
Abstract
Background
Linkage maps are useful tools for examining both the genetic architecture of quantitative traits and the evolution of reproductive incompatibilities. We describe the generation of two genetic maps using reciprocal interspecific backcross 1 (BC1) mapping populations from crosses between Iris brevicaulis and Iris fulva. These maps were constructed using expressed sequence tag (EST)- derived codominant microsatellite markers. Such a codominant marker system allowed for the ability to link the two reciprocal maps, and compare patterns of transmission ratio distortion observed between the two.
Results
Linkage mapping resulted in markers that coalesced into 21 linkage groups for each of the reciprocal backcross maps, presumably corresponding to the 21 haploid chromosomes of I. brevicaulis and I. fulva. The composite map was 1190.0-cM long, spanned 81% of the I. brevicaulis and I. fulva genomes, and had a mean density of 4.5 cM per locus. Transmission ratio distortion (TRD) was observed in 138 (48.5%) loci distributed in 19 of the 21 LGs in BCIB, BCIF, or both BC1 mapping populations. Of the distorted markers identified, I. fulva alleles were detected at consistently higher-than-expected frequencies in both mapping populations.
Conclusions
The observation that I. fulva alleles are overrepresented in both mapping populations suggests that I. fulva alleles are favored to introgress into I. brevicaulis genetic backgrounds, while I. brevicaulis alleles would tend to be prevented from introgressing into I. fulva. These data are consistent with the previously observed patterns of introgression in natural hybrid zones, where I. fulva alleles have been consistently shown to introgress across species boundaries.
Similar content being viewed by others


Background
The Louisiana Iris (Iridaceae) species complex has long been recognized as a study system for examining the evolutionary dynamics of natural hybridization and introgression [1]. It is now widely considered a model system for studying plant evolutionary/speciation genetics [2]. Four phenotypically diverse species comprise this complex: Iris brevicaulis, Iris hexagona, Iris fulva, and Iris nelsonii. The four species are broadly sympatric throughout the Mississippi River drainage of east-central North America, with the exception of I. nelsonii, which is locally endemic to a single parish in Southern Louisiana. When two or more of the Louisiana Iris species are locally sympatric, hybrid swarms form [e.g. [3, 4]], and this natural hybridization has resulted in the introgression of heterospecific DNA into plants that are phenotypically indistinguishable from the parental species [4–7]. Despite introgressive hybridization occurring in each of the three widely-distributed species, these taxa, for the most part, maintain their phenotypic integrity throughout their ranges, largely due to a number of sequentially acting prezygotic and postzygotic reproductive barriers that serve to reduce the probability of interspecific gene flow [for review see [8]]. Thus, this model system provides evolutionary biologists a unique opportunity to examine the reproductive barriers most important in preventing gene flow between hybridizing taxa, and to evaluate the evolutionary consequences when reproductive barriers are incomplete and natural hybridization takes place. Recent analyses of the I. brevicaulis/I. fulva species pair - using a quantitative qrait locus (QTL) mapping approach - have resolved the genetic architecture for a portion of the factors that limit and/or promote reproductive isolation and introgressive hybridization [9–15].
A study of the genetic architecture of speciation is necessarily a study of the genetics underlying reproductive isolating mechanisms that prevent gene flow between species. Iris fulva and I. brevicaulis have a number of such reproductive barriers that reduce the chance for interspecific gene flow. First, the two species' ranges reflect ecogeographic isolation [see [16] for an explanation], such that locally-allopatric populations are often encountered [4, 5]. Such ecogeographic isolation would result in increased intraspecific mating because a large proportion [but not all, see [17]] of pollinator flight movements occur between closely-spaced flowers [18, 19]. This ecogeographic isolation is likely due to the fact that I. fulva and I. brevicaulis are adapted to divergent microhabitats [20, 21]. Iris fulva is normally found in intermittently flooded, forested bayous and swamps, while I. brevicaulis most often occurs in drier, shaded riparian-typified hardwood forests [3, 20]. As suggested by their habitat associations in nature, Martin et al. [9, 10] found under experimental conditions that I. fulva is more flood-tolerant, while I. brevicaulis is a more drought-resistant species. In this regard, when locally sympatric populations are encountered [e.g. [3, 4, 6, 7, 22]], the microhabitat associations of the two species would be expected to reduce interspecific pollen transfer.
There are additional, divergent, reproductive components that interact to reduce the chance for interspecific gene flow between I. fulva and I. brevicaulis. For example, though the two species must overlap in their flowering times to produce the observed natural hybrid zones, I. fulva begins flowering, on average, one month earlier than I. brevicaulis [3, 23]. Furthermore, in experimental plots planted out into nature, no flowering overlap has been recorded between I. fulva and I. brevicaulis over three separate flowering seasons [[11], Martin et al. unpublished]. Yet, Cruzan and Arnold [23] did indeed record small windows of flowering overlap in naturally-occurring sympatric populations, indicating that this component leads to only partial isolation between these two taxa.
These two Iris species display divergent pollination syndromes as well [12], which results in the attraction of different suites of pollinators [13]. Iris brevicaulis possesses blue flowers with prominent white and yellow nectar guides, stiff erect sepals and petals, and short anthers, and is primarily bumblebee-pollinated [13, 14]. Iris fulva has red flowers with reflexed sepals and petals without nectar guides, protruding anthers, and is primarily hummingbird and butterfly-pollinated [13, 14, 18, 19]. These divergent flowering syndromes result in reduced interspecific foraging bouts between the two species [13]. Furthermore, due to the divergent anther positioning in flowers of the two species, pollen from the two species may be transferred from the anthers to different parts of the pollinators' bodies. Such differential placement reduces even further the chance for interspecific pollen transfer (studies currently underway by Martin et al.). Finally, when interspecific pollen transfer occurs, there is also evidence that conspecific pollen precedence reduces the incidence of F1 hybrid formation [24].
Due to these strong, sequentially acting prezygotic barriers, the formation of F1 hybrids between the two species has been shown to be extremely rare in nature [25]. However, once established as adult plants, F1 hybrids reveal extremely high fitness relative to genotypes of the parental species. These hybrids produce over twice as many asexual growth points in nature, flower at 2-3 times the rate and produce significantly more flowers and fruits than either I. fulva or I. brevicaulis [15]. Thus, despite their rare formation, these extremely fit F1 hybrids can and do backcross with the two pure-species plants, resulting in a number of genotypically diverse hybrid populations throughout the broadly-sympatric species ranges. Indeed, naturally occurring hybrid individuals have been confirmed by both phenotypic and molecular markers [20, 21, 25, 26]. Furthermore, population genetic analyses of hybrid zones have revealed a prominent role for assortative mating, conspecific pollen precedence, and selection in determining the ultimate genetic makeup of late-generation hybrid individuals [23], with adaptive introgression potentially contributing during the formation of natural hybrid zones [reviewed by [8, 27, 28]].
Genetic mapping is a powerful tool to identify the number, location, distribution, effect, and magnitude of the genetic factors underlying species differences, introgressive hybridization, reproductive barriers, and hybrid speciation [e.g. [2, 29–36]]. Using two reciprocal BC1 mapping populations between I. fulva and I. brevicaulis, Bouck et al. [14] produced independent BC1 linkage maps by scoring segregation patterns of dominant Iris retroelement (IRRE) markers. The use of these maps and QTL analyses made possible the determination of the underlying genetic architecture of many of the reproductive barriers described above [habitat isolation: [9, 10]; flowering phenology: [11]; pollinator isolation: [13, 14]; hybrid fitness: [15]]. These analyses have indicated that a complex genetic architecture underlies most barriers examined. In general, many QTLs contributed to the additive genetic variation observed in backcross hybrids, and these additive QTLs also varied with respect to the direction of their effects (i.e. introgressed I. fulva alleles may cause BCIB hybrids to either flower earlier or later, depending on which QTL is examined). Epistatic interactions between otherwise additive QTLs commonly contribute to phenotypic variation. In addition, QTLs have been detected that act epistatically (2 × 2 epistasis), yet do not contribute additive effects [15]. In sum, these findings provide support for the notion that the Iris genome is potentially a mosaic with respect to gene flow [e.g. see [37, 38]], with some regions of the genome being permeable to introgression because the QTLs contained within these regions promote a reduction in reproductive isolation. However, these studies also provide support for the "genic view of speciation" [2, 39, 40], wherein a small number of genes (or genomic regions), may be sufficient to prevent the complete fusion of hybridizing populations, even in the face of extensive gene flow.
The genetic maps developed by Bouck et al. [14] have been useful tools for examining the underlying genetic architecture of reproductive isolation and introgression between I. fulva and I. brevicaulis [9–15]. However, because the markers (i.e. Iris retroelement- IRRE); [14, 41] used to construct the maps were dominantly inherited, there were also some limitations for the QTL analyses. The maps were developed from each of two reciprocal hybrid populations (first-generation backcrosses to I. brevicaulis - hereafter referred to as BCIB, and first-generation backcrosses to I. fulva - hereafter referred to as BCIF), with dominant I. fulva markers segregating in the F1 to produce the BCIB map, and dominant I. brevicaulis markers segregating in the F1 to produce the reciprocal BCIF map. Because of the dominant inheritance patterns, the two maps obtained were unlinked and it is therefore unknown whether or not QTLs detected in each of these separate linkage maps are located on homologous linkage groups. Here, we present two new linkage maps based on expressed sequence tag (EST) - associated microsatellite loci. Given the codominant inheritance of microsatellites, homology of individual markers can be determined, and the two maps developed from the different reciprocal mapping populations can be linked. We report on patterns of transmission ratio distortion (TRD) of these two novel microsatellite maps, and comment as to whether such patterns promote or inhibit introgression of heterospecific alleles. We also note the utility of these new maps for future QTL mapping studies.
Methods
Description of Mapping Populations
Two reciprocal interspecific backcross 1 (BC1) mapping populations, BCIB and BCIF, were produced from crosses between I. brevicaulis genotype IB25 (previously referred to as IB72 by [15, 16, 19–22, 25] and I. fulva genotype IF174 [22]. The I. fulva individual, IF174, was collected from a wild population in Terrebonne Parish, Louisiana, USA, and the I. brevicaulis individual, IB25, was collected from a wild population in St. Martin Parish, Louisiana, USA. Clones from the same individuals (IF174 and IB25) were utilized to make the initial F1 parents of the backcross populations, using IB25 as the seed parent and IF174 as the pollen parent. Two different F1 individuals, designated as F1(2) and F1(3), were used as pollen parents to produce multiple BC1 hybrids. Separate F1 hybrids were used as pollen parents because flowering had ceased in the F1(2) parent prior to the initiation of I. brevicaulis flowering. The F1(2) plant was thus utilized to pollinate flowers from several clones of IF174, while the F1(3) plant was utilized to pollinate several flowers from a number of clones of IB25. Ultimately, several hundred seeds were generated for each reciprocal backcross mapping population. These BC1 hybrid seeds were planted in the greenhouse at the University of Georgia in 1999 and monitored for germination success. Successfully-germinated seeds were transplanted into six-inch azalea pots shortly after germination, and plants have been repotted annually from a single rhizome. The current BCIB population housed at the University of Georgia has 230 BC1 plants, while BCIF consists of 180 BC1 plants. Additional genotypes are located in field plots in Louisiana [described in [9–11, 13, 15]] as well as at Texas State University - San Marcos. A subset of 94 BCIB and 92 BCIF BC1 hybrids from the University of Georgia collection were used in the genetic map construction described herein. From these individuals, genomic DNA was isolated from leaves using a modified CTAB (cetyltrimethylammonium bromide) extraction method.
EST-SSR Marker Genotyping
Microsatellite marker development and genotyping was essentially the same as described by Tang et al. [42, 43] and Tang and Knapp [44]. A total of 1,447 microsatellites were identified from the EST database of I. brevicaulis and I. fulva at repeat number n ≥ 5, and 526 EST-microsatellite markers were developed [45]. These 526 markers were screened for utility, functionality, and length polymorphisms in the two mapping parents, IB25 and IF174. To facilitate multiplex genotyping, the expected lengths of the target amplicons were uniformly distributed in the 100 to 450 bp range, and the forward primers were labeled with one of the three fluorophores 6FAM, HEX, and TAMRA. PCR was performed by using 12 μL of reaction mixture containing 1.0 × PCR buffer, 2.5 mM Mg++, 0.2 mM each of the dNTPs, 5.0 pmol of each primer, 0.5 units of Taq polymerase, and 10 to 15 ng of genomic DNA. 'Touchdown' PCR [46] was used to reduce spurious amplification. The initial denaturation step was performed at 94°C for 1 min, followed by 1 cycle of 94°C for 25 s, 64°C for 25 s, and 72°C for 45 s. The annealing temperature was decreased 1°C per cycle in subsequent cycles until reaching 58°C. Products were subsequently amplified for 33 cycles at 94°C for 20 s, 58°C for 20 s, and 72°C for 45 s with a final extension at 72°C for 20 min.
Amplicon multiplexing was possible because fluorescence labels and allele sizes differed amongst the multiplexed microsatellite markers. Because no allele size information was available for the new microsatellites, we initially multiplexed only three SSR markers (each marker with different fluorescence labels) for parental genotyping. For mapping population genotyping, we were able to multiplex a minimum of eight markers of varying lengths and fluorescence labeling. Each PCR product was diluted 60-100 fold with distilled H2O, and pooled. Samples were prepared for genotyping by combining 0.7 to 1.0 uL of the diluted amplicons with 8 uL diluted GeneScan ROX500, the internal-lane size standard. The diluted ROX500 size standard was prepared by mixing 2 uL of original ROX500 size standard (Applied Biosystems, Foster City, Calif., USA) with 100 uL of 100% Formamide. Samples were heated to 92°C for 5 min, chilled on ice for 5 min, and loaded into an ABI 3700 XL Capillary Sequencer (Applied Biosystems, Foster City, CA) for GeneScan. GeneScan Filter Set D was used for data collection; the emission colors of 6FAM, HEX, TAMRA, and ROX were blue, green, yellow, and red, respectively. SSR allele lengths were scored using GeneMapper (Applied Biosystems, Foster City, CA) or Mapmarker (SoftGenetics LLC, State College, PA).
Genetic Mapping
The genetic maps were constructed using 94 BCIB and 92 BCIF BC1 hybrids. Chi-square tests for segregation distortion were performed for each EST-SSR marker using log-likelihood ratio statistics (G) of G-MENDEL 3.0 [47]. Genetic maps were constructed using Mapmaker 3.0 [48, 49]. The framework maps were constructed at a likelihood odds (LOD) threshold of 7.0 and a maximum recombination frequency threshold of 0.4. Then, we incorporated the unlinked marker loci to the framework maps at LOD scores 5.0 and 3.0. Using the group information from both BCIB and BCIF populations, we assembled 283 of the 285 EST-SSR marker loci into 21 linkage groups (LGs) at LOD threshold ≥ 3.0. Map distances (cM) were calculated using the Kosambi [50] mapping function. For the composite map, the raw genotyping data from both BCIB and BCIF populations was combined for map construction. Of the 285 EST-SSR marker loci genotyped, 222 were genotyped in both BCIB and BCIF, and 63 were genotyped in only one of the BCIB or BCIF populations. For the EST-SSR marker loci mapped in only one population, we used missing data for all of the BC1 hybrids from the other population in the composite map construction.
The inferred genome length was estimated by L + (2tL)/n, as proposed by Fishman et al. [33],

and as proposed by Chakravarti et al. [51], where L is the observed length of the genetic map (cM), n = k - t is the number of marker loci intervals, k i is the number of the framework marker loci on the ith linkage group, and i = 1, 2, ..., t, (t = 21). The proportion of the genome within d cM of a marker locus, assuming a random distribution of framework marker loci, was estimated by 1 - e-2dk/L[51].
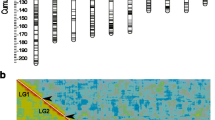
The linkage groups were designated from one to 21 according to the LG lengths in the composite map (Additional File 1: Supplemental Figure S1). A common prefix 'IM' ('Iris microsatellite') was used in naming the microsatellite markers. LG number suffixes were used to identify individual loci produced by multilocus markers, e.g., IM56-1 and IM56-14 are loci on LGs 1 and 14, respectively, amplified by the EST-SSR marker IM56. If duplicated loci were mapped to the same LG, then consecutive letters (A, B, C etc.) were used to identify individual loci within the LG, e.g., IM103-7A and IM103-7B are duplicated loci amplified by the IM103 primer pair and mapped at different positions on LG 7 (Figures 1, 2 and 3).

Transcript genetic linkage maps of I. brevicaulis and I. fulva based on 283 EST-SSR marker loci genotyped in 94 progeny from the backcross mapping population BCIB, and 92 progeny from the backcross mapping population BCIF. BCIB and BCIF were reciprocal backcross mapping populations derived from crosses between I. brevicaulis (IB25) × I. fulva (IF174). The genetic linkage groups were named from 1 to 21 (here groups 1-6) in the order of their genetic map lengths in the composite map (Additional File 1: Supplemental Figure 1). Marker loci showing significant segregation ratio distortion (α ≤ 0.05) in the mapping populations were highlighted with * (overrepresentation of the IF174 alleles) and # (overrepresentation of IB25 alleles).

Linkage groups 7-12. See Figure 1 for details.

Linkage groups 13-21. See Figure 1 for details.
Results
EST Microsatellite Marker Genotyping and Polymorphisms
The 526 EST microsatellite markers were screened for utility, functionality, and length polymorphisms in two mapping parents, IB25 and IF174. Of the 526 primer pairs, 399 (76%) amplified distinct bands in at least one of the parents. Of the 399 functional markers, 72 spanned introns larger than 200 bp, and amplified bands larger than 700 bp, which exceed the size range of the ABI 3700 XL Capillary Sequencer; allele sizes of these markers could not be determined and scored (Additional File 2: Supplemental Table S1). The parental individuals IB25 and IF174 were highly heterozygous, indicating that both individuals are members of outcrossing lineages. Of the 327 SSR markers with alleles scored, 213 (65.1%) were heterozygous in IB25, and 163 (49.8%) were heterozygous in IF174. Further, 275 (84%) were polymorphic between IB25 and IF174 (Additional File 2: Supplemental Table S1), and these markers were useful for the current mapping study. We selected 261 of the polymorphic markers and screened them in the two F1 hybrids, F1(2) and F1(3). Some of the polymorphic markers could not be used in genetic mapping because the markers amplified alleles shared by the two parents in addition to the polymorphic alleles, and both parents transferred the shared alleles to the F1 hybrids. For example, IM58 amplified 189- and 199-bp alleles in IB25, and 180- and 189-bp alleles in IF174; both parents transferred its 189-bp allele to the F1(2) and F1(3) hybrids (with homozygous 189-bp alleles), and it is therefore not possible to identify which parents contributed the 189-bp allele in BC1 hybrids (Additional File 2: Supplemental Table S1). We found that 24 and 29 polymorphic markers were rendered noninformative in the F1(2) and F1(3) hybrids, respectively for this reason. Thus, 237 markers in all were genotyped in the BCIB population, and 232 markers were genotyped in the BCIF population (253 different polymorphic markers in all).
Genetic Maps
Several mapping iterations were performed to produce the final map presented here. At LOD threshold of 7.0 and a maximum recombination frequency threshold of 0.4, the microsatellite markers were assembled into 29, 35 and 26 groups in BCIB, BCIF and composite populations, respectively. At a reduced LOD score of 5.0 (and 3.0), the EST-SSR markers were assembled into 25 (22 at LOD 3.0), 29 (26 at LOD 3.0) and 23 (20 at LOD 3.0) groups in BCIB, BCIF and composite populations, respectively. When the LOD threshold was dropped to 2.5, the marker loci were assembled into 22 LGs in BCIB, 22 LGs in BCIF, and 20 LGs in the composite map. In two cases, the EST-SSR markers from one group in one population were separated into two groups in the other population at LOD threshold ≥ 3.0. Using the group information from both BCIB and BCIF populations, we assembled 283 of the 285 EST-SSR marker loci into 21 linkage groups (LGs) at LOD threshold ≥ 3.0.
A total of 237 polymorphic EST-SSR markers were genotyped in 94 BCIB BC1 hybrids, which produced 258 usable marker loci. With the exception of a single locus IM37, all loci coalesced into 21 LGs, presumably corresponding to the 21 haploid chromosomes in both I. brevicaulis and I. fulva (Figures 1, 2 and 3; Table 1). The map was 1093.6-cM long. The LGs ranged from 2.2 (LG 21) to 99.9 cM (LGs 1 and 2) in length, and had four (LGs 20 and 21) to 23 (LGs 1 and 9) marker loci. The marker densities ranged from 0.7 cM/locus in LG 21 to 8.3 cM/locus in LG 2 with a mean of 4.6 cM/locus for the entire map. Gaps larger than 30.0 cM were observed in LG 2 (45.7 cM), LG 3 (34.3 cM), LG 4 (45.7 cM), LG 10 (32.7 cM), LG 14 (34.3 cM), and LG 15 (45.7 cM) (Figures 1, 2 and 3; Table 1). The inferred total map length ranged from 1414.8 cM [33] to 1419.6 cM [51]; the BCIB map covered 77% of the Louisiana Iris genome. Based on this map, 25.9% of the genome is within 1.0 cM and 95.0% of the genome is within 10.0 cM of a SSR marker locus in the BCIB map.
A total of 232 polymorphic EST-SSR markers were genotyped in 92 BCIF BC1 hybrids, which produced 249 usable marker loci. Except for IM37 and IM518U, all loci coalesced into 21 LGs, again presumably corresponding to the 21 haploid chromosomes in I. brevicaulis and I. fulva (Figures 1, 2 and 3; Table 1). The map was 1181.1-cM long. The LGs ranged from 3.3 (LG 21) to 127.4 cM (LG 1) in length, and marker numbers ranged from two (LG 20) to 23 (LG 6). The marker densities ranged from 1.1 cM/locus in LG 21 to 13.7 cM/locus in LG 13 with a mean of 5.2 cM/locus for the entire map. Gaps larger than 30.0 cM were observed in LG 1(53.2 cM), LG 2 (52.6 cM), LG 3 (47.4 cM), LG 5 (37.1 cM), LG 7 (42.9 cM), LG 10 (42.9 cM), and LG 17 (30.5 cM) (Figures 1, 2 and 3; Table 1). The inferred total map length ranged from 1535.4 cM [33] to 1549.0 cM [51]; the BCIF map covered 77% of the Louisiana Iris genome. Based on this map, 23.9% of the genome is within 1.0 cM and 93.5% of the genome is within 10.0 cM of a SSR marker locus in the BCIF map.
A total of 285 marker loci from 253 EST-SSR markers were genotyped in BCIB, BCIF, or both populations; 222 marker loci were genotyped in both BCIB and BCIF, and 63 were genotyped in only one of the BCIB or BCIF populations. The marker order was roughly the same in the reciprocal mapping populations. For the purposes of graphical display, we combined the raw genotyping data from both BCIB and BCIF populations, and constructed a composite genetic map (Additional File 1: Supplemental Figure S1, Figure S2). Of the 285 marker loci genotyped, 283 marker loci coalesced into 21 LGs (Additional File 1: Supplemental Figure S1). This composite map was 1190.0-cM long. The LGs ranged from 2.7 (LG 21) to 123.4 cM (LG 1) in length, and had four (LGs 20 and 21) to 25 (LGs 1 and 9) marker loci. The marker densities ranged from 0.9 cM/locus in LG 21 to 8.4 cM/locus in LG 13 with a mean of 4.5 cM/locus for the entire map. Gaps larger than 30.0 cM were observed in LG 1(39.9 cM), LG 2 (48.4 cM), LG 3 (40.2 cM), LG 4 (45.5 cM), LG 7 (34.8 cM), LG 10 (37.4 cM), and LG 15 (33.2 cM) (Additional File 1: Supplemental Figure S1; Table 1). The inferred total map length ranged from 1558.7 cM [33] to 1564.1 cM [51]; the composite map spanned 81% of the Louisiana Iris genome. Based on this map, 29.4% of the genome is within 1.0 cM and 96.9% of the genome is within 10.0 cM of a SSR marker locus in the composite map.
Transmission Ratio Distortion
Approximately one-third of the markers in each linkage map revealed significant transmission ratio distortion (TRD - α < 0.05). In the BCIB map, 92 (35.8%) of the 257 mapped marker loci showed significant TRD, while 76 (30.8%) of the 247 mapped marker loci showed significant TRD in the BCIF map (Fig. 4). In both linkage maps, TRD revealed directional bias, with I. fulva alleles being significantly overrepresented. In the BCIB map, 79.1% (72/92) of the distorted markers revealed a significant overrepresentation of introgressed I. fulva alleles (χ2 = 30.87, d.f. = 1, p < 0.001). In the BCIF map, 67.1% (51/76) of the distorted markers also revealed significant overrepresentation of recurrent I. fulva alleles (χ2 = 8.89, d.f. = 1, p = 0.003), at the expense of introgressed I. brevicaulis alleles. Significant transmission ratio distortion was thus observed in 138 loci distributed across 19 of the 21 linkage groups in BCIB, BCIF, or both mapping populations (Figure 4).

The observed frequencies of introgressed heterospecific alleles transmitted from F 1 (2) or F 1 (3) hybrids to backcross progeny in BCIB or BCIF populations. The X-axis indicates the genetic distances (cM) of the LGs in the composite map; the Y-axis indicates the transmission ratio of introgressed heterospecific alleles, the IF174 alleles in the BCIB population and the IB25 alleles in the BCIF population. Frequencies > 0.50 indicate an overrepresentation of heterospecific alleles. Frequencies < 0.50 indicate an overrepresentation of homospecific alleles. The expected frequency is 0.50. Data points above and below the stippled lines indicate significant deviations from 0.50 (α = 0.05).
A visual inspection of the patterns of segregation distortion reveals several regions of "clustering" of distorted markers (i.e. one or more adjacent markers showing significant transmission ratio distortion, Figure 4). The most striking pattern observed was one in which I. fulva alleles were significantly overrepresented across both mapping populations (i.e. LG 1: 2.7-3.7 cM, LG 5: 23.4-43.5 cM, LG 11: 41.5-60 cM, LG 12: 0-24.4 cM, LG 13: 0, 27.4-34.2 cM). Iris fulva alleles were also found to be significantly overrepresented in only one of the two mapping populations for a number of linkage groups (i.e. BCIB: LG 1: 90.7-123.4 cM, LG 3: 0-20.8 cM, LG 4: 38.5-84.0 cM, LG 5: 0-16.9, 70.6-75.5 cM, LG 11: 0.0, 21.9-34.4 cM, LG 12: 35.5-46.3 cM, LG 13: 20.4, 48.5-50.4 cM, LG 17: 0 cM, BCIF: LG 2: 105.2 cM, LG 7: 43.5-67.0 cM, LG 8: 38.4-63.6 cM, LG 10: 10.0-60.0 cM, LG 14: 33.8 cM, LG 16: 0.0 cM). In contrast, clusters of I. brevicaulis alleles were significantly overrepresented in both mapping populations in only two instances (i.e. LG 9: 62.4 cM, LG 20: 0 cM), and overrepresented in only one of the mapping populations in relatively few instances (i.e. BCIB: LG 6: 55.4-68.6 cM, LG 14: 0.0-3.8 cM, LG 19: 0.0-4.3 cM, BCIF: LG 17: 26.6-38.7, LG 18: 0.0-16.7 cM). Only a single region of segregation distortion was discovered in which heterospecific alleles were overrepresented in both mapping populations (LG 1: 63.6-78.1 cM). Strikingly, no regions of segregation distortion were found in which homospecific alleles were significantly overrepresented in both mapping populations (Figure 4).
Discussion
The Transcript Genetic Maps for Iris
We constructed the first sequence-based genetic maps for Iris using codominant (i.e. EST- microsatellite; [45]) markers. Our map construction was based on the same two reciprocal interspecific BC1 populations from crosses between I. brevicaulis and I. fulva utilized to generate the dominant IRRE-based maps described by Bouck et al. [14]. Because of the codominant nature of microsatellite markers, the current maps allow the identification of homologous linkage groups from I. fulva and I. brevicaulis. Thus, it will be possible to determine whether or not QTLs identified in one mapping population likewise influence quantitative traits in the reciprocal mapping population.
In the current map, more than 80% of the EST- microsatellite markers were polymorphic between IB25 and IF174 and were subsequently mapped in one or both of the mapping populations. The maps consisted of 283 marker loci distributed across 21 LGs, which corresponds to the number of chromosomes identified through karyotyping of these species [52]. The combined map had a length of 1190.0 cM, spanning 81% of the I. brevicaulis and I. fulva genome, and calculations of map length and map coverage were similar across both non-integrated maps. Based on the shared marker loci, the homology of the LGs in BCIB and BCIF genetic maps were well-established. The marker order was nearly identical in both reciprocal maps, and little evidence of potential genomic rearrangements was found.
The EST- microsatellite loci were not evenly distributed in either of the linkage maps. Substantial clustering was observed in most of the LGs, with complete co-segregation of some markers being observed in almost all of the 21 LGs, even though all the markers were developed from non-redundant unigenes or uniscripts (Figures 1, 2 and 3; Additional File 2: Supplemental Table S1). Significant marker clustering in the present map may be due to a non-random distribution of genes in the Iris genome. Since the EST-microsatellite loci are gene-based, the clustering of markers might thus reflect gene-rich regions. Another cause of such non-random distributions of markers could be reduced recombination. For example, centromeric regions of the genome usually reveal suppressed recombination [53–56], and regions of high marker clustering could be associated with such regions.
Only 18 of the 253 polymorphic EST-microsatellite markers produced multiple (2-7) marker loci in the mapping populations (Figures 1, 2 and 3; Additional File 2: Supplemental Table S1); this indicated that the vast majority of the markers were highly conserved throughout the Iris genome, and are thus excellent resources for comparative mapping. These 18 multi-locus markers all together resulted in 50 mapped loci in the two maps; 8, 7, 7 and 5 marker loci clustered on the LGs 1, 9, 10 and 7, respectively. We found no apparent syntenic linkage blocks of duplicated EST-SSR marker loci although LG 6 and LG 9, and LG 10 and LG 18 had linkage blocks with two duplicated loci shared (Figures 1, 2 and 3; Additional File 1: Supplemental Figure S1). BLAST indicated that the sequences of these EST-SSR marker belonged to the same gene or pseudogene families.
Implications of Transmission Ratio Distortion
Approximately 1/3 of all microsatellite markers were significantly distorted in each of the reciprocal backcross maps. This level of distortion is commonly observed in interspecific crosses [14, 33, 57, 58]. Since markers distributed across a linkage group are, by definition, not independent observations, the distorted markers were often found to be clustered in specific regions (Figure 4). These regions of transmission ratio distortion reveal a bias towards I. fulva, in that I. fulva alleles are largely overrepresented at the expense of I. brevicaulis alleles. For instance, in the BCIB mapping population, 18 separate regions were identified in which introgressed I. fulva alleles were significantly favored, while in the BCIF mapping population, recurrent I. fulva alleles were significantly favored in 12 genomic regions (see results and Figure 4). In contrast, I. brevicaulis alleles were significantly overrepresented in only five locations in the BCIB mapping population, and only five locations in the BCIF mapping population. This transmission ratio bias towards I. fulva alleles was significant or nearly so in both mapping populations (BCIB: χ2 = 7.35, P = 0.007, d.f. = 1; BCIF: χ2 = 7.35, P = 0.089, d.f. = 1). Thus, it appears that some causal factor(s) underlie this effect. Whatever the mechanism(s) involved, given that these two species hybridize in nature, this asymmetry in gene flow could have important implications for introgressive hybridization. Namely, we would expect that for a majority of the regions revealing transmission ratio distortion, I. fulva alleles might be favored to introgress into a predominately I. brevicaulis species-background, while the introgression of I. brevicaulis alleles into I. fulva would be retarded. Consistent with this prediction, asymmetrical isolation has been observed in natural hybrid zones between I. brevicaulis and I. fulva, with I. fulva, I. fulva-like hybrids, I. brevicaulis and I. brevicaulis-like hybrids all revealing extraordinarily high probabilities of being sired by I. fulva-like genotypes [23].
A number of biological processes may result in transmission ratio distortion in mapping populations. Due to the nature of our crossing design, in which F1 hybrids were backcrossed to their original parents, inbreeding depression could cause some instances of transmission ratio distortion. Both original parents were wild-collected, presumably outcrossed, individuals. The high levels of heterozygosity observed in the present analysis and in previous studies [14] corroborate this conclusion. Both parents could thus be carrying lethal or semi-lethal recessive alleles in a heterozygous state. In order for inbreeding depression to manifest as significant transmission ratio distortion, a deleterious allele from the recurrent parent must first be passed on to the F1 parent. Then, in producing a backcross individual, the F1 must pass on that allele to the offspring, and the recurrent parent must again provide the deleterious allele as well. It is an increase in these homozygous semi-lethal/lethal recessive homozygotes in a mapping population that can ultimately result in transmission ratio distortion. However, such inbreeding depression will only result in introgressed heterospecific alleles being overrepresented, and cannot explain the overrepresentation of recurrent homospecific alleles. In the BCIB mapping population, introgressed I. fulva alleles tend to be favored, suggesting that inbreeding depression could play a causal role in much of the observed transmission ratio distortion patterns in this mapping population. However, an examination of transmission ratio distortion patterns in the reciprocal BCIF mapping population indicates that this is likely not the case for many of the distorted regions identified. Were inbreeding depression causing overrepresentation of I. fulva alleles in the BCIB mapping population, the same mechanism would not cause such distortion in the BCIF mapping population. However, for six significantly distorted regions in the BCIB map (located on LGs 1, 5, 11, 12, and 13; see results and figure 4), I. fulva alleles were also significantly overrepresented in the reciprocal BCIF map. This suggests that some I. fulva alleles are selectively favored independent of the genetic background. In contrast, I. fulva alleles were overrepresented on LG 1 in the BCIB map, but underrepresented in the BCIF map (LG 1: 63.6-78.1 cM, Figure 4). This suggests selection for hybridity in this region. Neither of these patterns, where regions of transmission ratio distortion are corellated across both reciprocal maps, are consistent with the expected effects from inbreeding depression.
In other mapping studies, negative interactions between heterospecific nuclear genes have been implicated as the primary causal factor of transmission ratio distortion [14, 33, 59, 60]. Interestingly, the present study reveals little evidence supporting this hypothesis. Not a single instance was observed in which introgressed alleles were underrepresented in both populations, indicating that "hybridity" was not universally disfavored across different genetic backgrounds. This may be due largely to the fact that genes conferring postzygotic isolation act mostly in a recessive fashion [reviewed by [61, 62]], and loci that could potentially confer hybrid inviability are masked by the recurrent parent's alleles in backcross mapping populations. Cytonuclear incompatibilities can also cause transmission ratio distortion if introgressed nuclear alleles are incompatible with the cytoplasmic genome. Since the original F1 parent contains an I. brevicaulis cytoplasm, any cytonuclear incompatibilities that manifest as transmission ratio distortion should result in an under-representation of I. fulva alleles. Since the opposite was generally observed in this study (and in both maps), are likely not the primary cause of transmission ratio distortion, though in some cases they cannot be ruled out.
In fact, it seems likely that the general overrepresentation of I. fulva alleles in both mapping populations is likely not due to inbreeding depression or uniform selection against hybrid genotypes. Rather, a large number of I. fulva alleles appear to be selected for over the I. brevicaulis allelic counterparts, regardless of the genetic background. This overrepresentation of I. fulva alleles could result from a number of factors, including 1) meiotic anomalies in the F1, 2) inviabilities of certain gametophytes, 3) pollen competition among the F1 hybrid (i.e. the pollen parent in these crosses) gametes, leading to differential fertilization success, 4) differential survival among the resultant backcross hybrid zygotes, 5) differential germination success of the backcross hybrid seeds, and/or 6) differential long-term survival among the resultant hybrid plants.
All of the above factors may play at least some role in promoting segregation ratio distortion. For example, there is evidence suggesting that competition among the F1 pollen grains and differential fertilization success together play the most important role in causing the observed overrepresentation of I. fulva alleles. Iris fulva pollen is much more successful at producing F1 seeds than either I. brevicaulis or the related Iris hexagona [23, 24, 63–65]. This is partially attributed to the fact that I. fulva pollen tubes travel at much faster rates than either I. brevicaulis or I. hexagona pollen tubes [24, 63, 64]. Furthermore, I. fulva acts as a very restrictive seed parent, such that when reciprocal, competitive crosses are compared, significantly fewer F1 progeny are formed in I. fulva fruits relative to both I. brevicaulis fruits [23, 24] and I. hexagona fruits [63–66]. Thus, both pollen tube competition and the increased selectivity of conspecific pollen by I. fulva have been implicated in promoting asymmetric isolation, which may manifest as segregation ratio distortion favoring I. fulva alleles. Likewise, Cruzan and Arnold [23] also detected differential seed abortion which contributed to this same pattern of asymmetric introgression from I. fulva into I. brevicaulis in a natural hybrid zone.
Since plants were genotyped nine years after they were initially planted in the greenhouse, differential survival among the resultant "adult" hybrid plants could have contributed to the transmission ratio distortion as well. Indeed, Martin et al. [9], using the same mapping population as the current study, found significant differences between the BCIB and BCIF hybrids in survivorship rates after six years under greenhouse conditions. The mortality rate of all BCIF hybrids (25.1%) was roughly twice that of BCIB hybrids (12.3%). Thus, this "adult mortality" could result in significant transmission ratio distortion, and may in fact be responsible for some of the distorted regions in the present study. However, it is quite clear that I. fulva genotypes were largely selected against [9], which cannot account for the fact that I. fulva alleles were generally found to be favored in the present study.
Conclusions
Transmission ratio distortion in plants can be caused by any number of post-pollination factors that favor certain hybrid genotypes that act prior to the point at which the mapping populations are assayed. Since reproductive barriers act in a sequential order, early-acting barriers (such as those that cause transmission ratio distortion) have the potential to be more effective at restricting gene flow than later acting barriers, even if the absolute strength of the barriers is the same [8, 61, 67–69]. As already mentioned, natural Louisiana Iris hybrid zones reveal strong asymmetries with respect to gene flow, with I. fulva alleles being much more likely to introgress into I. brevicaulis than the reverse [23]. All of our experimental data, the present data set included, suggest that this is likely due mainly to the presence of early-acting barriers. To test this hypothesis, we will soon be re-analyzing the genetic architecture of all of the previously-analyzed components of pre- and post-zygotic reproductive isolation [9–15] using the maps described in the current study. We will thus be able to test whether or not QTLs underlying the same phenotypes occur on the same or different linkage groups in I. fulva and I. brevicaulis. These QTLs (specifically the markers closely linked to those QTLs) will then serve as important testable hypotheses that will allow us to determine what specific regions of the genome (underlying which type of QTLs) are involved in introgression between I. fulva and I. brevicaulis in natural hybrid zones.
References
Anderson E: Introgressive Hybridization. John Wiley and Sons: New York; 1949.
Lexer C, Widmer A: The genic view of plant speciation: recent progress and emerging questions. Phil Trans R Soc B. 2008, 363: 3023-3036. 10.1098/rstb.2008.0078.
Viosca P: The irises of southeastern Louisiana - a taxonomic and ecological interpretation. Bulletin of the Am Iris Soc. 1935, 57: 3-56.
Arnold ML, Bennett BD, Zimmer EA: Natural hybridization between Iris fulva and I. hexagona: pattern of ribosomal DNA variation. Evolution. 1990, 44: 1512-1521. 10.2307/2409333.
Arnold ML, Buckner CM, Robinson JJ: Pollen mediated introgression and hybrid speciation in Louisiana irises. Proc Nat Acad Sci. 1991, 88: 1398-1402. 10.1073/pnas.88.4.1398.
Arnold ML, Robinson JJ, Buckner CM, Bennett BD: Pollen dispersal and interspecific gene flow in Louisiana irises. Heredity. 1992, 68: 399-404.
Arnold ML: Iris nelsonii (Iridaceae): origin and genetic composition of a homoploid hybrid species. Am J Bot. 1993, 80: 577-583. 10.2307/2445375.
Arnold ML: Evolution Through Genetic Exchange. Oxford University Press: Oxford; 2006.
Martin NH, Bouck AC, Arnold ML: Loci affecting long-term hybrid survivorship in Louisiana Iris es: implications for reproductive isolation and introgression. Evolution. 2005, 59: 2116-2124.
Martin NH, Bouck AC, Arnold ML: Detecting adaptive trait introgression between Iris fulva and I. brevicaulis in highly selective field conditions. Genetics. 2006, 172: 2481-2489. 10.1534/genetics.105.053538.
Martin NH, Bouck AC, Arnold ML: The genetic architecture of reproductive isolation in Louisiana Iris es: flowering phenology. Genetics. 2007, 175: 1803-1812. 10.1534/genetics.106.068338.
Bouck AC, Wessler SR, Arnold ML: QTL analysis of floral traits in Louisiana Iris hybrids. Evolution. 2007, 61: 2308-2319. 10.1111/j.1558-5646.2007.00214.x.
Martin NH, Sapir Y, Arnold ML: The genetic architecture of reproductive isolation in Louisiana Irises: Pollination syndromes and pollinator preferences. Evolution. 2008, 62: 740-752. 10.1111/j.1558-5646.2008.00342.x.
Bouck AC, Peeler R, Arnold ML, Wessler SR: Genetic mapping of species boundaries in Louisiana Iris es using IRRE retrotransposon display markers. Genetics. 2005, 171: 1289-1303. 10.1534/genetics.105.044552.
Taylor SJ, Arnold ML, Martin NH: The genetic architecture of reproductive isolation in Louisiana Irises: hybrid fitness in nature. Evolution. 2009, 63: 2581-2594. 10.1111/j.1558-5646.2009.00742.x.
Ramsey J, Bradshaw HD, Schemske DW: Components of reproductive isolation between the Monkeyflowers Mimulus lewisii and M. cardinalis (Phrymaceae). Evolution. 2003, 57: 1520-1534.
Cornman RS, Burke JM, Wesselingh RA, Arnold ML: Contrasting genetic structure of adults and progeny in a Louisiana Iris hybrid population. Evolution. 2004, 58: 2669-2681.
Emms SK, Arnold ML: Site-to-site differences in pollinator visitation patterns in a Louisiana Iris hybrid zone. Oikos. 2000, 91: 568-578. 10.1034/j.1600-0706.2000.910319.x.
Wesselingh RA, Arnold ML: Pollinator behavior and the evolution of Louisiana Iris hybrid zones. J Evol Biol. 2000, 13: 171-180. 10.1046/j.1420-9101.2000.00153.x.
Cruzan MB, Arnold ML: Ecological and genetic associations in an Iris hybrid zone. Evolution. 1993, 47: 1432-1445. 10.2307/2410158.
Johnston JA, Wesselingh RA, Bouck AC, Donovan LA, Arnold ML: Intimately linked or hardly speaking? The relationship between genotype and environmental gradients in a Louisiana Iris hybrid population. Mol Ecol. 2001, 10: 673-681. 10.1046/j.1365-294x.2001.01217.x.
Riley HP: A character analysis of colonies of Iris fulva, Iris hexagona var. giganticaerulea and natural hybrids. Am J Bot. 1938, 25: 727-738. 10.2307/2436599.
Cruzan MB, Arnold ML: Assortative mating and natural-selection in an Iris hybrid zone. Evolution. 1994, 48: 1946-1958. 10.2307/2410519.
Emms SK, Hodges SA, Arnold ML: Pollen-tube competition, siring success, and consistent asymmetric hybridization in Louisiana Irises. Evolution. 1996, 50: 2201-2206. 10.2307/2410691.
Arnold ML: Anderson's paradigm: Louisiana Iris es and the study of evolutionary phenomena. Mol Ecol. 2000, 9: 1687-1698. 10.1046/j.1365-294x.2000.01090.x.
Johnston JA, Arnold ML, Donovan LA: High hybrid fitness at seed and seedling life history stages in Louisiana Iris es. J Ecol. 2003, 91: 438-446. 10.1046/j.1365-2745.2003.00774.x.
Arnold ML: Natural Hybridization and Evolution. 1997, Oxford University Press. Oxford
Arnold ML, Bouck AC, Cornman RS: Verne Grant and Louisiana Irises: Is there anything new under the sun?. New Phytol. 2004, 161: 143-149. 10.1046/j.1469-8137.2003.00856.x.
Tanksley SD: Mapping polygenes. Ann Rev Genet. 1993, 27: 205-233. 10.1146/annurev.ge.27.120193.001225.
Rieseberg LH, Linder CR, Seiler GJ: Chromosomal and genic barriers to introgression in Helianthus. Genetics. 1995, 141: 1163-1171.
Rieseberg LH, Raymond O, Rosenthal DM, Lai Z, Livingstone K, Nakazato T, Durphy JL, Schwarzbach AE, Donovan LA, Lexer C: Major ecological transitions in wild sunflowers facilitated by hybridization. Science. 2003, 301: 1211-1216. 10.1126/science.1086949.
Rieseberg LH: Hybrid origins of plant species. Annu Rev Ecol Syst. 1997, 28: 359-389. 10.1146/annurev.ecolsys.28.1.359.
Fishman L, Kelly AJ, Morgan E, Willis JH: A genetic map in the Mimulus guttatus species complex reveals transmission ratio distortion due to heterospecific interactions. Genetics. 2001, 159: 1701-1716.
Lai Z, Nakazato T, Salmaso M, Burke J, Tang S, Knapp SJ, Rieseberg L: Extensive chromosomal repatterning and the evolution of sterility barriers in hybrid sunflower species. Genetics. 2005, 171: 291-303. 10.1534/genetics.105.042242.
Lexer C, Rosenthal DM, Raymond O, Donovan LA, Rieseberg LH: Genetics of species differences in the wild annual sunflowers, Helianthus annuus and H. petiolaris. Genetics. 2005, 169: 2225-2239. 10.1534/genetics.104.031195.
Yatabe Y, Kane NC, Scotti-Saintagne C, Rieseberg LH: Rampant gene exchange across a strong reproductive barrier between the annual sunflowers, Helianthus annuus and H. petiolaris. Genetics. 2007, 175: 1883-1893. 10.1534/genetics.106.064469.
Besansky NJ, Krzywinski J, Lehmann T, Simard F, Kern M, Mukabayire O, Fontenille D, Tourél Y, Sagnon N'F: Semipermeable species boundaries between Anopheles gambiae and Anopheles arabiensis: evidence from multilocus DNA sequence variation. Proc Nat Acad Sci USA. 2003, 100: 10818-10823. 10.1073/pnas.1434337100.
Machado CA, Kliman RM, Markert JA, Hey J: The causes of phylogenetic conflict in a classic Drosophila species group. Proc Roy Soc London - B Biol Sci. 2003, 270: 1193-1202. 10.1098/rspb.2003.2333.
Wu C-I: The genic view of the process of speciation. J Evol Biol. 2001, 14: 851-865. 10.1046/j.1420-9101.2001.00335.x.
Wu C-I: Genes and speciation. J Evol Biol. 2001, 14: 889-891. 10.1046/j.1420-9101.2001.00351.x.
Kentner EK, Arnold ML, Wessler SR: Characterization of high copy number retrotransposons from the large genomes of the Louisiana Iris species and their use as molecular markers. Genetics. 2003, 164: 685-697.
Tang S, Yu JK, Slabaugh MB, Shintani DK, Knapp SJ: Simple sequence repeat map of the sunflower genome. Theor Appl Genet. 2002, 105: 1124-1136. 10.1007/s00122-002-0989-y.
Tang S, Kishore VK, Knapp SJ: PCR-multiplexes for a genome-wide framework of simple sequence repeat marker loci in cultivated sunflower. Theor Appl Genet. 2003, 107: 6-19.
Tang S, Knapp SJ: Microsatellites uncover extraordinary diversity in Native American land races and wild populations of cultivated sunflower. Theor Appl Genet. 2003, 106: 990-1003.
Tang S, Okashah RA, Cordonnier-Pratt MM, Pratt LH, Johnson VE, Taylor CA, Knapp SJ, Arnold ML: EST and EST-SSR marker resources for Iris. BMC Plant Biol. 2009, 9: 72. 10.1186/1471-2229-9-72.
Don RH, Cox PT, Wainwright BJ, Baker K, Mattick JS: 'Touchdown' PCR to circumvent spurious priming during gene amplification. Nucleic Acids Res. 1991, 19: 4008. 10.1093/nar/19.14.4008.
Holloway JL, Knapp SJ: G-MENDEL 3.0 user's guide. Oregon State University, Corvallis, OR; 1993.
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Newburg L: MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics. 1987, 1: 174-181. 10.1016/0888-7543(87)90010-3.
Lincoln SE, Daly MJ, Lander ES: Constructing genetic maps with MAPMAKER/EXP 3.0 Manual. Whitehead Institute, Cambridge, MA; 1992.
Kosambi DD: The estimation of map distance from recombination values. Ann Eugen. 1944, 12: 172-175.
Chakravarti A, Lasher LK, Reefer JE: A maximum-likelihood method for estimating genome length using genetic-linkage data. Genetics. 1991, 128: 175-182.
Randolph LF, Mitra J, Nelson IS: Cytotaxonomic studies of Louisiana Irises. Bot Gazette. 1961, 123: 125-133. 10.1086/336137.
Choo KHA: Why is the centromere so cold?. Genome Res. 1998, 8: 81-82.
Geraldes A, Ferrand N, Nachman MW: Contrasting patterns of introgression at X-linked loci across the hybrid zone between subspecies of the European Rabbit (Oryctolagus cuniculus). Genetics. 2006, 173: 919-933. 10.1534/genetics.105.054106.
Borodin PM, Karamysheva TV, Belonogova NM, Torgasheva AA, Rubtsov NB, Searle JB: Recombination map of the common shrew, Sorex araneus (Eulipotyphla, Mammalia). Genetics. 2008, 178: 621-632. 10.1534/genetics.107.079665.
Carneiro M, Ferrand N, Nachman MW: Recombination and speciation: loci near centromeres are more differentiated than loci near telomeres between subspecies of the European Rabbit (Oryctolagus cuniculus). Genetics. 2009, 181: 593-606. 10.1534/genetics.108.096826.
Myburg AA, Vogl C, Griffin AR, Sederoff RR, Whetten RW: Genetics of postzygotic isolation in Eucalyptus: whole-genome analysis of barriers to introgression in a wide interspecific cross of Eucalyptus grandis and E. globules. Genetics. 2004, 166: 1405-1418. 10.1534/genetics.166.3.1405.
Niehuis O, Judson AK, Gadau J: Cytonuclear genic incompatibilities cause increased mortality in male F2 hybrids of Nasonia giraulti and N. vitripennis. Genetics. 2008, 178: 413-426. 10.1534/genetics.107.080523.
Hall MC, Willis JH: Transmission ratio distortion in intraspecific hybrids of Mimulus guttatus: implications for genomic divergence. Genetics. 2005, 170: 375-386. 10.1534/genetics.104.038653.
Moyle LC, Graham EB: Genome-wide associations between hybrid sterility QTL and marker transmission ratio distortion. Mol Biol and Evol. 2006, 23: 973-980. 10.1093/molbev/msj112.
Coyne J, Orr HA: Speciation. Sinauer Asociates, Sunderland, MA; 2004.
Orr HA: The genetic basis of reproductive isolation: insights from Drosophila. Proc Nat Acad Sci. 2005, 102: 6522-6526. 10.1073/pnas.0501893102.
Carney SE, Cruzan MB, Arnold ML: Reproductive interactions between hybridizing irises: analyses of pollen tube growth and fertilization success. Am J Bot. 1994, 81: 1169-1175. 10.2307/2445479.
Carney SE, Hodges SA, Arnold ML: Effects of pollen-tube growth and ovule position on hybridization in the Louisiana Irises. Evolution. 1996, 50: 1871-1878. 10.2307/2410745.
Hodges SA, Burke JM, Arnold ML: Natural formation of Iris hybrids: experimental evidence on the establishment of hybrid zones. Evolution. 1996, 50: 2504-2509. 10.2307/2410717.
Arnold ML, Hamrick JL, Bennett BD: Interspecific pollen competition and reproductive isolation in Iris. J Hered. 1993, 84: 13-16.
Schemske DW: Understanding the origin of species. Evolution. 2000, 54: 1069-1073.
Husband BC, Sabara HA: Reproductive isolation between autotetraploids and their diploid progenitors in fireweed, Chamerion angustifolium (Onagraceae). New Phytol. 2004, 161: 703-713. 10.1046/j.1469-8137.2004.00998.x.
Martin NH, Willis JH: Ecological divergence associated with mating system causes nearly complete reproductive isolation between sympatric Mimulus species. Evolution. 2007, 61: 68-82. 10.1111/j.1558-5646.2007.00006.x.
Acknowledgements
Our research was supported by grants from the National Science Foundation (DEB-0345123 to MLA and DEB-0816905 to NHM) from the American Iris Society Foundation, the Society for Louisiana Iris, and the Research Enhancement Program at Texas State University (to NHM) and from the Office of the Vice President for Research, University of Georgia to SJK. and MLA.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
ST developed the cDNA libraries, produced the ESTs, developed and screened the DNA markers, performed molecular and statistical genetic analyses, and assisted with drafting the manuscript. RAO assisted with the molecular analyses. SJK, MLA, and NHM designed and coordinated the study, and assisted with statistical analyses drafting the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12870_2009_542_MOESM1_ESM.PPT
Additional file 1: Composite linkage map. Composite genetic linkage map of I. brevicaulis and I. fulva based on 283 EST-SSR marker loci genotyped in 94 progeny of backcross mapping population BCIB, and 92 progeny of backcross mapping population BCIF. The genetic linkage groups were labeled from 1 to 21 in the order of their genetic map lengths in cM. (PPT 508 KB)
12870_2009_542_MOESM2_ESM.XLS
Additional file 2: EST genotyping data. Polymorphisms and map locations of the 526 EST-SSR markers genotyped in the mapping parents IB25 and IF174. (XLS 214 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Tang, S., Okashah, R.A., Knapp, S.J. et al. Transmission ratio distortion results in asymmetric introgression in Louisiana Iris. BMC Plant Biol 10, 48 (2010). https://doi.org/10.1186/1471-2229-10-48
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2229-10-48