regulator of SID2 expression, EIN3 [45, 47]. Interestingly, numerous clusters appear to be enriched for SR1 binding sites with some overlapping GAAATT enrichment (Additional file 6: Table S4). Cluster 8 revealed several genes up-regulated in the absence of CBP60g and SARD1 and multiple members of this cluster act as inhibitors of defence responses (BON1, BAP1, GILP, NUDT7) [44, 48, 49]. Furthermore, PBP1 mediates auxin signalling, a potential inhibitor of SA-mediated defence signalling [50, 51]. Another important defence component in this cluster is MPK11. While MPK11 is significantly down-regulated in cbp60g sard1 plants 24 hpi with Pma ES4326, it may be that the moderate increase in the mock inoculated or basal state is more important since MPK11 is activated within minutes of biotic challenge and steady-state transcript levels may be important [41]. Another feature of this cluster is the significant over-abundance of calcium binding proteins including BON1, BAP1, PBP1, CML37 and At3g10830, along with the calmodulin dependent kinase CPK substrate CZF [52]. These features combine to make this particular regulon an intriguing target for further investigation.
The process involved in the identification of WRKY28 as a regulator of SID2 led us to expect WRKY28 would form some part of the SID2 cluster. The clear evidence that WRKY28’s association with SID2 exists as a subset of the conditions under which there is a strong and significant association between CBP60g and / or SARD1 and SID2 implies some specific role for WRKY28. However, there was no clear trend in the conditions under which WRKY28 was correlated with SID2. One trend appearing to emerge from the conditions where SID2 was correlated with CBP60g and/or SARD1 but not WRKY28 were experiments in which the treatment was an exogenous application of SA or some analogue of SA such as benzothiadiazole (BTH), 3,5-dichloroanthranilic acid (DCA) or 2,6-dichloroisonicotinic acid (INA). For example, in experiments with exogenous SA application, NASCARRAYS192 and NASCARRAYS365, the WRKY28:SID2 correlation is −0.17 and 0.18 while the SARD1:SID2 correlation is 0.71 and 0.68. In experiments with BTH application, GSE9955 and NASCARRAYS392, WRKY28:SID2 correlation scores are −0.48 and −0.03 while SARD1:SID2 scores 0.86 and 0.89. In experiment GSE13833 [53], where DCA and INA are applied, the scores are 0.16 and 0.85 respectively. While these experiments are not sufficient to construct a stable co-expression matrix they suggest a role for SARD1 in amplifying an existing SA-mediated signal through a feed-forward loop that is independent of WRKY28.
Conclusions
Co-expression analysis has facilitated the identification of an SA-mediated defence signalling regulon at two different degrees of resolution. The promoters of these genes are enriched for a fragment of an oligomer demonstrated to bind to CBP60g and SARD1, indicating that some members of these clusters are likely targets for regulation by CBP60g and SARD1. Other putative targets have been identified in separate clusters and intriguingly some genes downstream of GAAATT-abundant promoters have been shown to be repressed by CBP60g and SARD1, indicating a potentially complex role in the control of defence gene expression responses. This co-expression analysis has also shed light on the relationship between WRKY28 and SID2 which may allow fine-tuning of regulatory models.
Methods
Plant growth conditions and pathogen cultures
Arabidopsis thaliana accession Col-0 was used as the wildtype control and cbp60g-1 (SALK_023199) [18] and sard1-2 (SALK_052422) [19] as the mutant lines. Plants were grown on autoclaved BM2 Germinating Mix (Berger) in a growth chamber with a 12 h photoperiod at 22°C with 75% humidity. Plants were grown for 4–5 weeks before bacterial infection or MAMPs treatment. Pma ES4326 cultures were grown overnight in King’s B medium with 100 μg ml-1 streptomycin at room temperature. Cultures were centrifuged, washed and resuspended in 5 mM MgSO4 to a density of OD600=0.01. Flg22 peptide was purchased from EZBiolab and prepared to 1 μM. MAMPs and bacterial inoculations were made with a needless syringe; mock treatments were 5 mM MgSO4.
Quantitative RT-PCR analysis
RNA was purified using Trizol (Invitrogen) and treated with DNaseI (NEB). Quantitative RT-PCR experiments were performed using a Lightcycler 480 Real-Time PCR system (Roche). 24 ng of total RNA was used for each 10 μl reaction with the SuperScript III Platinum SYBR Green One-Step quantitative RT-PCR kit (Invitrogen) according to the protocols of the manufacturers. The thermal cycling program was 50°C for 10 min, 95°C for 10 min followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. For each reaction amplification curve the crossing point (Cp) was calculated using the 2nd derivative max method provided with the Lightcycler software. Each reaction was run with two technical replicates and the Cp values for these replicates were averaged. Either four or five independent biological replicates were included for each gene and Actin2 (At3g18780) was used as the internal reference. The following model was fit to the Cp value data using the lme function in the nlme package in the R environment: Cp gytr = GYT gyt +R r +ε gytr , where GYT is the gene:genotype:treatment interaction as a fixed effect, and R (replicate) and ε (residual) are random effects. Once modelled, the mean estimate of the gene:genotype:treatment interaction was used as the Cp value and relative log2 expression values were obtained by subtracting Cp values of the Actin2 gene. For two-tailed t-tests, the standard error appropriate for each comparison was calculated using the variance and covariance values obtained from the model fitting. A full list of the primers used can be found in Additional file 8: Table S5.
Microarray data analysis
Affymetrix ATH1 whole genome microarray datasets were downloaded in the form of .CEL files from the following data repositories: NASCArrays (http://affymetrix.arabidopsis.info/narrays/experimentbrowse.pl); NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/); EBI ArrayExpress (http://www.ebi.ac.uk/arrayexpress/); Ausubel lab IMDS (http://ausubellab.mgh.harvard.edu/imds/). A list of the experiments included in this study can be found in Additional file 9: Table S6. Data quality of the individual arrays were assessed for each experiment using the affyQCReport package, part of the Bioconductor suite of programs within the R environment [54]. Outlying arrays which appeared to distort the normalisation of the experiment were discarded. Each experiment was pre-processed and quantile normalised using the RMA algorithm as implemented by the justRMA function of the gcrma R package [55]. For each experiment the Spearman rank correlation coefficient, and corresponding p-value for this test, between probesets representing SID2 (262177_at) and CBP60g (246821_at) or SARD1 (260046_at, 260068_at) were determined using the cor and cor.test R functions. Due to prior misannotation of SARD1, two probesets report SARD1 expression on the ATH1 chip but analysis of selected experiments revealed that the 260046_at probeset was the more reliable and sensitive of the two and is used in Figure 2. Correlation was scored using the Spearman rank correlation coefficient on the presumption that a non-parametric test would be more robust in the face of presumably widely varying data structures and that it would provide a more conservative measure of correlation, particularly in analyzing smaller datasets. Experiments were filtered based on the maximum significant correlation (Spearman correlation ≥ 0.7, p-value ≤ 0.05) of either CBP60g:SID2 or SARD1:SID2.
Co-expression analysis
For each of the selected microarray experiments the log2 expression values for each probeset were normalised such that the median value was set to 0. Datasets were merged and each probeset across all arrays was normalised such that the standard deviation was set to 1. 45 genes were identified as significantly differentially expressed in the custom mini-array experiments previously described (GEO: GSE18865 [20]) in cbp60g, sard1 or cbp60g sard1 plants relative to wildtype inoculated with either virulent Pma ES4326 or disarmed Pto DC3000 hrcC- with at least 2 fold down-regulation and a false discovery rate of 0.05 or less. Genes co-expressed with these 45 seeds were selected based on the Spearman rank correlation coefficient with a threshold of 0.8 for experiment #1 and 0.7 for experiment #2. Correlation matrices were calculated for both experiments and connections between genes set at the 0.8 and 0.7 thresholds, respectively. DPClus was used to cluster both experiments with the following parameters: minimum cluster density of 0.75; CP threshold of 0.75; minimum cluster size of 3 for experiment #1 and 5 for experiment #2; allowing overlapping clusters to form [29]. Clusters were visualised using CYTOSCAPE [56].
For the abiotic stress co-expression network constructed for Additional file 7: Figure S3, 27 experiments comprising 501 arrays were selected based on the induction of CBP60g in response to an abiotic stress without strong correlation between CBP60g and SID2 (≤0.5) (Additional file 9: Table S6). Genes co-expressed with either CBP60g or SARD1 above a threshold of 0.7 across these experiments were included in the network.
For hierarchical clustering of array experiment correlation coefficients the CLUSTER program was used to organise experiments into self organising maps, subsequently complete linkage hierarchical cluster using an uncentered correlation metric was applied. Clustering was visualised using TREEVIEW [57].
Promoter analysis
Promoter sequences were retrieved from the RSAT database (http://rsat.ulb.ac.be/) with fixed 1500 bp sequences upstream of the transcription start site used in all analyses except Additional file 1: Table S1 [58]. 1500 bp promoters were chosen, despite the enrichment of GAATT motifs being more pronounced in 1000 bp promoters, in order to include the original SID2 site identified by Zhang et al. [19]. Five gene promoter sequences were not available on the RSAT server as they are considered pseudogenes (At4g13900, At1g21525, At4g14610, At3g48650, At2g24160). Nevertheless, these genes were included, with the promoter sequences retrieved from the TAIR database (http://www.arabidopsis.org/) [59]. The over-representation of known promoter cis-elements and motifs was assessed using the POBO application (http://ekhidna.biocenter.helsinki.fi/poxo/pobo/pobo) [31]. For each set of promoters, 1000 pseudoclusters of a size equal to the cluster in question were generated both from within the genes in question and the Arabidopsis genomic background (clean). Statistical significance of the t-values generated by POBO was calculated using the linked Graphpad application for a two-tailed comparison. For selected clusters, motif finding algorithms were used to uncover additional potential cis-elements using the MEME and RSAT tool suites [60, 61].
Abbreviations
- BTH:
-
Benzothiadiazole
- CaM:
-
Calmodulin
- Cp:
-
Crossing point
- DCA:
-
3,5-dichloroanthranilic acid
- ETI:
-
Effector triggered immunity
- EMSA:
-
Electrophoretic mobility shift assay
- HPI:
-
Hours post inoculation
- INA:
-
2,6-dichloroisonicotinic acid
- MAMP:
-
Microbe associated molecular pattern
- OG:
-
Oligogalacturonide
- Pma Pseudomonas syringae :
-
pv maculicola
- PTI:
-
Pattern triggered immunity
- SA:
-
Salicylic acid
- SAR:
-
Systemic acquired resistance.
References
Jones JDG, Dangl JL: The plant immune system. Nature. 2006, 444: 323-329.
Tsuda K, Sato M, Stoddard T, Glazebrook J, Katagiri F: Network properties of robust immunity in plants. PLoS Genet. 2009, 5: e1000772.
Glazebrook J: Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu Rev Phytopathol. 2005, 43: 205-227.
Métraux JP, Signer H, Ryals J, Ward E, Wyss-Benz M, Gaudin J, Raschdorf K, Schmid E, Blum W, Inverardi B: Increase in salicylic Acid at the onset of systemic acquired resistance in cucumber. Science. 1990, 250: 1004-1006.
Loake G, Grant M: Salicylic acid in plant defence–the players and protagonists. Curr Opin Plant Biol. 2007, 10: 466-472.
Wiermer M, Feys BJ, Parker JE: Plant immunity: the EDS1 regulatory node. Curr Opin Plant Biol. 2005, 8: 383-389.
Wang L, Mitra RM, Hasselmann KD, Sato M, Lenarz-Wyatt L, Cohen JD, Katagiri F, Glazebrook J: The genetic network controlling the Arabidopsis transcriptional response to Pseudomonas syringae pv. maculicola: roles of major regulators and the phytotoxin coronatine. Mol Plant Microbe Interact. 2008, 21: 1408-1420.
Wildermuth MC, Dewdney J, Wu G, Ausubel FM: Isochorismate synthase is required to synthesize salicylic acid for plant defence. Nature. 2001, 414: 562-565.
Nawrath C, Heck S, Parinthawong N, Métraux J-P: EDS5, an essential component of salicylic acid-dependent signaling for disease resistance in Arabidopsis, is a member of the MATE transporter family. Plant Cell. 2002, 14: 275-286.
Fan W, Dong X: In vivo interaction between NPR1 and transcription factor TGA2 leads to salicylic acid-mediated gene activation in Arabidopsis. Plant Cell. 2002, 14: 1377-1389.
Mou Z, Fan W, Dong X: Inducers of plant systemic acquired resistance regulate NPR1 function through redox changes. Cell. 2003, 113: 935-944.
Tada Y, Spoel SH, Pajerowska-Mukhtar K, Mou Z, Song J, Wang C, Zuo J, Dong X: Plant immunity requires conformational changes [corrected] of NPR1 via S-nitrosylation and thioredoxins. Science. 2008, 321: 952-956.
Wu Y, Zhang D, Chu JY, Boyle P, Wang Y, Brindle ID, De Luca V, Després C: The Arabidopsis NPR1 protein Is a receptor for the plant defense hormone salicylic acid. Cell Rep. 2012, 1: 639-647.
Fu ZQ, Yan S, Saleh A, Wang W, Ruble J, Oka N, Mohan R, Spoel SH, Tada Y, Zheng N, Dong X: NPR3 and NPR4 are receptors for the immune signal salicylic acid in plants. Nature. 2012, 486: 228-232.
van Verk MC, Bol JF, Linthorst HJM: WRKY transcription factors involved in activation of SA biosynthesis genes. BMC Plant Biol. 2011, 11: 89.
Chen H, Xue L, Chintamanani S, Germain H, Lin H, Cui H, Cai R, Zuo J, Tang X, Li X, Guo H, Zhou J-M: Ethylene insensitive3 and Ethylene insensitive3-LIKE1 repress Salicylic acid induction deficient2 expression to negatively regulate plant innate immunity in Arabidopsis. Plant Cell. 2009, 21: 2527-2540.
Zheng X-Y, Spivey NW, Zeng W, Liu P-P, Fu ZQ, Klessig DF, He SY, Dong X: Coronatine promotes Pseudomonas syringae virulence in plants by activating a signaling cascade that inhibits salicylic acid accumulation. Cell Host Microbe. 2012, 11: 587-596.
Wang L, Tsuda K, Sato M, Cohen JD, Katagiri F, Glazebrook J: Arabidopsis CaM binding protein CBP60g contributes to MAMP-induced SA accumulation and is involved in disease resistance against Pseudomonas syringae. PLoS Pathog. 2009, 5: e1000301.
Zhang Y, Xu S, Ding P, Wang D, Cheng YT, He J, Gao M, Xu F, Li Y, Zhu Z, Li X, Zhang Y: Control of salicylic acid synthesis and systemic acquired resistance by two members of a plant-specific family of transcription factors. Proc Natl Acad Sci USA. 2010, 107: 18220-18225.
Wang L, Tsuda K, Truman W, Sato M, Nguyen LV, Katagiri F, Glazebrook J: CBP60g and SARD1 play partially redundant critical roles in salicylic acid signaling. Plant J. 2011, 67: 1029-1041.
Persson S, Wei H, Milne J, Page GP, Somerville CR: Identification of genes required for cellulose synthesis by regression analysis of public microarray data sets. Proc Natl Acad Sci USA. 2005, 102: 8633-8638.
Yonekura-Sakakibara K, Tohge T, Niida R, Saito K: Identification of a flavonol 7-O-rhamnosyltransferase gene determining flavonoid pattern in Arabidopsis by transcriptome coexpression analysis and reverse genetics. J Biol Chem. 2007, 282: 14932-14941.
Hirai MY, Sugiyama K, Sawada Y, Tohge T, Obayashi T, Suzuki A, Araki R, Sakurai N, Suzuki H, Aoki K, Goda H, Nishizawa OI, Shibata D, Saito K: Omics-based identification of Arabidopsis Myb transcription factors regulating aliphatic glucosinolate biosynthesis. Proc Natl Acad Sci USA. 2007, 104: 6478-6483.
Han X, Yin L, Xue H: Co-expression analysis identifies CRC and AP1 the regulator of Arabidopsis fatty acid biosynthesis. J Integr Plant Biol. 2012, 54: 486-499.
van Verk MC, Bol JF, Linthorst HJM: Prospecting for genes involved in transcriptional regulation of plant defenses, a bioinformatics approach. BMC Plant Biol. 2011, 11: 88.
Ferrari S, Galletti R, Denoux C, De Lorenzo G, Ausubel FM, Dewdney J: Resistance to Botrytis cinerea induced in Arabidopsis by elicitors is independent of salicylic acid, ethylene, or jasmonate signaling but requires PHYTOALEXIN DEFICIENT3. Plant Physiol. 2007, 144: 367-379.
Albrecht V, Simková K, Carrie C, Delannoy E, Giraud E, Whelan J, Small ID, Apel K, Badger MR, Pogson BJ: The cytoskeleton and the peroxisomal-targeted snowy cotyledon3 protein are required for chloroplast development in Arabidopsis. Plant Cell. 2010, 22: 3423-3438.
Nishimura N, Okamoto M, Narusaka M, Yasuda M, Nakashita H, Shinozaki K, Narusaka Y, Hirayama T: ABA hypersensitive germination2-1 causes the activation of both abscisic acid and salicylic acid responses in Arabidopsis. Plant Cell Physiol. 2009, 50: 2112-2122.
Altaf-Ul-Amin M, Shinbo Y, Mihara K, Kurokawa K, Kanaya S: Development and implementation of an algorithm for detection of protein complexes in large interaction networks. BMC Bioinformatics. 2006, 7: 207.
McCormack E, Braam J: Calmodulins and related potential calcium sensors of Arabidopsis. New Phytol. 2003, 159: 585-598.
Kankainen M, Holm L: POBO, transcription factor binding site verification with bootstrapping. Nucleic Acids Res. 2004, 32: W222-W229.
Tanaka H, Osakabe Y, Katsura S, Mizuno S, Maruyama K, Kusakabe K, Mizoi J, Shinozaki K, Yamaguchi-Shinozaki K: Abiotic stress-inducible receptor-like kinases negatively control ABA signaling in Arabidopsis. Plant J. 2012, 70: 599-613.
Glazebrook J, Rogers EE, Ausubel FM: Isolation of Arabidopsis mutants with enhanced disease susceptibility by direct screening. Genetics. 1996, 143: 973-982.
Parker JE, Holub EB, Frost LN, Falk A, Gunn ND, Daniels MJ: Characterization of eds1, a mutation in Arabidopsis suppressing resistance to Peronospora parasitica specified by several different RPP genes. Plant Cell. 1996, 8: 2033-2046.
Bonardi V, Tang S, Stallmann A, Roberts M, Cherkis K, Dangl JL: Expanded functions for a family of plant intracellular immune receptors beyond specific recognition of pathogen effectors. Proc Natl Acad Sci USA. 2011, 108: 16463-16468.
Gao M, Wang X, Wang D, Xu F, Ding X, Zhang Z, Bi D, Cheng YT, Chen S, Li X, Zhang Y: Regulation of cell death and innate immunity by two receptor-like kinases in Arabidopsis. Cell Host Microbe. 2009, 6: 34-44.
Usadel B, Nagel A, Steinhauser D, Gibon Y, Bläsing OE, Redestig H, Sreenivasulu N, Krall L, Hannah MA, Poree F, Fernie AR, Stitt M: PageMan: an interactive ontology tool to generate, display, and annotate overview graphs for profiling experiments. BMC Bioinformatics. 2006, 7: 535.
Falk A, Feys BJ, Frost LN, Jones JD, Daniels MJ, Parker JE: EDS1, an essential component of R gene-mediated disease resistance in Arabidopsis has homology to eukaryotic lipases. Proc Natl Acad Sci USA. 1999, 96: 3292-3297.
Jirage D, Tootle TL, Reuber TL, Frost LN, Feys BJ, Parker JE, Ausubel FM, Glazebrook J: Arabidopsis thaliana PAD4 encodes a lipase-like gene that is important for salicylic acid signaling. Proc Natl Acad Sci USA. 1999, 96: 13583-13588.
Zhang Z, Feechan A, Pedersen C, Newman M-A, Qiu J, Olesen KL, Thordal-Christensen H: A SNARE-protein has opposing functions in penetration resistance and defence signalling pathways. Plant J. 2007, 49: 302-312.
Bethke G, Pecher P, Eschen-Lippold L, Tsuda K, Katagiri F, Glazebrook J, Scheel D, Lee J: Activation of the Arabidopsis thaliana mitogen-activated protein kinase MPK11 by the flagellin-derived elicitor peptide, flg22. Mol Plant Microbe Interact. 2012, 25: 471-480.
Aoki K, Ogata Y, Shibata D: Approaches for extracting practical information from gene co-expression networks in plant biology. Plant Cell Physiol. 2007, 48: 381-390.
Wan D, Li R, Zou B, Zhang X, Cong J, Wang R, Xia Y, Li G: Calmodulin-binding protein CBP60g is a positive regulator of both disease resistance and drought tolerance in Arabidopsis. Plant Cell Rep. 2012, 31: 1269-1281.
Bartsch M, Gobbato E, Bednarek P, Debey S, Schultze JL, Bautor J, Parker JE: Salicylic acid-independent ENHANCED DISEASE SUSCEPTIBILITY1 signaling in Arabidopsis immunity and cell death is regulated by the monooxygenase FMO1 and the Nudix hydrolase NUDT7. Plant Cell. 2006, 18: 1038-1051.
Du L, Ali GS, Simons KA, Hou J, Yang T, Reddy ASN, Poovaiah BW: Ca(2+)/calmodulin regulates salicylic-acid-mediated plant immunity. Nature. 2009, 457: 1154-1158.
Doherty CJ, Van Buskirk HA, Myers SJ, Thomashow MF: Roles for Arabidopsis CAMTA transcription factors in cold-regulated gene expression and freezing tolerance. Plant Cell. 2009, 21: 972-984.
Nie H, Zhao C, Wu G, Wu Y, Chen Y, Tang D: SR1, a calmodulin-binding transcription factor, modulates plant defense and ethylene-induced senescence by directly regulating NDR1 and EIN3. Plant Physiol. 2012, 158: 1847-1859.
Yang H, Li Y, Hua J: The C2 domain protein BAP1 negatively regulates defense responses in Arabidopsis. Plant J. 2006, 48: 238-248.
He S, Tan G, Liu Q, Huang K, Ren J, Zhang X, Yu X, Huang P, An C: The LSD1-interacting protein GILP is a LITAF domain protein that negatively regulates hypersensitive cell death in Arabidopsis. PLoS One. 2011, 6: e18750.
Benjamins R, Ampudia CSG, Hooykaas PJJ, Offringa R: PINOID-mediated signaling involves calcium-binding proteins. Plant Physiol. 2003, 132: 1623-1630.
Robert-Seilaniantz A, Grant M, Jones JDG: Hormone crosstalk in plant disease and defense: more than just jasmonate-salicylate antagonism. Annu Rev Phytopathol. 2011, 49: 317-343.
Kanchiswamy CN, Takahashi H, Quadro S, Maffei ME, Bossi S, Bertea C, Zebelo SA, Muroi A, Ishihama N, Yoshioka H, Boland W, Takabayashi J, Endo Y, Sawasaki T, Arimura G: Regulation of Arabidopsis defense responses against Spodoptera littoralis by CPK-mediated calcium signaling. BMC Plant Biol. 2010, 10: 97.
Knoth C, Salus MS, Girke T, Eulgem T: The synthetic elicitor 3,5-dichloroanthranilic acid induces NPR1-dependent and NPR1-independent mechanisms of disease resistance in Arabidopsis. Plant Physiol. 2009, 150: 333-347.
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JYH, Zhang J: Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004, 5: R80.
Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP: Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003, 4: 249-264.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T: Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13: 2498-2504.
Eisen MB, Spellman PT, Brown PO, Botstein D: Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998, 95: 14863-14868.
van Helden J: Regulatory sequence analysis tools. Nucleic Acids Res. 2003, 31: 3593-3596.
Huala E, Dickerman AW, Garcia-Hernandez M, Weems D, Reiser L, LaFond F, Hanley D, Kiphart D, Zhuang M, Huang W, Mueller LA, Bhattacharyya D, Bhaya D, Sobral BW, Beavis W, Meinke DW, Town CD, Somerville C, Rhee SY: The Arabidopsis Information Resource (TAIR): a comprehensive database and web-based information retrieval, analysis, and visualization system for a model plant. Nucleic Acids Res. 2001, 29: 102-105.
Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS: MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009, 37: W202-W208.
Defrance M, Janky R, Sand O, van Helden J: Using RSAT oligo-analysis and dyad-analysis tools to discover regulatory signals in nucleic sequences. Nat Protoc. 2008, 3: 1589-1603.
Acknowledgements
This work was funded by grant IOS-0925375 from the U.S. National Science Foundation to JG. Thanks to Kenichi Tsuda for creating R scripts for statistical analysis of qPCR data and assistance with promoter analysis.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
WT carried out all analyses and wrote the paper. JG provided helpful suggestions and edited the manuscript. Both authors read and approved the final manuscript.
Electronic supplementary material
12870_2012_1149_MOESM1_ESM.docx
Additional file 1: Table S1. Significance of various motifs within promoters of genes clustered with SID2 Motifs are derived from the 10mer oligo found to bind CBP60g and SARD1 in vitro [19]. POBO analyses of a cluster of 11 genes including SID2 identified during pilot co-expression analysis are reported as t-values from two-tailed t-tests. Positive values indicate an enrichment of motifs compared to the genome background and negative values an under-representation with increasing magnitude indicating greater significance. Promoter regions were defined as starting at the transcription start site. (DOCX 13 KB)
12870_2012_1149_MOESM4_ESM.pptx
Additional file 4: Figure S1. CBP60g and SARD1 exert antagonist effects on the expression of phylogenetically related calmodulin-like genes. qRT-PCR measurement of gene expression 24 hpi Pma ES4326 (OD600 =0.01). Data from five biological replicates were merged using a mixed linear model and the mean log2 ratio to Actin2 expression plotted along with the standard error. Asterisks denote a significant differential expression between wildtype and the cbp60g sard1 mutant with p-value ≤ 0.05 from a two-tailed t-test. (PPTX 46 KB)
12870_2012_1149_MOESM5_ESM.pptx
Additional file 5: Figure S2. Distribution of selected motifs in the SID2 regulon. Visualisation of the distribution of GAAATT and CCTN7TCC motifs throughout cluster 2 of experiment #2. Plot created using the feature map function of the RSAT suite of tools (http://rsat.ulb.ac.be/). Red ticks denote CCTN7TCC and blue ticks represent GAAATT motifs, ticks above the promoter line are in the sense orientation and ticks below the line antisense. There is a significant bias of the CCTN7TCC motif towards the 750 bp proximal to the transcription start site (p-value=0.005). (PPTX 77 KB)
12870_2012_1149_MOESM6_ESM.xlsx
Additional file 6: Table S4. POBO analysis of motif enrichment in the promoters of clusters from experiment #1 and experiment #2. POBO analysis of the abundance of assorted motifs in the 1500 bp promoter region of clustered genes. 1000 pseudoclusters of size matched to the given cluster were generated both from within the cluster and the genome background. Significance is recorded as t-values from two-tailed t-tests. Positive values indicate an enrichment of motifs compared to the genome background and negative values an under-representation with increasing magnitude indicating greater significance. Motifs are derived from various sources. (1) The 10mer oligo found to bind CBP60g and SARD1. (2) Known W-box consensus sequences involved in binding WRKY transcription factors. (3) A novel dyad motif identified in a cluster containing SARD1 and SID2. (4) An uncharacterised motif enriched in several clusters containing CBP60g. (5) The binding site for the SID2 repressor EIN3. (6) The core binding site for NAC transcription factors including the negative regulator of SID2, ANA019. (7) The binding site for SR1 (CAMTA3), the negative regulator of EDS1 and other defence signalling components. (XLSX 16 KB)
12870_2012_1149_MOESM7_ESM.pptx
Additional file 7: Figure S3. CBP60g forms a SID2 independent co-expression network in response to abiotic stress. A network was created from the genes co-expressed with CBP60g and SARD1 across 27 selected abiotic stress microarray datasets where CBP60g was induced by stress but not strongly correlated with SID2 expression. Edges represent a Spearman rank correlation coefficient of at least 0.7; the width is proportional to the correlation between two genes. (PPTX 384 KB)
12870_2012_1149_MOESM9_ESM.xlsx
Additional file 9: Table S6. Overview of microarray data used for co-expression analysis. For each experiment the Spearman rank correlation coefficient between SID2 and WRKY28, CBP60g and the two SARD1 probesets is followed by the p-values associated with these correlations. Experiments used for the abiotic stress co-expression network are marked. (XLSX 43 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Truman, W., Glazebrook, J. Co-expression analysis identifies putative targets for CBP60g and SARD1 regulation. BMC Plant Biol 12, 216 (2012). https://doi.org/10.1186/1471-2229-12-216
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2229-12-216