
Abstract
Central to the pathogenesis of Alzheimer's disease (AD) is the conversion of normal, soluble β-amyloid (sAβ) to oligomeric, fibrillar Aβ. This process of conformational conversion can be influenced by interactions with other proteins that can stabilize the disease-associated state; these proteins have been termed 'pathological chaperones'. In a number of AD models, intervention that block soluble Aβ aggregation, including β-sheet breakers, and compounds that block interactions with pathological chaperones, have been shown to be highly effective. When combined with early pathology detection, these therapeutic strategies hold great promise as effective and relatively toxicity free methods of preventing AD related pathology.
Similar content being viewed by others
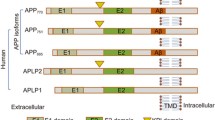
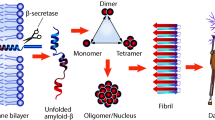
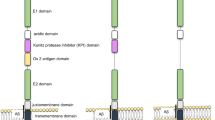
Introduction
Formation of β-amyloid (Aβ) fibrils and deposition of Aβ in the brain parenchyma, or in the brain's vessels, occurs in the setting of increased Aβ peptide concentrations [1, 2]. Initially, conditions do not favor aggregation of fibrils, but once a critical nucleus has been formed, conditions change to favor aggregation in an exponential manner. Any available monomer becomes instantly entrapped in an aggregate or fibril. Several compounds – for example, Congo red [3], anthracycline [4], rifampicin [5], anionic sulphonates [6], and melatonin [7] – can interact with Aβ and prevent its aggregation into oligomers and fibrils in vitro, reducing toxicity. These oligomeric structures have been associated with the greatest toxicity [8]. Several Aβ homologous peptides have been identified that have amino acid substitutions using residues such as proline and can bind to Aβ oligomers and fibril structures, leading to disruption of the β-sheet conformation [9–12]. These peptides have been termed β-sheet breakers. An advantage of such compounds, in comparison to other putative therapeutic approaches for AD, such as vaccination, is that they specifically target the abnormal conformation of Aβ and will not disrupt any possible normal function of the soluble Aβ peptide. Several modifications have been used to extend the serum half-life and increase the blood-brain barrier (BBB) permeability of these β-sheet breakers. Permanne et al. [13], using a BBB permeable pentapeptide (iAβ5), were able to demonstrate a reduction of Aβ load in AD Tg mice compared to an age-matched control group. Of interest, a similar concept of β-sheet breakers appears to be applicable to other protein conformational disorders caused by prions [14].
Pathological chaperone inhibitors
Aβ homologous peptides can spontaneously aggregate and form fibrils in vitro; however, in vivo this process appears more dependant on Aβ pathological chaperones. This group of proteins actively promotes conformational transformation by increasing the β-sheet content of these disease-specific proteins, stabilizing their abnormal structure [15–17]. Examples in Alzheimer's disease (AD) include apolipoprotein E (apoE), especially its E4 isoform [16, 18], α1-antichymotrypsin [19], and C1q complement factor [20, 21]. In their presence, the formation of Aβ fibrils in a solution of soluble Aβ monomers becomes much more efficient [16, 19]. These 'pathological chaperone' proteins have been found histologically and biochemically in association with fibrillar Aβ deposits [15, 22–24], but not in preamyloid aggregates, which are not associated with neuronal toxicity [25–27]. Inheritance of the apoE4 isoform has been identified as the major genetic risk factor for sporadic, late-onset AD [28] and correlates with an earlier age of onset and greater Aβ deposition in an allele-dose-dependent manner [28, 29]. On the other hand, epidemiological data suggest that inheritance of the E2 allele has a protective effect. In vitro all apoE isoforms can propagate the β-sheet content of Aβ peptides promoting fibril formation [16, 23], with apoE4 being the most efficient [16]. The critical dependence of Aβ deposition in plaques on the presence of apoE has also been confirmed in AD Tg amyloid precursor protein (APP)V717F/apoE-/- mice, which have a delayed onset of Aβ deposition, a reduced Aβ load, and no fibrillar Aβ deposits compared with APPV717F/apoE+/+ Tg mice. APPV717F/apoE+/- mice demonstrate an intermediate level of pathology [30–33]. Neutralization of the chaperoning effect of apoE would therefore potentially have a mitigating effect on Aβ accumulation. ApoE binds hydrophobically to amino acids 12–28 of Aβ, forming SDS-insoluble complexes [34–36]. Ma et al. [37] have demonstrated that a synthetic peptide homologous to this sequence of Aβ can be used as a competitive inhibitor of the binding of full length Aβ to apoE, resulting in reduced fibril formation in vitro and increased survival of cultured neurons. Several modifications to Aβ12–28, including the replacement of a valine for proline at position 18 (Aβ12–28P), made this peptide non-toxic, non-fibrillogenic, and prevented any potential for co-deposition on existing Aβ plaques. Further modifications included protection of its amino and carboxyl termini, and using D-amino acids resulted in an extended serum half-life (62 ± 18 minutes, mean ± standard deviation. These modifications did not limit its ability to block the apoE-Aβ interaction (K i = 11.37 nM) [38, 39]. Although Aβ12–28P had a limited serum half-life, it was able to cross the BBB, exerting a therapeutically prolonged effect. Treatment of APPK670N/M671L/PS1M146L, and APPK670N/M671L AD Tg mice with Aβ12–28P resulted in a significant reduction of Aβ deposition in brain parenchyma and in brain vessels [39]. Furthermore, treatment with Aβ12–28P prevented memory decline in single APP Tg mice. Measurement of Aβ levels in the brain homogenate revealed a significant reduction in the absolute Aβ level while the concentrations of the soluble Aβ fraction and Aβ oligomers remained stable during treatment [39]. This observation is important in light of a concern regarding the blocking of the apoE-Aβ interaction and that this form of therapeutic interference could destabilize fibrillar Aβ assemblies, and impair apoE-mediated Aβ clearance, potentially increasing brain levels of soluble Aβ, thus favoring the formation of toxic oligomers [8, 40]. During treatment with Aβ12–28P, no signs of toxicity, including systemic amyloidosis or disturbance of vascular integrity leading to cerebral hemorrhages (as observed with some vaccination approaches [39]), were noted. The treatment outcome with Aβ12–28P could not be attributed to an immunization effect since there were no significant changes in the serum titer of anti-Aβ antibodies before and after the treatment of AD Tg animals. Thus, interference with the apoE-Aβ interaction in vivo appears to have a net effect of increasing Aβ clearance and reducing the absolute Aβ level in the brain, producing a cognitive benefit. Further development of ligands inhibiting the apoE-Aβ interaction, potentially making them suitable for clinical application, should be aggressively pursued.
Of interest, Pepys and co-workers [41] have developed a ligand that chelates serum amyloid-P component. Serum amyloid-P component, along with apoE, binds to amyloid fibrils in vivo in amyloid-A primary systemic amyloidosis, and inhibits the normal degradative process of amyloid-A [42]. Both disease model animals and affected humans treated with this compound in phase I/II clinical trials demonstrated reduced amyloid-A deposition [41].
A similar treatment concept is being developed based on inhibiting the interaction between glycosaminoglycans and Aβ fibrils [6]. Neurochem Inc. is conducting clinical trials to determine the efficacy of 3-amino-1-propanesulfonic acid (3APS), a synthetic sulphated glycosaminoglycan mimetic designed to compete with naturally occurring glycosaminoglycans in their binding to Aβ to prevent its deposition http://www.neurochem.com/ResearchActivities.htm. The efficacy of this compound is being tested for AD and cerebral amyloid angiopathy under two different brand names, Alzhemed™ and Cerebril™, respectively. The phase II clinical trial of Alzhemed™ (tramiprosate) demonstrated a good safety profile, with the most frequently reported adverse events being nausea and vomiting, the occurrence of which appeared to be dose related. This multi-centre, double-blind, placebo-controlled study recruited 58 patients who received either tramiprosate (100 mg, 200 mg or 300 mg daily) or placebo. Results of this trial indicated that tramiprosate reduced the Aβ1–42 concentration in the cerebrospinal fluid and stabilized cognitive decline (monitored using the Mini-Mental State Examination (MMSE) and Alzheimer's Disease Assessment Scale-cognitive subscale (ADAS-cog) scales) compared with the control group [43]. Unfortunately, the North American phase III clinical trial of tramiprosate, which finished in late 2007, did not provide clear enough benefit in a large population. The clinical data gathered by this trial are being subjected to secondary statistical analysis. Currently, the prospect of FDA approval for tramiprosate remains unclear.
Another class of compounds characterized by inherent anti-Aβ aggregation properties in vivo comprises the cyclohexanehexol (inositol) stereoisomers. These compounds are naturally occurring sugars characterized by good gastrointestinal absorption, making them attractive candidates for clinical development. When administered orally to AD Tg mice, the cyclohexanehexol stereoisomers scyllo-cyclohexenehexol and epi- cyclohexenehexol reduced several disease features, including impaired cognition, altered synaptic physiology, and cerebral Aβ deposition [44]. Clinical trials of scyllo-cyclohexanehexol are currently entering a multi-center phase II trial under the name ELND005.
The US phase II clinical trial of NC-758 (Cerebril™) for the prevention of hemorrhagic stroke due to cerebral amyloid angiopathy has recently finished. This multi-center, randomized, double-blind and parallel-designed study was conducted at five centers, enrolled 24 cerebral amyloid angiopathy (CAA) patients, and found no safety concerns related to the use of NC-758. Neurochem Inc. has also recently successfully finished a phase II/III clinical trial of 1,3-propanedisulfonate (Fibrillex™), a compound designed to treat amyloidosis A, which is associated with a number of systemic, chronic inflammatory diseases, including rheumatoid arthritis and Crohn's disease. Its mechanism of action is analogous to that of tramiprosate. Based on the results of this trial the company has filed for market approval.
Necessity for early diagnosis and treatment
For maximal benefit, it is likely that all of the interventions discussed above will need to be initiated at the earliest stages of AD pathology [45]. From AD models, therapeutic strategies to prevent and remove amyloid deposition using methods such as vaccination are very effective at preventing cognitive deficits when initiated at the time deposition first starts. However, when the same treatment is administered after deposits are well-established there are no cognitive benefits [46]. In AD patients it is estimated that mild cognitive impairment slowly develops over 10–30 years prior to the presentation of pathology [47, 48]. In the active vaccination study, which targeted early clinical stage AD, patients showed a very modest cognitive benefit [49, 50] despite the fact that autopsies on four study participants indicated that the vaccine had produced a significant reduction in amyloid plaques [51]. Positron emission tomography (PET) imaging of amyloid binding ligands can detect amyloid deposition at early stages [52] and similar methods are being developed for amyloid detection by magnetic resonance imaging (MRI) [53]. These methods will be critical in identifying patients who are at risk of developing clinical AD and who might benefit from preventing Aβ aggregation and deposition therapies.
Conclusion
Numerous therapeutic interventions are currently being developed for AD. It is likely that, in the future, multi-modal therapies will be tailored to individual patients based on their genotype, immune status, and pathological stage of their disease. However, β-sheet breakers and pathological chaperone inhibitors are likely to be among the most effective and safest strategies.
Abbreviations
- Aβ:
-
β-amyloid
- AD:
-
Alzheimer's disease
- apoE:
-
apolipoprotein E
- APP:
-
amyloid precursor protein
- BBB:
-
blood-brain barrier
- CAA:
-
cerebral amyloid angiopathy.
References
Barrow CJ, Yasuda A, Kenny PT, Zagorski MG: Solution conformations and aggregational properties of synthetic amyloid beta-peptides of Alzheimer's disease. Analysis of circular dichroism spectra. J Mol Biol. 1992, 225: 1075-1093. 10.1016/0022-2836(92)90106-T.
Wisniewski T, Ghiso J, Frangione B: Biology of Aβ amyloid in Alzheimer's disease. Neurobiol Dis. 1997, 4: 313-328. 10.1006/nbdi.1997.0147.
Lorenzo A, Yankner BA: Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc Natl Acad Sci USA. 1994, 91: 12243-12247. 10.1073/pnas.91.25.12243.
Merlini G, Ascari E, Amboldi N, Bellotti V, Arbustini E, Perfetti V, Ferrari M, Zorzoli I, Marinone MG, Garini P: Interaction of the anthracycline 4'-iodo-4'-deoxydoxorubicin with amyloid fibrils: inhibition of amyloidogenesis. Proc Natl Acad Sci USA. 1995, 92: 2959-2963. 10.1073/pnas.92.7.2959.
Tomiyama T, Shoji A, Kataoka K, Suwa Y, Asano S, Kaneko H, Endo N: Inhibition of amyloid β protein aggregation and neurotoxicity by rifampicin – Its possible function as a hydroxyl radical scavenger. J Biol Chem. 1996, 271: 6839-6844. 10.1074/jbc.271.12.6839.
Kisilevsky R, Lemieux LJ, Fraser PE, Kong X, Hultin PG, Szarek WA: Arresting amyloidosis in vivo using small-molecule anionic sulphonates or sulphates: implications for Alzheimer's disease. Nat Med. 1995, 1: 143-148. 10.1038/nm0295-143.
Pappolla M, Bozner P, Soto C, Shao H, Robakis NK, Zagorski M, Frangione B, Ghiso J: Inhibition of Alzheimer beta-fibrillogenesis by melatonin. J Biol Chem. 1998, 273: 7185-7188. 10.1074/jbc.273.13.7185.
Walsh DM, Selkoe DJ: Abeta oligomers – a decade of discovery. J Neurochem. 2007, 101: 1172-1184. 10.1111/j.1471-4159.2006.04426.x.
Hilbich C, Kisters-Woike B, Reed J, Masters CL, Beyreuther K: Substitutions of hydrophobic amino acids reduce the amyloidogenicity of Alzheimer's disease βA4 peptides. J Mol Biol. 1992, 228: 1-14. 10.1016/0022-2836(92)90835-8.
Soto C, Kindy MS, Baumann M, Frangione B: Inhibition of Alzheimer's amyloidosis by peptides that prevent β-sheet conformation. Biochem Biophys Res Commun. 1996, 226: 672-680. 10.1006/bbrc.1996.1413.
Soto C, Sigurdsson EM, Morelli L, Kumar A, Castaño EM, Frangione B: β-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: implications for Alzheimer's therapy. Nat Med. 1998, 4: 822-826. 10.1038/nm0798-822.
Sigurdsson EM, Permanne B, Soto C, Wisniewski T, Frangione B: In vivo reversal of amyloid β lesions in rat brain. J Neuropath Exp Neurol. 2000, 59: 11-17.
Permanne B, Adessi C, Saborio GP, Fraga S, Frossard MJ, Van Dorpe J, Dewachter I, Banks WA, Van Leuven F, Soto C: Reduction of amyloid load and cerebral damage in a transgenic mouse model of Alzheimer's disease by treatment with a β-sheet breaker peptide. FASEB J. 2002, 16: 860-862.
Soto C, Kascsak RJ, Saborio GP, Aucouturier P, Wisniewski T, Prelli F, Kascsak R, Mendez E, Harris DA, Ironside J, Tagliavini F, Carp RI, Frangione B: Reversion of prion protein conformational changes by synthetic β-sheet breaker peptides. Lancet. 2000, 355: 192-197. 10.1016/S0140-6736(99)11419-3.
Wisniewski T, Frangione B: Apolipoprotein E: a pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci Lett. 1992, 135: 235-238. 10.1016/0304-3940(92)90444-C.
Wisniewski T, Castaño EM, Golabek AA, Vogel T, Frangione B: Acceleration of Alzheimer's fibril formation by apolipoprotein E in vitro. Am J Pathol. 1994, 145: 1030-1035.
Sadowski M, Wisniewski T: Disease modifying approaches for Alzheimer's pathology. Curr Pharmaceutic Design. 2007, 13: 1943-1954. 10.2174/138161207781039788.
Sanan DA, Weisgraber KH, Russell SJ, Mahley RW, Huang D, Saunders A, Schmechel D, Wisniewski T, Frangione B, Roses AD, Strittmatter WJ: Apolipoprotein E associates with beta amyloid peptide of Alzheimer's disease to form novel monofibrils. Isoform apoE4 associates more efficiently than apoE3. J Clin Invest. 1994, 94: 860-869. 10.1172/JCI117407.
Ma J, Yee A, Brewer HB, Das S, Potter H: Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature. 1994, 372: 92-94. 10.1038/372092a0.
Johnson LV, Leitner WP, Rivest AJ, Staples MK, Radeke MJ, Anderson DH: The Alzheimer's A beta-peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc Natl Acad Sci USA. 2002, 99: 11830-11835. 10.1073/pnas.192203399.
Boyett KW, DiCarlo G, Jantzen PT, Jackson J, O'Leary C, Wilcock D, Morgan D, Gordon MN: Increased fibrillar beta-amyloid in response to human C1q injections into hippocampus and cortex of APP+PS1 transgenic mice. Neurochem Res. 2003, 28: 83-93. 10.1023/A:1021600212829.
Wisniewski T, Lalowski M, Golabek AA, Vogel T, Frangione B: Is Alzheimer's disease an apolipoprotein E amyloidosis?. Lancet. 1995, 345: 956-958. 10.1016/S0140-6736(95)90701-7.
Golabek AA, Soto C, Vogel T, Wisniewski T: The interaction between apolipoprotein E and Alzheimer's amyloid β-peptide is dependent on β-peptide conformation. J Biol Chem. 1996, 271: 10602-10606. 10.1074/jbc.271.52.33623.
Permanne B, Perez C, Soto C, Frangione B, Wisniewski T: Detection of apolipoprotein E dimeric soluble amyloid β complexes in Alzheimer's disease brain supernatants. Biochem Biophys Res Commun. 1997, 240: 715-720. 10.1006/bbrc.1997.7727.
Wisniewski HM, Sadowski M, Jakubowska-Sadowska K, Tarnawski M, Wegiel J: Diffuse, lake-like amyloid-beta deposits in the parvopyramidal layer of the presubiculum in Alzheimer disease. J Neuropathol Exp Neurol. 1998, 57: 674-683. 10.1097/00005072-199807000-00004.
Kida E, Golabek AA, Wisniewski T, Wisniewski KE: Regional differences in apolipoprotein E immunoreactivity in diffuse plaques in Alzheimer's disease brain. Neurosci Lett. 1994, 167: 73-76. 10.1016/0304-3940(94)91030-8.
Gallo G, Wisniewski T, Choi-Miura NH, Ghiso J, Frangione B: Potential role of apolipoprotein-E in fibrillogenesis. Am J Pathol. 1994, 145: 526-530.
Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, Pericak-Vance MA, Goldgaber D, Roses AD: Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA. 1993, 90: 9649-9653. 10.1073/pnas.90.20.9649.
Rebeck GW, Reiter JS, Strickland DK, Hyman BT: Apolipoprotein E in sporadic Alzheimer's disease: allelic variation and receptor interactions. Neuron. 1993, 11: 575-580. 10.1016/0896-6273(93)90070-8.
Bales KR, Verina T, Dodel RC, Du YS, Altstiel L, Bender M, Hyslop P, Johnstone EM, Little SP, Cummins DJ, Piccardo P, Ghetti B, Paul SM: Lack of apolipoprotein E dramatically reduces amyloid β-peptide deposition. Nat Genet. 1997, 17: 263-264. 10.1038/ng1197-263.
Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, Fishman CE, DeLong CA, Piccardo P, Petegnief V, Ghetti B, Paul SM: Apolipoprotein E is essential for amyloid deposition in the APPV717F transgenic mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 1999, 96: 15233-15238. 10.1073/pnas.96.26.15233.
Holtzman DM, Bales KR, Wu S, Bhat P, Parsadanian M, Fagan AM, Chang LK, Sun Y, Paul SM: Expression of human apolipoprotein E reduces amyloid-beta deposition in a mouse model of Alzheimer's disease. J Clin Invest. 1999, 103: R15-R21. 10.1172/JCI6179.
Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, Paul SM: Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2000, 97: 2892-2897. 10.1073/pnas.050004797.
Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD: Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA. 1993, 90: 1977-1981. 10.1073/pnas.90.5.1977.
Wisniewski T, Golabek AA, Matsubara E, Ghiso J, Frangione B: Apolipoprotein E: binding to soluble Alzheimer's beta-amyloid. Biochem Biophys Res Commun. 1993, 192: 359-365. 10.1006/bbrc.1993.1423.
Naslund J, Thyberg J, Tjernberg LO, Wernstedt C, Karlstrom AR, Bogdanovic N, Gandy SE, Lannfelt L, Terenius L, Nordstedt C: Characterization of stable complexes involving apolipoprotein E and the amyloid beta peptide in Alzheimer's disease brain. Neuron. 1995, 15: 219-228. 10.1016/0896-6273(95)90079-9.
Ma J, Brewer BH, Potter H, Brewer HB: Alzheimer Aβ neurotoxicity: promotion by antichymotrypsin, apoE4; inhibition by Aβ-related peptides. Neurobiol Aging. 1996, 17: 773-780. 10.1016/0197-4580(96)00112-1.
Sadowski M, Pankiewicz J, Scholtzova H, Ripellino JA, Li Y, Schmidt SD, Mathews P, Fryer JD, Holtzman DM, Sigurdsson EM, Wisniewski T: Blocking the apolipoprotein E/β-amyloid interaction reduces β-amyloid toxicity and decreases β-amyloid load in transgenic mice. Am J Pathol. 2004, 165: 937-948.
Sadowski M, Pankiewicz J, Scholtzova H, Mehta P, Prelli F, Quartermain D, Wisniewski T: Blocking the apolipoproteinE/Amyloid β interaction reduces the parenchymal and vascular amyloid-β deposition and prevents memory deficit in AD transgenic mice. Proc Natl Acad Sci USA. 2006, 103: 18787-18792. 10.1073/pnas.0604011103.
Kirkitadze MD, Bitan G, Teplow DB: Paradigm shifts in Alzheimer's disease and other neurodegenerative disorders: the emerging role of oligomeric assemblies. J Neurosci Res. 2002, 69: 567-577. 10.1002/jnr.10328.
Pepys MB, Herbert J, Hutchinson WL, Tennent GA, Lachmann HJ, Gallimore JR, Lovat LB, Bartfai T, Alanine A, Hertel C, Hoffmann T, Jakob-Roetne R, Norcross RD, Kemp JA, Yamamura K, Suzuki M, Taylor GW, Murray S, Thompson D, Purvis A, Kolstoe S, Wood SP, Hawkins PN: Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nature. 2002, 417: 254-259. 10.1038/417254a.
Gillmore JD, Hawkins PN, Pepys MB: Amyloidosis: a review of recent diagnostic and therapeutic developments. Br J Haematol. 1997, 99: 245-256. 10.1046/j.1365-2141.1997.303194.x.
Aisen PS, Mehran M, Poole R, Lavoie E, Gervais F, Laurin J, Braind R, Garceau L: Clinical data on Alzhemed after 12 months of treatment in patients with mild to moderate Alzheimer's disease. Neurobiol Aging. 2004, 25: 20-10.1016/S0197-4580(04)80065-4.
McLaurin J, Kierstead ME, Brown ME, Hawkes CA, Lambermon MH, Phinney AL, Darabie AA, Cousins JE, French JE, Lan MF, Chen F, Wong SS, Mount HT, Fraser PE, Westaway D, St George-Hyslop P: Cyclohexanehexol inhibitors of Abeta aggregation prevent and reverse Alzheimer phenotype in a mouse model. Nat Med. 2006, 12: 801-808. 10.1038/nm1423.
DeKosky S: Early intervention is key to successful management of Alzheimer disease. Alzheimer Dis Assoc Disord. 2003, 17 (Suppl 4): S99-104. 10.1097/00002093-200307004-00004.
Asuni A, Boutajangout A, Scholtzova H, Knudsen E, Li Y, Quartermain D, Frangione B, Wisniewski T, Sigurdsson EM: Aβ derivative vaccination in alum adjuvant prevents amyloid deposition and does not cause brain microhemorrhages in Alzheimer's model mice. Eur J Neurosci. 2006, 24: 2530-2542. 10.1111/j.1460-9568.2006.05149.x.
Mondadori CR, Buchmann A, Mustovic H, Schmidt CF, Boesiger P, Nitsch RM, Hock C, Streffer J, Henke K: Enhanced brain activity may precede the diagnosis of Alzheimer's disease by 30 years. Brain. 2006, 129: 2908-2922. 10.1093/brain/awl266.
Schmitt FA, Davis DG, Wekstein DR, Smith CD, Ashford JW, Markesbery WR: "Preclinical" AD revisited: neuropathology of cognitively normal older adults. Neurology. 2000, 55: 370-376.
Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Boada Rovira M, Forette F, Orgogozo JM: Clinical effects of Aβ immunization (AN1792) in patients with AD in an interupted trial. Neurology. 2005, 64: 1553-1562. 10.1212/01.WNL.0000159740.16984.3C.
Hock C, Konietzko U, Straffer JR, Tracy J, Signorell A, Muller-Tillmanns B, Lemke U, Henke K, Moritz E, Garcia E, Axel Wollmar M, Umbricht D, de Quervain DJF, Hofmann M, Maddalena A, Papassotiropoulos A, Nitsch RM: Antibodies against β-amyloid slow cognitive decline in Alzheimer's disease. Neuron. 2003, 38: 547-554. 10.1016/S0896-6273(03)00294-0.
Wisniewski T, Sigurdsson EM: Therapeutic approaches for prion and Alzheimer's diseases. FEBS J. 2007, 274: 3784-3798. 10.1111/j.1742-4658.2007.05919.x.
Klunk WE, Price JC, Mathis CA, Tsopelas ND, Lopresti BJ, Ziolko SK, Bi W, Hoge JA, Cohen AD, Ikonomovic MD, Saxton JA, Snitz BE, Pollen DA, Moonis M, Lippa CF, Swearer JM, Johnson KA, Rentz DM, Fischman AJ, Aizenstein HJ, DeKosky ST: Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. J Neurosci. 2007, 27: 6174-6184. 10.1523/JNEUROSCI.0730-07.2007.
Sigurdsson E, Wadghiri YZ, Mosconi L, Blind JA, Knudsen E, Asuni A, Tsui WH, Sadowski M, Turnbull D, de Leon M, Wisniewski T: A non-toxic ligand for voxel-based MRI analysis of plaques in AD transgenic mice. Neurobiol Aging. 2008, 29: 836-847. 10.1016/j.neurobiolaging.2006.12.018.
Acknowledgements
This manuscript was supported by NIH grants AG15408, AG24847 and AG31221.
This article has been published as part of BMC Neuroscience Volume 9 Supplement 2: 2008 Proceedings of the 8th International Conference on Alzheimer's Disease Drug Discovery The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2202/9?issue=S2.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
Both authors carried out a literature review. TW wrote the draft, which was modified and corrected by MS.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wisniewski, T., Sadowski, M. Preventing β-amyloid fibrillization and deposition: β-sheet breakers and pathological chaperone inhibitors. BMC Neurosci 9 (Suppl 2), S5 (2008). https://doi.org/10.1186/1471-2202-9-S2-S5
Published:
DOI: https://doi.org/10.1186/1471-2202-9-S2-S5