
Abstract
Background
Overexpression and abnormal accumulation of aggregated α-synuclein (αS) have been linked to Parkinson's disease (PD) and other synucleinopathies. αS can misfold and adopt a variety of morphologies but recent studies implicate oligomeric forms as the most cytotoxic species. Both genetic mutations and chronic exposure to neurotoxins increase αS aggregation and intracellular reactive oxygen species (ROS), leading to mitochondrial dysfunction and oxidative damage in PD cell models.
Results
Here we show that curcumin can alleviate αS-induced toxicity, reduce ROS levels and protect cells against apoptosis. We also show that both intracellular overexpression of αS and extracellular addition of oligomeric αS increase ROS which induces apoptosis, suggesting that aggregated αS may induce similar toxic effects whether it is generated intra- or extracellulary.
Conclusions
Since curcumin is a natural food pigment that can cross the blood brain barrier and has widespread medicinal uses, it has potential therapeutic value for treating PD and other neurodegenerative disorders.
Similar content being viewed by others

Background
Parkinson's disease (PD) affects 1% of the population over the age of 65 and is the second most common progressive neurodegenerative disorder after Alzheimer's disease (AD) [1, 2]. The classical symptoms of PD include resting tremor, muscular rigidity and bradykinesia [2, 3] resulting from the progressive loss of dopaminergic neurons in the substantia nigra region of the brain [3, 4]. Intracellular inclusions known as Lewy bodies (LB) and Lewy neurites (LN), composed primarily of insoluble aggregates of ubiquitin and α-synuclein (αS), are neuropathological hallmarks of PD found in many regions of the brain and central nervous system (CNS) [4–6]. Point mutations and multiplication of the αS gene are associated with rare early onset familial forms of the disease, further implicating the role of αS in PD [7–10]. The increased degeneration of dopaminergic neurons in the substantia nigra of PD animal models correlates with increased levels of LBs and LNs in this region of the brain and strongly suggests that overexpression of αS selectively targets dopaminergic neurons [11–13]. While it is unclear why dopaminergic neurons are more susceptible to degeneration by αS, the oxidation of dopamine and exposure to neurotoxins such as rotenone [14, 15] and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) [16–19] generate excessive reactive oxygen species (ROS), promoting mitochondrial complex I dysfunction [15, 20, 21] and depleting glutathione levels [22, 23] ultimately causing acute Parkinsonism in animal and cell models. In addition, overexpression of both wild type (WT) and mutant αS results in formation of cytoplasmic inclusions and degeneration of dopaminergic neurons in mouse and Drosophila models [11–13, 24].
αS is a presynaptic protein expressed at synaptic terminals in the CNS [25, 26]. While αS is a natively unfolded protein, the monomeric form can misfold and aggregate into larger oligomeric and fibrillar forms which are linked to the pathogenesis of PD. Recent studies have implicated small soluble oligomeric and protofibrillar forms of αS as the most neurotoxic species [27–30]. While previous studies provide good evidence for the intracellular toxicity of αS in PD, there is also evidence showing an extracellular component as well [27–29, 31, 32]. Monomeric and oligomeric forms of αS have been detected in blood plasma and cerebrospinal fluid of PD patients [27, 31–33], and exposure to extracellular pre-aggreated αS induces cytotoxicity in primary mesencephalic neuron-glia and human neuroblastoma cell cultures [28, 29, 34, 35].
Since generation of ROS has been correlated with onset of PD, anti-oxidants may have therapeutic value. Curcumin, a polyphenolic compound commonly used as food additives in Asian cuisine, has anti-oxidant properties and suppresses inflammatory responses of brain microglial cells [36–38]. Curcumin was also shown to have protective effects in neurodegenerative disease by either reducing inflammation and oxidative damage in AD [36–39], or by inhibiting protein misfolding and aggregation in Creutzfeld-Jakob disease [40] and PD [41, 42].
Given these numerous beneficial properties, curcumin shows promise as a therapeutic agent for neurodegenerative diseases. We show that curcumin can provide protection against αS-induced cytotoxicity in SH-SY5Y neuroblastoma cells by decreasing cytotoxicity of aggregated αS, reducing intracellular ROS, inhibiting caspase-3 activation and ameliorating signs of apoptosis. We also show that either extracellular addition of oligomeric αS and intracellular overexpression of αS increases generation of intracellular ROS in SH-SY5Y cells and both have similar cytotoxic effects resulting in induced caspase-3 activity and apoptosis.
Results
Curcumin protects SH-SY5Y cells against extracellular αS-induced cytotoxicity
Extracellular incubation of SH-SY5Y cells with oligomeric but not monomeric or fibrillar αS induced significant cytotoxicity (Fig. 1) in agreement with previous studies implicating oligomeric αS as the toxic species [27–30]. While co-incubation of curcumin does not alter the monomeric and pre-formed oligomeric αS morphologies, it does destabilize pre-formed αS fibrils (Fig. 1A, Additional file 1), consistent with previous results [41]. PAGE and AFM size distribution data also confirm that curcumin does not alter the molecular weight or size of the oligomeric αS species (Fig. 1B and 1C). Toxicity assays show that addition of curcumin significantly reduces the αS-induced toxicity induced by pre-formed oligomeric αS while co-incubation of curcumin with pre-formed αS fibrils shows a significant increase in toxicity (Fig. 1D). Co-incubation of curcumin with monomeric αS does not alter cytotoxicity (Fig. 1D) similar to incubation with curcumin and Tris buffer alone (Table 1). Toxicity studies of curcumin alone towards SH-SY5Y cells showed no toxic effects at concentrations below 5 μM (data not shown). Since only oligomeric αS aggregates induced toxicity in SH-SY5Y cells, subsequent experiments were performed with oligomeric αS to determine the protective effects of curcumin against αS.

Oligomeric αS induces cytotoxicity in SH-SY5Y cells. Conformation and cytotoxicity of αS with and without curcumin addition observed by AFM imaging, PAGE and LDH assay. (A) AFM images of αS alone: (i) monomeric αS, (ii) pre-formed oligomeric αS and (iii) fibrillar αS; and αS co-incubated with curcumin for 2-h: (iv) monomeric αS, (v) pre-formed oligomeric αS and (vi) and fibrillar αS. Scale bar = 1 μm. (B) Pre-formed oligomeric αS samples, with and without curcumin were separated on a 10% Tris/Tricine native PAGE gel and analyzed using silver staining. (C) Height distribution of oligomeric αS samples. Particle heights of pre-formed oligomeric αS samples, with (--) and without curcumin (-) were analyzed using AFM and SPIP software. (D) LDH activity of SH-SY5Y cells incubated with different morphologies of αS with or without co-incubation with curcumin. LDH release was expressed as a percentage of the Tris control samples. Data was reported as mean ± SE, n = 4. **p < 0.01 compared with the untreated control samples.
Extracellular addition of αS generates excessive ROS
When oligomeric αS was added extracellularly to SH-SY5Y cells, the intracellular ROS level significantly increased from 100 ± 3.8 (control) to 165.4 ± 11.8 (Fig. 2A), indicating that extracellular αS enhances ROS levels in SH-SY5Y cells. Treatment with curcumin substantially reduces this increase in intracellular ROS levels to 118.5 ± 4.6 (Fig. 2A). Curcumin and Tris buffer alone did not affect ROS levels (Fig. 2A). The ability of curcumin to reduce ROS levels generated by oligomeric αS is consistent with results obtained using anti-oxidants in MPP+ PD models [17, 20, 43] providing further evidence that ROS plays a central role in the selective degeneration of dopaminergic neurons in PD.

Curcumin reduces αS-induced intracellular ROS generation and inhibits caspase-3 activation in SH-SY5Y cells. SH-SY5Y cells were incubated with Tris buffer, oligomeric αS, αS+curcumin and curcumin and the intracellular ROS and caspase-3 activity were determined using cell based assays. (A) Intracellular ROS was determined by DCF fluorescence. DCFH-DA was then added to each well and the plate was incubated at 37°C for an additional 1 hr. The fluorescence intensity of DCF was measured at Ex485 nm and Em535 nm, respectively. The increase in DCF fluorescence was expressed as a percentage of the control and is a direct measurement of intracellular ROS due to the oxidation of DCFH-DA to DCF by intracellular ROS. (B) Caspase-3 activity was determined by the absorbance of pNA substrate. After 24 h of treatment, the cells were detached, lysed and an equal protein loading was added to the 2× reaction buffer with DTT and DEVD-pNA substrate. After 1 hr of incubation at 37°C, the absorbance intensity was measured at 405 nm and the caspase-3 activity was reported percentage of the Tris buffer control. Data was analyzed using one way ANOVA followed by Bonferroni post-hoc test and reported as mean ± SE, n = 4. **p < 0.01, ***p < 0.001, compared with the untreated samples; and ##p < 0.01, ##p < 0.001, compared with αS-treated samples
Curcumin inhibits caspase-3 activity and apoptosis induced by extracellular αS
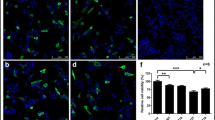
In addition to increasing ROS levels, extracellular incubation of SH-SY5Y cells with oligomeric αS also activates caspase-3 activity and triggers apoptosis. Caspase-3 activity in the oligomeric αS-treated sample increased by 2.4-fold compared to the control, while pre-incubation of curcumin with αS reduced the increase in caspase-3 activation by almost half (Fig. 2B). Addition of curcumin and Tris buffer alone had no significant effects on caspase-3 activity (Fig. 2B). Extracellular addition of oligomeric αS to SH-SY5Y cells also induced apoptosis in the cells as marked by the changes in morphologies of the cell nuclei. While the control SH-SY5Y cells had regular nuclei with uniformly dispersed chromatin and intact cell membrane (Fig. 3A), cells incubated with oligomeric αS showed signs of apoptosis as indicated by condensed nuclei and intense fluorescence staining with Hoechst dye (Fig. 3B). Pre-incubation of curcumin with αS reduced nuclear damage induced by extracellular oligomeric αS (Fig. 3C) while curcumin alone did not affect the nuclear morphology of the cells (Fig. 3D).

Curcumin ameliorates αS-induced morphological changes in SH-SY5Y cells evaluated by fluorescence microscopy. After 48 hr incubation, cells were fixed with 4% paraformaldehyde, stained with Hoechst 33342 (5 mg/mL) and analyzed using a Nikon TE300 fluorescence microscope. Fluorescence micrographs (100× magnification) of the (A) control cells, (B) cells exposed to αS, (C) cells exposed to αS+curcumin and (D) curcumin.
Curcumin reduces ROS and cytotoxicity induced by intracellular overexpression of αS
While curcumin provided substantial protection against extracellular αS-induced toxicity, PD pathology includes intracellular aggregation and accumulation of αS. We evaluated whether extracellularly added curcumin can also protect against intracellularly induced αS toxicity by overexpressing αS in SH-SY5Y cells by transient transfection with a WTsynEGFP gene. The αS-transfected cells showed intracellular eGFP fluorescence, indicating αS expression (Fig. 4A). The level of αS overexpression was estimated to be around 10% from 3 independent experiments, which was typical for this cell line [9]. The eGFP fluorescence intensity and the αS levels were markedly reduced in the curcumin treated sample, suggesting a suppressive effect of curcumin on αS expression (Fig. 4A). Intracellular ROS levels in the αS-transfected cells increased over 2-fold compared to untransfected cells, while treatment with curcumin reduced the increase in ROS level to just 40% over the control value (Fig. 4B). Similarly, intracellular αS increased LDH release by 40% compared to the control cells and addition of curcumin reduced the LDH increase to just 20% (Fig. 4C). The overexpression of αS significantly increased ROS and LDH levels and addition of curcumin alleviated these effects, even with the low expression levels studied. Curcumin therefore has similar potent protective effects against αS cytotoxicity regardless of whether αS toxicity is induced extra- or intracellularly.

Curcumin reduces intracellular ROS and cytotoxicity of transiently transfected WTαS-EGFP SH-SY5Y cells. Localization of WTαS-EGFP and protective effects of curcumin in transiently transfected SH-SY5Y cells. (A) Representative images of WTαS-EGFP transfected cells in the absence (i-iii) and presence of curcumin (iv-vi) were captured using a Nikon TE300 fluorescence microscope. Scale bar = 50 μm. (B) ROS levels and (C) cytotoxicity of the untransfected control, αS-transfected and αS-transfected+curcumin were measured. For LDH assay, cell culture media was collected after 48 hr and the LDH release was expressed as a percentage of the untransfected samples. For ROS measurements, DCFH was added to each well and the DCF fluorescence was measured at Ex485 nm and Em535 nm after an additional incubation at 37°C for 30 min. DCF fluorescence was expressed as a percentage of the untransfected control reported as mean ± SD, n = 3. **p < 0.01; ***p < 0.001, compared with the untransfected control, #p < 0.05; ###p < 0.001 compared with αS-transfected sample analyzed using one way ANOVA followed by Bonferroni post-hoc test.
Discussion
A key pathological feature of PD is the formation of cytoplasmic inclusions containing ubiquitin and αS known as LBs and LNs in the dopaminergic neurons of the substantia nigra region of the brain [3, 4]. The many factors that influence αS aggregation and the subsequent downstream cytotoxic events that lead to neuronal cell death are being actively studied. Several point mutations in the αS gene which correlate to rare familial early-onset PD and rapid progression of the disease [7–9] accelerate aggregation of αS and favor formation of nonfibrillar oligomeric forms. Recent studies have suggested that soluble oligomeric and protofibrillar structures are the toxic species [27–30], and that these forms can permeabilize plasma membranes, alter intracellular function, induce oxidative stress and trigger apoptosis in cells [30, 44, 45]. Epidemiological studies have also suggested that exposure to environmental agents such as neurotoxins and pesticides [16, 17] cause an increase in oxidative damage to the cells by suppressing mitochondrial complex I activity and reducing glutathione levels [22, 23], thereby increasing the risk for PD.
Oxidative stress plays a major role in aging and is associated with several neurodegenerative diseases including PD [46], where an increase in ROS accompanies αS aggregation and degeneration of dopaminergic neurons [15, 47, 48]. Intracellular overexpression of αS generates excess ROS and causes oxidative stress to the cells [23, 46], leading to disruption in redox homeostasis cell metabolism, free radical generation, lipid peroxidation, cholesterol and protein oxidation [46, 49]. Excess ROS causes plasma membrane damage, mitochondrial dysfunction, defects in the glutathione peroxidase expression and reduction in glutathione levels, all of which render the brain more susceptible to oxidative stress [46, 49, 50]. In this study, we find that extracellular addition of oligomeric αS and intracellular overexpression of αS in SH-SY5Y cells both increase ROS levels by almost 2-fold. The αS-induced increase in ROS levels in our current study shows similar oxidative damage to the SH-SY5Y cell as previous MPP+ PD cell models, where MPP+ selectively targets and degenerates dopaminergic neurons due to excess generation of ROS [13, 15, 18]. Prolonged exposure to MPP+ and other neurotoxins has been shown to activate caspase-3 [16, 19, 51], an important effector caspase in the final apoptotic cascade leading to cell death. If oxidative stress exacerbates the etiology of PD, then agents that can simultaneously attenuate ROS damage and suppress caspase-3 activation may hold promise for the treatment of PD and other neurodegenerative diseases.
Here we show that curcumin, a natural phenolic food additive, effectively inhibits activation of caspase-3 (Fig. 2B) and ameliorates signs of apoptosis (Fig. 3) induced by extracellular addition of oligomeric αS to SH-SY5Y cells. We also demonstrated that curcumin reduces intracellular overexpression of αS and reduces ROS generation [15, 46, 48].
Conclusions
Overexpression and abnormal accumulation of oligomeric αS is key in the pathogenesis of PD [14, 48, 52], and numerous studies suggest that there is both an intra- and extracellular component to αS toxicity in PD [12, 24, 31, 32, 53]. We recently demonstrated that an anti-oligomeric αS antibody fragment binds oligomeric αS on the surface of SH-SY5Y cells, verifying the presence of intracellularly produced oligomeric αS on external cell membrane surfaces [29]. Here we show that extracellular addition of oligomeric αS induces similar cytotoxic effects as intracellular overexpression of αS, and that these αS-induced cytotoxic effects are similar to those reported in MPTP Parkinsonian models. We also show that curcumin can significantly reduce the cytotoxicity induced by extracellular or intracellular αS aggregates, suggesting it may have value for treating PD. Since extracellularly added curcumin provides protection even against intracellularly induced αS toxicity, our results suggest that there is a significant extracellular or cell surface component of αS-induced toxicity in PD models, which is consistent with a recently published report of interneuronal transmission of extracellular αS pathology in neuronal cells [53]. However, additional studies are needed to further elucidate the mechanism of αS-induced cytotoxicity and its subsequent pathogenesis and progression to induced-apoptosis in PD.
Methods
α-synuclein aggregation
αS was prepared and purified in our lab as previously described [28, 54]. Purified αS was lyophilized and stored at -80°C until further use. Stocks of the lyophillized αS were first dissolved in DI water and subsequent dilutions were made in Tris buffer (25 mM Tris, 150 mM NaCl, pH 7.4). The various forms of αS samples (70 μM) were prepared by dissolving the αS stock in Tris buffer. Monomeric αS samples were utilized immediately after dilution with Tris buffer, oligomeric αS were generated by incubating the samples at 37°C for 5-7 days (without shaking) while predominantly fibrillar morphologies of αS were generated by incubation at 37°C for up to 30 days (without shaking). αS morphologies were verified by AFM before use. All other chemicals were purchased from Sigma-Aldrich (Sigma-Aldrich, MO) and used as is without further treatment unless otherwise specified.
Co-incubation of curcumin with pre-formed αS samples
Curcumin stocks (1 mg/mL) were prepared in dimethyl sulfoxane (DMSO) and stored at -20°C in dark conditions until use. Curcumin was diluted to 140 μM with Tris buffer in a 2:1 molar ratio of curcumin to pre-formed αS sample.
Atomic Force Microscopy
A 10 uL aliquot of each sample was applied to a piece of freshly cleaved mica, incubated at room temperature for 10 minutes, rinsed with DI water and dried under a gentle stream of N2 gas. Topographic AFM images were acquired using OTESPA tips (k = 40 N/m, fo = 300-kHz) (Veeco, Santa Barbara, CA) at scan rates of 2 Hz with 512 × 512 pixel resolution on a Nanoscope IIIa TM-AFM (Veeco, Santa Barbara, CA). AFM images were analyzed with the scanning probe imaging processor software (SPIP, Image Metrology) to generate height distribution plots, as previously described [55].
PAGE and silver staining
Oligomeric αS samples, with and without curcumin were separated on a 10% Tris/Tricine native PAGE and developed using Pierce silver stain kit according to manufacturer's protocol. (Thermo Scientific, Rockford, IL).
Cell culture and transient transfection of SH-SY5Y cells
SH-SY5Y-human neuroblastoma cells were maintained and grown as described previously [28, 29]. Transient transfection of SH-SY5Y cells was performed using TransFast™ transfection reagent according to the manufacturer's protocol (Promega, Madison, WI) with slight modification. SH-SY5Y cells were grown for 4 days (50-65% confluency) in a 6-well plate in vitro before transfection. A transfection mixture consisting of a 1 μg aliquot of wildtype α-synuclein/eGFP (WTsynEGFP) fusion protein plasmid DNA (Clontech, Palo Alto, CA) and TransFast™ reagent (1:2 v/v) in serum free media was pre-incubated in the dark for 15 min at room temperature before addition to the cells. Cell culture media was removed and the transfection mixture (500 μL) was added to each well and incubated for 1 hr at 37°C, followed by addition of complete media with serum (500 μL). The culture plates were incubated and grown in a 5% CO2 atmosphere at 37°C for 48 hrs. A 10 μL aliquot of curcumin (4 μM final concentration) was added 48 hr post-transfection and the cells were incubated for another 24 hr before analysis. Previous studies have shown that the α-synuclein fusion protein aggregates similarly to α-synuclein alone, and that eGFP expression does not induce toxicity [56].
Cytotoxicity by lactate dehydrogenase assay
Cytotoxicity of samples towards SH-SY5Y cells was measured using a lactate dehydrogenase (LDH) assay as described [57]. Cells were seeded (2 × 104 cells/mL) in a 96-well plate 24 hr prior to the following treatment conditions: (a) pre-formed oligomeric αS (2 μM), (b) co-incubated samples of αS (2 μM) with curcumin (4 μM), (c) curcumin (4 μM) and (d) Tris buffer control. After incubating cells with each treatment for 48 hr, cytotoxicity of each sample was determined by measuring the reduction of iodonitrotetrazolium salt by LDH enzyme using a Wallac 1420 plate reader (Perkin Elmer, USA) at 490 nm and 650 nm. The values were expressed as a percentage of the Tris buffer control. Experiments were repeated a minimum of three times.
Cell viability by resazurin reduction assay
Cell viability was determined using a resazurin reduction assay [58]. Viable cells convert resazurin (blue) to resorufin (pink), and the degree of cell death can be measured directly by either absorbance or fluorescence spectrometry. Resaruzin stocks (10 mM) were made in DMSO and kept at -20°C until use when they were diluted to a 100 μM working solution with Tris buffer. Cells were seeded (5 × 104 cells/mL) in a 48-well plate 24 hr prior to exposure to the treatment conditions described above. Following treatment for 48 hr, cell culture media was removed and the cells were resuspended with 200 μL Tris buffer. An aliquot (10 μL) of resaruzin (20 μM final concentration) was added to each well and incubated at 37°C for an additional 3 hr. Absorbance of resorufin was measured at 560 nm and 600 nm. Cell viability of each sample was calculated by subtracting the background OD600 nm from OD560 nm and reported as a percentage of the Tris buffer control.
Measurement of intracellular ROS formation
The formation of intracellular ROS was measured using a fluorescent probe, 2,7-dichlorofluorescein diacetate (DCFH-DA) as described [59]. The cells were seeded (2 × 104 cells/mL) in a 96-well plate and were incubated for 48 hrs prior to ROS measurement with the conditions described above. After treatment, the cells were washed twice and resuspended in 100 μL Tris buffer. DCFH-DA (10 μM final concentration) was added to each well and the cells were incubated for 1 hr at 37°C in dark conditions. The fluorescence intensity of dichlorofluorescein (DCF, the oxidized species of DCFH-DA) was measured using a fluorescence spectrophotometer with excitation wavelength of 485 nm and emission wavelength of 535 nm.
Determination of caspase-3 activity
Caspase-3 activity was determined using the Caspase-3/CPP32 colorimetric assay kit following the manufacturer's protocol (BioVision, Inc., CA). Since caspase-3 is a pre-apoptotic marker, measurements of caspase-3 activity were taken after 24 hr incubation with the various treatments to ensure proper detection. Briefly, cells (106 cells/mL) were exposed to different treatments as described above for 24 hr, detached and lysed on ice for 10 min. The supernatant was removed and the total protein concentration of each sample was determined using a bicinchoninic acid assay (BCA, Pierce, Rockford, IL). Cell lysate was then diluted to 150 μg with lysis buffer for each assay. An equal loading amount of lysate (50 μL) was mixed with 50 μL of 2× reaction buffer with 10 mM dithiothreitol (DTT) and 5 μL DEVD-pNA substrate (200 μM) and incubated at 37°C for 1 hr. The absorbance of released p-nitroanilide (p NA) was measured at 405 nm using a plate reader. The increase in caspase-3 activity was determined by comparing the absorbance of the treated sample with the absorbance of the Tris buffer control sample.
Fluorescence microscopy and nuclear staining
WTsynEGFP-transfected cells were evaluated 48 hr post-transfection using a Nikon TE300 fluorescence microscope at an excitation wavelength of 488 nm with a 40× magnification objective. For nuclear staining, untransfected SH-SY5Y cells were seeded on glass coverslips and allowed to attach for 24 hr. The cells were fixed with 4% paraformaldehyde for 25 min, washed twice in cold Tris buffer, and stained with Hoechst 33342 (10 μg/mL) for 15 min. Nuclear morphology was observed using a 100× magnification objective. Images were captured and processed by MetaMorph software (Molecular Devices, USA). Cells stained by Hoechst 33342 with diffused nuclei were scored as viable, while cells with reduced nuclei, condensed chromatin, and increased fluorescence were considered apoptotic.
Statistical Analysis
Data was presented as mean ± SE from at least three independent experiments. Statistical analysis was evaluated using either Student's t-test or using a one-way ANOVA followed by Bonferoni post-hoc test for all pair-wise comparison. A p-value of < 0.05 was considered as significant.
References
Hoehn MM: Parkinsonism: Onset progression and mortality - Commentary. Neurology. 1998, 50 (2): 318-318.
Recchia A, Debetto P, Negro A, Guidolin D, Skaper SD, Giusti P: alpha-synuclein and Parkinson's Disease. Faseb J. 2004, 18 (6): 617-626. 10.1096/fj.03-0338rev.
Lotharius J, Brundin P: Pathogenesis of Parkinson's disease: dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci. 2002, 3 (12): 932-942. 10.1038/nrn983.
Gomez-Tortosa E, Newell K, Irizarry MC, Albert M, Growdon JH, Hyman BT: Clinical and quantitative pathologic correlates of dementia with Lewy bodies. Neurology. 1999, 53 (6): 1284-1291.
Spillantini M, Crowther R, Jakes R, Cairns N, Lantos P, Goedert M: Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson's disease and dementia with Lewy bodies. Neurosci Lett. 1998, 251 (3): 205-208. 10.1016/S0304-3940(98)00504-7.
Spillantini M, Crowther R, Jakes R, Hasegawa M, Goedert M: alpha-synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. P Natl Acad Sci USA. 1998, 95 (11): 6469-6473. 10.1073/pnas.95.11.6469.
Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT: Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson's disease: Implications for pathogenesis and therapy. P Natl Acad Sci USA. 2000, 97 (2): 571-576. 10.1073/pnas.97.2.571.
Li J, Uversky VN, Fink AL: Effect of familial Parkinson's disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human alpha-synuclein. Biochemistry. 2001, 40 (38): 11604-11613. 10.1021/bi010616g.
Pandey N, Schmidt R, Galvin J: The alpha-synuclein mutation E46K promotes aggregation in cultured cells. EXP NEUROL. 2006, 197 (2): 515-520. 10.1016/j.expneurol.2005.10.019.
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, et al.: alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003, 302 (5646): 841-10.1126/science.1090278.
Auluck PK, Chan HYE, Trojanowski JQ, Lee VMY, Bonini NM: Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science. 2002, 295 (5556): 865-868. 10.1126/science.1067389.
Feany MB, Bender WW: A Drosophila model of Parkinson's disease. Nature. 2000, 404 (6776): 394-398. 10.1038/35006074.
Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L: Dopaminergic loss and inclusion body formation in alpha-synuclein mice: Implications for neurodegenerative disorders. Science. 2000, 287 (5456): 1265-1269. 10.1126/science.287.5456.1265.
Ahmadi FA, Grammatopoulos TN, Poczobutt AM, Jones SM, Snell LD, Das M, Zawada WM: Dopamine selectively sensitizes dopaminergic neurons to rotenone-induced apoptosis. Neurochem Res. 2008, 33 (5): 886-901. 10.1007/s11064-007-9532-5.
Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno-Yagi A, Greenamyre JT: Mechanism of toxicity in rotenone models of Parkinson's disease. J Neurosci. 2003, 23 (34): 10756-10764.
Kalivendi SV, Cunningham S, Kotamraju S, Joseph J, Hillard CJ, Kalyanaraman B: alpha-synuclein up-regulation and aggregation during MPP+-induced apoptosis in neuroblastoma cells - Intermediacy of transferrin receptor iron and hydrogen peroxide. J Biol Chem. 2004, 279 (15): 15240-15247. 10.1074/jbc.M312497200.
Pettifer KM, Jiang SC, Bau C, Ballerini P, D'Alimonte I, Werstiuk ES, Rathbone MP: MPP+-induced cytotoxicity in neuroblastoma cells: Antagonism and reversal by guanosine. Purinerg Signal. 2007, 3 (4): 399-409. 10.1007/s11302-007-9073-z.
Przedborski S, Vila M: MPTP: a review of its mechanisms of neurotoxicity. Clin Neurosci Res. 2001, 1 (6): 407-418. 10.1016/S1566-2772(01)00019-6.
Turmel H, Hartmann A, Parain K, Douhou A, Srinivasan A, Agid Y, Hirsch EC: Caspase-3 activation in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated mice. MOVEMENT DISORD. 2001, 16 (2): 185-189. 10.1002/mds.1037.
Chen J, Tang XQ, Zhi JL, Cui Y, Yu HM, Tang EH, Sun SN, Feng JQ, Chen PX: Curcumin protects PC12 cells against 1-methyl-4-phenylpyridinium ion-induced apoptosis by Bcl-2-mitochondria-ROS-iNOS pathway. Apoptosis. 2006, 11 (6): 943-953. 10.1007/s10495-006-6715-5.
Srinivas BMM, Mythri R, Jagatha B, Vali S: Neuroprotective effect of curcumin against inhibition of mitochondrial complex I in vitro and in vivo: Implications for Parkinson's disease explained via in silico studies. J Neurochem. 2008, 106: 25-25.
Jagatha B, Mythri RB, Vali S, Bharath MMS: Curcumin treatment alleviates the effects of glutathione depletion in vitro and in vivo: Therapeutic implications for Parkinson's disease explained via in silico studies. Free Radical Bio Med. 2008, 44 (5): 907-917. 10.1016/j.freeradbiomed.2007.11.011.
Junn E, Mouradian MM: Human alpha-synuclein over-expression increases intracellular reactive oxygen species levels and susceptibility to dopamine. Neurosci Lett. 2002, 320 (3): 146-150. 10.1016/S0304-3940(02)00016-2.
Lee VMY, Trojanowski JQ: Mechanisms of Parkinson's disease linked to pathological alpha-synuclein: New targets for drug discovery. Neuron. 2006, 52 (1): 33-38. 10.1016/j.neuron.2006.09.026.
Iwai A: Properties of NACP/alpha-synuclein and its role in Alzheimer's disease. BBA-MOL BASIS DIS. 2000, 1502 (1): 95-109.
Uversky VN, Li J, Souillac P, Millett IS, Doniach S, Jakes R, Goedert M, Fink AL: Biophysical properties of the synucleins and their propensities to fibrillate: inhibition of alpha-synuclein assembly by beta- and gamma-synucleins. J Biol Chem. 2002, 277 (14): 11970-11978. 10.1074/jbc.M109541200.
El-Agnaf OM, Salem SA, Paleologou KE, Curran MD, Gibson MJ, Court JA, Schlossmacher MG, Allsop D: Detection of oligomeric forms of alpha-synuclein protein in human plasma as a potential biomarker for Parkinson's disease. Faseb J. 2006, 20 (3): 419-425. 10.1096/fj.03-1449com.
Emadi S, Barkhordarian H, Wang MS, Schulz P, Sierks MR: Isolation of a human single chain antibody fragment against oligomeric alpha-synuclein that inhibits aggregation and prevents alpha-synuclein-induced toxicity. J Mol Biol. 2007, 368 (4): 1132-1144. 10.1016/j.jmb.2007.02.089.
Emadi S, Kasturirangan S, Wang MS, Schulz P, Sierks MR: Detecting morphologically distinct oligomeric forms of alpha-synuclein. J Biol Chem. 2009, 284 (17): 11048-11058. 10.1074/jbc.M806559200.
Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT: Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002, 418 (6895): 291-10.1038/418291a.
Borghi R, Marchese R, Negro A, Marinelli L, Forloni G, Zaccheo D, Abbruzzese G, Tabaton M: Full length alpha-synuclein is present in cerebrospinal fluid from Parkinson's disease and normal subjects. Neurosci Lett. 2000, 287 (1): 65-67. 10.1016/S0304-3940(00)01153-8.
El-Agnaf OM, Salem SA, Paleologou KE, Cooper LJ, Fullwood NJ, Gibson MJ, Curran MD, Court JA, Mann DM, Ikeda S, et al.: Alpha-synuclein implicated in Parkinson's disease is present in extracellular biological fluids, including human plasma. Faseb J. 2003, 17 (13): 1945-1947.
Jakowec MW, Petzinger GM, Sastry S, Donaldson DM, McCormack A, Langston JW: The native form of alpha-synuclein is not found in the cerebrospinal fluid of patients with Parkinson's disease or normal controls. Neurosci Lett. 1998, 253 (1): 13-16. 10.1016/S0304-3940(98)00599-0.
Walsh DM, Selkoe DJ: Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein Pept Lett. 2004, 11 (3): 213-228. 10.2174/0929866043407174.
Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhang W, Zhou Y, Hong JS, et al.: Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson's disease. Faseb J. 2005, 19 (6): 533-542. 10.1096/fj.04-2751com.
Begum AN, Jones MR, Lim GP, Morihara T, Kim P, Heath DD, Rock CL, Pruitt MA, Yang FS, Hudspeth B, et al.: Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer's disease. J Pharmacol Exp Ther. 2008, 326 (1): 196-208. 10.1124/jpet.108.137455.
Cole GM, Morihara T, Lim GP, Yang FS, Begum A, Frautschy SA: NSAID and antioxidant prevention of Alzheimer's disease lessons from in vitro and animal models. Ann N Y Acad Sci. 2004, 1035: 68-84. 10.1196/annals.1332.005.
Frautschy SA, Hu W, Kim P, Miller SA, Chu T, Harris-White ME, Cole GM: Phenolic anti-inflammatory antioxidant reversal of A beta-induced cognitive deficits and neuropathology. Neurobiol Aging. 2001, 22 (6): 993-1005. 10.1016/S0197-4580(01)00300-1.
Lim GP, Chu T, Yang FS, Beech W, Frautschy SA, Cole GM: The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci. 2001, 21 (21): 8370-8377.
Hafner-Bratkovic I, Gaspersic J, Smid LM, Bresjanac M, Jerala R: Curcumin binds to the alpha-helical intermediate and to the amyloid form of prion protein - a new mechanism for the inhibition of PrPSc accumulation. J Neurochem. 2008, 104 (6): 1553-1564. 10.1111/j.1471-4159.2007.05105.x.
Ono K, Yamada M: Antioxidant compounds have potent anti-fibrillogenic and fibril-destabilizing effects for alpha-synuclein fibrils in vitro. J Neurochem. 2006, 97 (1): 105-115. 10.1111/j.1471-4159.2006.03707.x.
Pandey N, Strider J, Nolan WC, Yan SX, Galvin JE: Curcumin inhibits aggregation of alpha-synuclein. Acta Neuropathol. 2008, 115 (4): 479-489. 10.1007/s00401-007-0332-4.
Wang XH, Xu JX: Salvianic acid A protects human neuroblastoma SH-SY5Y cells against MPP+-induced cytotoxicity. Neurosci Res. 2005, 51 (2): 129-138. 10.1016/j.neures.2004.10.001.
Caughey B, Lansbury PT: Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003, 26: 267-298. 10.1146/annurev.neuro.26.010302.081142.
Volles MJ, Lee SJ, Rochet JC, Shtilerman MD, Ding TT, Kessler JC, Lansbury PT: Vesicle permeabilization by protofibrillar alpha-synuclein: Implications for the pathogenesis and treatment of Parkinson's disease. Biochemistry. 2001, 40 (26): 7812-7819. 10.1021/bi0102398.
Jenner P: Oxidative mechanisms in nigral cell death in Parkinson's disease. MOVEMENT DISORD. 1998, 13: 24-34.
Betarbet R, Sherer TB, Di Monte DA, Greenamyre JT: Mechanistic approaches to Parkinson's disease pathogenesis. Brain Pathol. 2002, 12 (4): 499-510.
Quilty MC, King AE, Gai WP, Pountney DL, West AK, Vickers JC, Dickson TC: Alpha-synuclein is upregulated in neurones in response to chronic oxidative stress and is associated with neuroprotection. EXP NEUROL. 2006, 199 (2): 249-256. 10.1016/j.expneurol.2005.10.018.
Olanow CW: An Introduction to the Free-Radical Hypothesis in Parkinsons-Disease. Ann Neurol. 1992, 32: S2-S9. 10.1002/ana.410320703.
Mancuso C, Scapagnini G, Curro D, Stella AMG, De Marco C, Butterfield DA, Calabrese V: Mitochondrial dysfunction, free radical generation and cellular stress response in neurodegenerative disorders. Front Biosci. 2007, 12: 1107-1123. 10.2741/2130.
Bo J, Ming BY, Gang LZ, Lei C, Jia AL: Protection by puerarin against MPP+-induced neurotoxicity in PC12 cells mediated by inhibiting mitochondrial dysfunction and caspase-3-like activation. Neurosci Res. 2005, 53 (2): 183-188. 10.1016/j.neures.2005.06.014.
Zhou ZD, Yap BP, Gung AYT, Leong SM, Ang ST, Lim TM: Dopamine-related and caspase-independent apoptosis in dopaminergic neurons induced by overexpression of human wild type or mutant alpha-synuclein. Exp Cell Res. 2005, 312 (2): 156-170. 10.1016/j.yexcr.2005.10.012.
Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ: Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. P Natl Acad Sci USA. 2009, 106 (31): 13010-13015. 10.1073/pnas.0903691106.
Volles MJ, Lansbury PT: Relationships between the sequence of alpha-synuclein and its membrane affinity, fibrillization propensity, and yeast toxicity. J Mol Biol. 2007, 366 (5): 1510-1522. 10.1016/j.jmb.2006.12.044.
Wang MS, Zameer A, Emadi S, Sierks MR: Characterizing Antibody Specificity to Different Protein Morphologies by AFM. LANGMUIR. 2009, 25 (2): 912-918. 10.1021/la8025914.
Yuan B, Sierks MR: Intracellular targeting and clearance of oligomeric alpha-synuclein alleviates toxicity in mammalian cells. Neurosci Lett. 2009, 459 (1): 16-18. 10.1016/j.neulet.2009.04.046.
Decker T, Lohmann-Matthes ML: A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J Immunol Methods. 1988, 115 (1): 61-69. 10.1016/0022-1759(88)90310-9.
Shahan TA, Siegel PD, Sorenson WG, Kuschner WG, Lewis DM: A Sensitive New Bioassay for Tumor-Necrosis-Factor. Journal of Immunological Methods. 1994, 175 (2): 181-187. 10.1016/0022-1759(94)90361-1.
Molina-Jimenez MF, Sanchez-Reus MI, Andres D, Cascales M, Benedi J: Neuroprotective effect of fraxetin and myricetin against rotenone-induced apoptosis in neuroblastoma cells. Brain Res. 2004, 1009 (1-2): 9-16. 10.1016/j.brainres.2004.02.065.
Acknowledgements
This research was supported in part by grants from the Michael J. Fox Foundation, the Arizona Alzheimer's Research Consortium, American Health Assistance Foundation and the ASU Graduate and Professional Student Association Research Grant. The authors would like to thank Dr. Page Baluch and the WM Keck Imaging facility at ASU for the assistance with fluorescence microscopy and Mr. Philip Schulz for protein purification.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
MSW participated in the experimental design of the study, prepared pre-aggregated αS samples, conducted in vitro cell-based assays, performed fluorescence microscopy and statistical analysis and drafted the manuscript. SB maintained the untransfected SH-SY5Y cells, plated and added samples for all the in vitro assays and edited the manuscript. SE conducted transient transfection of SH-SY5Y cells and edited the manuscript. MRS supervised the design of study, provided technical assistance in data interpretation, and played a major part in revision of the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12868_2009_1616_MOESM1_ESM.PDF
Additional file 1: Curcumin alters αS aggregation kinetics. Aggregation kinetics were monitored using Thioflavin T assay. (A) monomeric αS and monomeric αS + curcumin, (B) pre-formed oligomeric αS and pre-formed oligomeric αS + curcumin, (C) pre-formed fibrillar αS and pre-formed fibrillar αS + curcumin. Samples were taken at the indicated time and mixed with Thioflavin T. The observed Thioflavin T fluorescence (Ex = 450 nm, Em = 482 nm) was normalized to each untreated αS sample at its maximum value. Data are presented as mean ± SE from 4 sets of experiments. (PDF 745 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wang, M.S., Boddapati, S., Emadi, S. et al. Curcumin reduces α-synuclein induced cytotoxicity in Parkinson's disease cell model. BMC Neurosci 11, 57 (2010). https://doi.org/10.1186/1471-2202-11-57
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2202-11-57