
Abstract
The double-stranded conformation of cellular DNA is a central aspect of DNA stabilisation and protection. The helix preserves the genetic code against chemical and enzymatic degradation, metabolic activation, and formation of secondary structures. However, there are various instances where single-stranded DNA is exposed, such as during replication or transcription, in the synthesis of chromosome ends, and following DNA damage. In these instances, single-stranded DNA binding proteins are essential for the sequestration and processing of single-stranded DNA. In order to bind single-stranded DNA, these proteins utilise a characteristic and evolutionary conserved single-stranded DNA-binding domain, the oligonucleotide/oligosaccharide-binding (OB)-fold. In the current review we discuss a subset of these proteins involved in the direct maintenance of genomic stability, an important cellular process in the conservation of cellular viability and prevention of malignant transformation. We discuss the central roles of single-stranded DNA binding proteins from the OB-fold domain family in DNA replication, the restart of stalled replication forks, DNA damage repair, cell cycle-checkpoint activation, and telomere maintenance.
Similar content being viewed by others


Introduction
DNA exists primarily as a duplex to stabilise and protect our genome. However, as a result of many cellular processes, such as replication and transcription, single-stranded DNA (ssDNA) is exposed. While a necessary metabolic intermediate, these exposed stretches are vulnerable to both chemical and enzymatic degradation, and as such must be sequestered. In this process, the single-stranded DNA binding protein family (SSBs) are essential cellular components [1–4]. In addition to this role, SSBs function in the correct processing of ssDNA, including the recruitment of appropriate functional enzymes. In the current review, we discuss the roles of a subset of human SSBs in the maintenance of genomic stability, an essential consideration in the prevention of malignant transformation and loss of cellular viability.
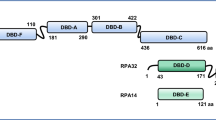
The characteristic functional unit of the SSBs is the oligonucleotide/oligosaccharide-binding (OB)-fold, a protein domain that facilitates binding to ssDNA, as well as various protein-protein interactions (Figure 1). As we have described previously [1], the SSB family consists of two core sub-groups; the simple SSBs, which contain one OB-fold per polypeptide, and the higher order SSBs, which contain multiple OB-folds (which may be on different polypeptides). The human genome encodes both simple and higher order SSBs: the simple SSBs are represented by human single-stranded DNA binding proteins 1 and 2 (hSSB1 and 2) and the mitochondrial SSB (mtSSB), while higher order SSBs are represented by heterotrimeric RPA. In addition, other proteins have also adopted the ssDNA-binding-OB-fold structure within their polypeptides and may be considered members of the SSB family. For instance the serine/threonine kinase receptor associated protein (Strap) structurally contains one DNA binding OB fold as do the simple SSBs, while the TPP1 - protection of telomeres 1 (POT1) breast cancer 2, early onset (BRCA2) and the CST complex form complexes reminiscent of higher order SSBs. While containing only a single OB-fold, the majority of simple SSBs, including all human simple SSBs, do however assemble as higher order multiple OB-fold containing oligomers [5–7]. This is exemplified by hSSB1, which is predominantly dimeric in solution and may shift to a stable tetrameric conformation following activation by the ataxia telangiectasia mutated kinase (ATM) [unpublished data from within our group].

Human OB-fold containing proteins are essential for multiple aspects of genome stability maintenance. Schematic representation of human OB-fold containing proteins, illustrating their predicted domains and central function. Domains are not drawn to scale.
DNA binding
The ssDNA-binding characteristic of the SSBs is largely facilitated by the OB-fold [4, 8]. This domain comprises five antiparallel β-strands arranged as a cylindrical β-barrel, capped at one end by an α-helix between the third and fourth β-strands [8]. OB-folds interact with their ssDNA substrates through nucleotide base stacking with aromatic residues in the binding cleft and electrostatic interactions with the phosphodiester backbone [9–20]. While ssDNA-binding of the OB-fold is in most cases largely non-specific, OB-fold mediated DNA binding of the telomere associated protein POT1 is seen to be highly specific for telomeric repeats [21]. This may be due to the high pyrimidine content of the telomeric single strand where POT1 binds. Thermodynamic studies of Escherichia coli SSB-ssDNA interactions show that purines (due to the double-ring) may not sterically fit into the SSB ssDNA-binding cleft compared to pyrimidines [22, 23]; a preference for binding to pyrimidine-rich sequences has also been noted for hSSB1 [7]. Telomere specific binding is also observed for the STN1 protein, however this seems to be due to two C-terminal winged helix-turn-helix domains, rather than specificity of the OB-fold [24].
Eukaryotic RPA is a heterotrimeric complex composed of ~70 kDa, ~32 kDa, and ~14 kDa subunits (RPA1, RPA2 and RPA3, respectively) (Figure 1). Between them, the subunits contain 6 OB-folds, which are homologous to those of the simple SSBs; these are designated A-F, in order of their respective DNA-binding affinities. Of the OB-folds in the RPA complex, four bind ssDNA. To date, two RPA DNA binding conformations have been designated [25, 26]. In one, facilitated by the A and B OB-folds of RPA1, RPA binds ssDNA with low-affinity, occluding a region of ~8 nucleotides (nt) [27, 28]. The additional contribution of the C and D OB-folds then allows RPA to bind ssDNA with a high-affinity, where ~30 nt are occluded [29]. Associated with these discrete binding modes is a difference in protein-protein interactions; while the A-OB-fold interacts with various proteins in the low-affinity mode, there is a considerable decrease following transition to the high-affinity state [30]. Furthermore, in the high-affinity state, RPA binds ssDNA with a 5′ to 3′ polarity, whereby RPA1 is positioned 5′ to RPA2; this arrangement has a considerable effect on the functionality of the RPA complex [26, 31–33].
The recently identified hSSB1 and 2 exemplify simple SSBs in the human genome [7]. Both proteins are structurally similar, possessing a single N-terminal OB-fold, as well as a basic C-terminus [1]. For hSSB1, the C-terminus is involved in a protein-protein interaction with NBS1, a component of the MRE11-NBS1-RAD50 (MRN) repair complex [34]. Agarose gel shift analysis using virion phiX174 ssDNA as a substrate has indicated that the hSSB1 dimer occludes a region of ~12 nt (5–6 nt per monomer) [1]. As yet, DNA binding activity has not been described for hSSB2. As well as binding DNA, the OB-fold of hSSB1 (and presumably hSSB2) has been shown to interact with the integrator complex subunit 3 (INTS3) [35].
Recent data has suggested the interaction with INTS3 leads to diminished hSSB1 ssDNA-binding, as demonstrated by an increase in observed Kd for a ssDNA substrate (15 nm [36] to 45 nm [34]) when in complex. Interestingly, a similar ssDNA-binding affinity has been observed between unbound hSSB1 and the RPA heterotrimer [36]. While both RPA and hSSB1 show high specificity for ssDNA, hSSB1 has also recently been shown to bind short (33 nt) duplex DNA constructs, as well as duplex DNA with short 6 bp overhangs [37]. In this instance, hSSB1 was suggested to bind ssDNA at natural breathing sites in the constructs rather than binding the dsDNA. As a number of SSBs are capable of melting dsDNA, such as the SSB from Sulfolobus solfataricus[38], this may suggest a similar activity for hSSB1. This is consistent with data that show the minimal ssDNA length required for binding of hSSB1 is reduced when it is adjacent to duplex DNA [36]. Interestingly, unlike the bacterial SSBs, which wrap 65 nt of DNA around their tetrameric structure [39], both RPA and hSSB1 bind ssDNA in an extended fashion, presumably to allow access of other DNA interacting factors [1, 36, 40].
SSBs are required for DNA replication
The accurate replication of DNA is a central process in the maintenance of a stable genome. For this, RPA has a central role in both replication initiation and progression. Despite the central importance of this process, RPA appears to function as the sole required SSB, and neither hSSB1 nor 2 have been found to co-localise with replication foci or to influence S-phase progression [7, 37]. In the earliest stage of eukaryotic DNA replication, sites of initiation are bound by origin recognition complexes (ORC), which stimulate the accumulation of downstream replicative factors. These include Cdc6 and Cdt1, both of which are required for the loading of minichromosome maintenance proteins (MCMs), which form the pre-initiation complex [41].
Following MCM mediated ssDNA exposure, RPA binds these stretches, where the 70 kDa subunit interacts with and stabilises the DNA polymerase α-primase complex (Pol α) [42, 43]. Pol α is required for the synthesis of short oligonucleotide primers from which the more processive DNA polymerases δ and ε (Pol δ and ε) can synthesise the new lagging and leading strands, respectively [44]. Therefore, Pol α is required for initiating leading strand synthesis, as well as for the production of the ~ 20 million Okazaki fragment primers [45]. Here the primase subunit of Pol α firstly synthesises an RNA strand of 7 – 12 nt, which is extended with an additional short DNA chain (~ 20 nt) by the polymerase subunit [46, 47]. The efficiency of Pol α chain extension appears reliant on RPA, where it has been reported to act as a enhancer of polymerase processivity and fidelity [48].
To allow access of Pol δ and ε, the Pol α complex must be removed following primer synthesis. This is at least partially facilitated by replication factor C (RFC), which competitively binds RPA, and disrupts its interaction with Pol α [42]. In addition, RFC is an essential clamp loader of the proliferating cell nuclear antigen (PCNA) sliding clamp [49]. PCNA is a central protein in the initiation and persistence of DNA replication, where it functions as a processivity factor for DNA polymerase δ [50]. PCNA encircles the duplex DNA by virtue of its ring-like structure, formed by three PCNA monomers; the inner surface of the homotrimeric clamp is rich in basic residues, allowing interaction with the DNA phosphodiester backbone, while the outer surface of PCNA interacts with various replication factors, thereby tethering them to sites of replication [51, 52]. The absolute requirement of RPA and PCNA in mediating primer extension has lead to the co-localisation of these proteins being widely used as a marker of replication [53, 54].
The discontinuous synthesis of the lagging strand means that each section will eventually collide with the initiating primer of the downstream fragment; this process results in displacement of short oligonucleotide flaps, which must be removed. In eukaryotic cells, this is facilitated by two major pathways, depending on the length of the displaced section [55]. In the first, short displaced ssDNA fragments of 3–5 nt are recognised and cleaved by the flap endonuclease (FEN1), leaving behind a nick in the phosphodiester backbone, which is sealed by DNA ligase [56–58]. In vitro data have however suggested that while the majority of these flaps are removed by FEN1, a small portion are missed, such that they form lengthier regions [59, 60]. In this instance, the second pathway seems to be initiated where the exposed ssDNA is bound by RPA [61, 62]. This event appears necessary for the recruitment of the Dna2 helicase/nuclease, which cleaves the flap and generates shorter 5–7 nt overhangs to be processed by FEN1 [61, 62].
The replicative activity of RPA is partially governed by phosphorylation events. During S and G2/M of the cell cycle, the N-terminus of RPA2 is phosphorylated at S23 by cyclin dependent kinase 2 (CDK2)-cyclin B. The functional consequence of this modification is however unclear, and contradictory findings regarding the role of this event have been reported. Indeed, while addition of purified CDK2-cyclin B has been shown to stimulate replication in vitro[63], S23 mutated RPA2 is still able to support DNA synthesis [64, 65]. At the initiation of mitosis, S29 of RPA2 is also phosphorylated by CDK2 [66]. This modification appears to deactivate RPA, as the DNA binding affinity of the complex purified from mitotic cells appears reduced when compared to the de-phosphorylated form [67]. This is supported by the finding that purified CDK2 is able to disassemble RPA foci from interphase chromatin [68]. Phosphorylation of RPA2 has also been demonstrated following DNA damage induction as a means of inhibiting RPA replicative activity [69–71]. This was observed after both oxidative [71] and replication-mediated damage [72] during S-phase, as well as throughout the cell cycle as a result of double-strand DNA break formation [73]. In these cases, damage activates the phosphatidylinositol 3-kinase related kinases: ATM, ATM and Rad3 related kinase (ATR) and DNA-dependent protein kinase (DNA-PK); each of these kinases phosphorylate RPA2 at five N-terminal residues [73–82]. Following DNA damage repair, or migration out of M phase, RPA2 must then be de-phosphorylated to reset replicative potential [83, 84]. This has been suggested to occur via the 1A/2A protein phosphatases (PP1A and PP2A, respectively), as RPA de-phosphorylation is suppressed following inhibition of these enzymes using okadaic acid [84]. This has been further supported by RNA interference data, which has indicated a role for PP2A in de-phosphorylation of the RPA T21 and S33 ATM/ATR phosphorylated residues [85].
SSBs are essential for the stabilisation and restart of stalled replication forks
As mentioned above, RPA is essential for the replication of DNA during S-phase, functioning both in the establishment and elongation of the replicative fork [1]. In the event of DNA damage, these processes may however be interrupted, causing the fork to stall [86]. Here SSBs also play an important role both in the stabilisation and protection of exposed ssDNA, as well as in re-initiation of fork migration. Additionally, the collapse or disassembly of the replicative machinery may result in the formation of single- or double-stranded DNA breaks, the repair of which requires the concerted effort of SSBs. In a typical scenario, replication fork migration is disrupted by a bulky lesion, which inhibits polymerase activity. Alternatively, certain regions of genetic material are apparently more difficult to replicate, and as such are subject to high rates of fork stalling. These sites include so-called ‘fragile sites’, regions of the human genome associated with increased genetic instability [87].
Following fork stalling, repair of the damaged region and restart of replication may be facilitated through two main mechanisms: homology directed repair (HDR) and translesion synthesis (TLS). While HDR makes use of a homologous region found in an opposing strand, TLS instead utilises a set of low-fidelity polymerases to replicate through the damage site, potentially introducing base mismatches [88].
Homology directed repair
One of the most immediate requirements in the prevention of stalled replication fork collapse is the stabilisation of exposed ssDNA; this is in part facilitated by the rapid accumulation of RPA [89, 90]. Here, the requirement of ssDNA-binding is further amplified by helicase uncoupling from the replicative machinery [91, 92]. In this role, RPA functions both to protect the exposed ssDNA, as well as to interact with a number of proteins involved in both stabilising and remodelling the replication fork. An example of this is the localisation of RPA with the MRN complex at sites of replicative stress [90, 93]. Here the RPA1 N-terminal OB-fold has also been shown to interact directly with Mre11, an association required for S-phase checkpoint activation [94–96]. In addition, NBS1 of the MRN complex appears necessary for hyper-phosphorylation of the 32 kDa RPA subunit [93], a modification mediated by the central DNA repair kinase ATR [97, 98]. Recently hyper-phosphorylation of RPA has been shown as stimulatory for RAD51 foci formation at replication forks stalled by hydroxyurea treatment [99]. The formation of RAD51 nucleofilaments is an essential aspect of homology directed strand invasion. Stabilisation of the RAD51 coated strand is also promoted by an interaction between RAD51 and the C-terminus of BRCA2, an interaction that is dispensable for the repair of double-strand DNA breaks but appears important in the prevention of fork collapse [100].
The RPA2 subunit is also known to interact directly with Timeless-interacting protein (Tipin) [101, 102]. Tipin is a component of the replication pausing complex (RPC), a protein complex that is involved in the coordinated pausing of both polymerase and helicase migration at stalled replication forks [103–112]. Following replication fork disruption, RPA stabilises the Timeless (Tim1)-Tipin heterodimer, a central unit of the RPC, at sites of DNA damage [102]. Furthermore, this interaction appears to promote the stabilisation of claspin, a Tipin binding partner required for efficient S-phase checkpoint activation [102]. In agreement with these findings, Tim1-Tipin depleted cells show deficient intra-S checkpoint activation, including deficient Chk1 phosphorylation [101, 112–114].
A central aspect of replication restart is remodelling of the stalled replicative fork; this is largely facilitated by the concerted effort of DNA helicases, a number of which have been shown to interact with RPA following replication fork stalling. Of these, the RecQ family members Werner’s syndrome protein (WRN) and Bloom’s syndrome protein (BLM) appear to play an important role [115–118]. This is highlighted by the cancer-prone syndromes for which they are named, where cells deficient of these proteins display high rates of genetic instability. In functional cells, both BLM and WRN are suggested to suppress excessive homologous recombination by inhibition of RAD51-mediated strand exchange, and the promotion of fork regression [119, 120]. In vitro data have suggested that this is largely facilitated by the interaction with RPA, which is seen to promote helicase activity [118, 121–123]. Interestingly, three OB-folds have recently been identified in the RMI heterodimer, a central component of the Bloom’s helicase complex [124–126]. RMI consists of two proteins, RMI1, which contains 2 OB-folds, and RMI2, which contains 1 OB-fold [124, 126]. Here, dimerisation appears to be facilitated by protein-protein interactions between the RMI1 C-terminal OB-fold and the OB-fold of RMI2 [124]. While no detectable DNA-binding activity has been observed for RMI [124], depletion experiments have indicated potential roles in the stabilisation, chromatin localisation, and activity of the BLM complex, including dissolution of Holliday junctions [124–126].
In response to replication fork disruption, RPA also interacts directly with the FANCJ helicase (also known as BRCA1-associated C-terminal helicase; BACH1) [127]. FANCJ is one of 15 known Fanconi anaemia (FA) proteins, the deficiency of which is associated with Fanconi anaemia syndrome [128–130]; like Bloom’s and Werner’s syndrome, FA is characterised by genetic instability [131]. In addition, FANCJ deficiency has been associated with early-onset breast cancer in cells normal for BRCA1 and BRCA2 [132]. Support for a role of FANCJ in the response to replicative stress is provided by sensitisation of cells to hydroxyurea treatment following its depletion [133]. At stalled replication forks, RPA promotes FANCJ processivity and helicase activity [127]. Interestingly, depletion of FANCJ or its binding partner TopBP1 [134, 135] decreases RPA-chromatin loading in cells [134]. In addition, FANCJ and TopBP1 depleted cells also show decreased Rad9 and ATR accumulation following hydroxyurea treatment, presumably due to disrupted RPA binding [134]. Recent findings have also demonstrated a direct interaction between the FANCJ C-terminus and BLM helicase following replicative stress [133]. A substantial decrease in BLM protein levels was observed when FANCJ was depleted, which could be rescued by treatment with the proteasome inhibitor MG132, suggesting a potential role for FANCJ in the stabilisation of BLM [133].
RPA has also been seen to localise with SWI/SNF-related, matrix associated, actin-dependent regulator of chromatin, subfamily A-like 1 (SMARCAL1) at stalled replication forks [136–139]. This seems to be largely facilitated by a SMARCAL1 RPA-binding domain similar to that found in TIPIN; this interaction appears necessary for the in vivo activity of the protein [137]. Interestingly, SMARCAL1 does not show conventional DNA unwinding helicase activity, but instead functions in the ATPase-mediated re-annealing of unwound ssDNA stretches (known as annealing helicase activity) [140]. In this way, SMARCAL1 appears to function in the regression and remodelling of stalled replication forks [136].
A potential role for hSSB1 in DNA repair at replication forks has also recently been proposed [141]. These data indicate that while Obfc2b, the murine homologue of hSSB1, is essential for maintenance of genomic stability, this is through a means other than double-strand DNA break recognition or cell cycle checkpoint activation. Specifically, Obfc2b is suggested to function directly in the suppression of replication-associated DNA damage. Additionally, a similar role has been suggested for Obfc2a (murine homologue of hSSB2), and indeed a compensatory effect for Obfc2a is observed following Obfc2b depletion [141, 142]. These data potentially represent a role for these homologues in the response to stalled replication forks, and as such, it will be of interest to determine if a similar role exists for hSSB1 and 2.
Translesion repair
The choice to repair by HDR or TLS appears largely governed by mono-ubiquitination of the PCNA sliding clamp. Such modification is generally considered to promote recruitment of TLS polymerases to sites of DNA damage [143–145] and indeed, while association between TLS polymerases and non-ubiquitinated PCNA has been demonstrated [146, 147], this is with decreased efficiency [148]. Complementary to this event is the poly-ubiquitin-mediated degradation of PCNA during HDR, presumably inhibiting TLS [149]. Recently it has been suggested the switch between PCNA mono- and poly-ubiquitination may be modulated by RPA. This comes from the observation that in cells, RPA interacts directly with RAD18, the E3 ubiquitin ligase which mono-ubiquitinates PCNA following damage [150]. In addition, RPA is seen to interact directly with DNA repair polymerase λ, and to promote correct nucleotide incorporation [151]. Together, these data suggest RPA may play at least two regulatory functions in translesion repair of replicative damage.
SSBs function in double-strand DNA break repair by homologous recombination
Double-strand DNA breaks (DSBs) are amongst the most cytotoxic DNA damage lesions encountered by the cell, and must be repaired to prevent chromosomal fragmentation. In eukaryotes, one of the major mechanisms through which these lesions are repaired is homologous recombination with a sister chromatid. In this process, DSBs are detected and signalling pathways initiated by the ATM and ATR DNA repair kinases. These signalling cascades serve to recruit repair proteins, including nucleases that resect DNA in a 5′-3′ direction. This resection generates stretches of ssDNA, which during homologous recombination may invade into a sister chromatid, forming an intermediate structure known as a D loop; this allows the sister chromatid to act as a template for polymerase-mediated extension of the invading strand. In addition, the non-invading strand can be extended using the displaced sister chromatid portion, present in the D loop. Resolution of this mobile junction (the Holliday junction) then yields two intact and identical DNA molecules [152–155].
In cells, the majority of DSBs generated by endogenous events, such as the collapse of replication forks, contain short ssDNA ‘sticky ends’ [156]. When compared with blunt-ended DSBs, the presence of these overhangs is seen to enhance activation of the ATM kinase. ATM is a central DNA repair kinase with downstream substrates involved in cell cycle checkpoint activation (e.g., CHK1, CHK2, p53) and directly in DSB repair (e.g., NBS1, EXO1 and the histone variant H2AX) [157–159]. The recruitment of ATM to sites of DNA damage is largely reliant on DSB recognition by the MRN complex [157, 159–161]. MRN is structurally conserved in higher eukaryotes; it contains a MRE11 flexible dimer enfolded by a single NBS1 polypeptide and two RAD50 polypeptides that function to bridge the DNA break [162–165]. Structural analysis has indicated that while the MRE11-RAD50 clamp interacts directly with DNA at DSBs, NBS1 interaction is important in modulating its DNA-binding function [162, 166]. Furthermore, crystallography data of the Saccharomyces pombe complex has indicated the NBS1 N-terminus is orientated in such a way as to interact directly with DSB foci proteins located near the MRE11-RAD50 binding cleft [162]. Of these interacting proteins, hSSB1 is seen to have a central role in DSB repair by homologous recombination [7, 34]. Here, hSSB1 rapidly interacts with ssDNA overhangs at sites of DSBs, where it functions as an initial recogniser of these breaks [34, 37] (Figure 2). This has been supported by in vitro data, where hSSB1 was found to bind duplex DNA with 6 bp ssDNA overhangs, as well as, to a lesser degree, natural breathing sites of a short 33 bp dsDNA oligo [34]. Additionally, laser micro-irradiation of cells has demonstrated hSSB1 binding at sites of DSBs within 10 seconds of DNA damage in all interphase cells [37]. hSSB1 recruitment to DSBs is also independent of CtIP, MDC1 or MRN [37]. Furthermore, MRN recruitment at sites of DSBs is substantially inhibited in cells deficient of hSSB1 [37]. As the hSSB1 C-terminus interacts directly with NBS1, this suggests that hSSB1 may participate in the immediate recruitment of the MRN complex to DSB foci [34]. Further in vitro data has supported this, demonstrating that MRN nuclease activity, which is known to be weak, is substantially stimulated in the presence of hSSB1 [34].

hSSB1, RPA, and BRCA2 are essential SSBs in the repair of double strand DNA breaks (DSBs) by homologous recombination. A potential model for human SSBs in the repair of DSBs: The MRN complex is initially recruited to sites of DSBs by hSSB1, allowing for resection of the 5′ strand. ssDNA stretches are rapidly bound by RPA, which, following BRCA2-DNA binding, is removed to allow for RAD51 nucleofilament formation. Homologous recombination is then facilitated by RAD51–mediated strand invasion into a sister chromatid. hSSB1 has also been found to interact directly with RAD51 and to facilitate strand invasion in vitro, however its precise function at these later stages is unclear.
Both hSSB1 and 2 have recently been shown to form separate heterotrimeric protein complexes (sensor of single-stranded DNA 1 and 2; SOSS1 and 2) with the integrator complex subunit 3 (INTS3) and hSSB-interacting protein 1 (hSSBIP1) [35, 167, 168]. These complexes are both structurally and functionally unlike the RPA heterotrimer, and neither INTS3 nor hSSBIP1 have yet been shown to have ssDNA-binding affinity. In addition, while hSSB1 and hSSBIP1 co-localisation has been observed at sites of DNA damage [167], inconsistent findings have been reported regarding the localisation of INTS3 and MRN at DNA damage sites. Interestingly, the addition of SOSS1 components has however recently been shown to promote exonuclease 1 (EXO1) DSB resection in vitro[36], although it remains unclear whether this is due to activity of the SOSS1 complex, or dissociated hSSB1. Furthermore, while reduced clonogenic survival of cells depleted of SOSS1 and SOSS2 components and exposed to DNA damaging agents has been observed, INTS3 and hSSBIP1 depletion caused only a slight reduction in homologous recombination-dependent DSB repair, when compared to depletion of hSSB1 or 2 [167]. As there is compounding evidence to suggest INTS3 associates at DNA damage sites at later time points (4–6 hr following damage) [35, 169], one explanation for these findings may be that the SOSS1 complex functions in DSB repair only during the later phases. Alternatively, as INTS3 depletion has been shown to destabilise hSSB1 and 2 [167, 168], as well as to decrease the transcriptional rate of hSSB1 [35], this may suggest INTS3 modulates DSB repair through the regulation of hSSB1 and 2, as opposed to a direct means. Interestingly, while most known SSBs bind ssDNA with four OB-folds, recent data has indicated hSSB1 may exist as part of the SOSS1 complex in a monomeric form [36]. Furthermore, while the complex contains a single hSSB1 OB-fold, this domain is also known to interact with INTS3 [35, 169], and so may indeed not be available for DNA binding.
An essential role of the MRN complex is the resection of DSB 5′ strands to expose stretches of ssDNA for RAD51-mediated strand invasion. While the MRE11 component has both exo- and endo-nuclease activity, only the endonuclease function seems to be required for DSB processing [170, 171]. For this, MRE11 nicks the target strand up to 300 bases from the DSB site, and resects the strand in the 3′-5′ direction. This is complemented by the 5′-3′ exonuclease activity of EXO1, which concurrently digests the target strand away from the DSB site [171]. The activation of MRN nuclease activity is stimulated by interaction with C-terminal binding protein interacting protein (CtIP) [172]. Interestingly, MRN and CtIP have been found to interact directly with BRCA1, an association necessary for efficient end resection and loading of RPA onto ssDNA [173–175]. The association of RPA with ssDNA is likely important for the protection of the otherwise exposed ssDNA strand, preventing it from damage and digestion, or the formation of disruptive secondary structure [176].
RPA ssDNA-binding is also necessary for the recruitment of downstream proteins involved in active DNA repair and checkpoint signalling. Of central importance is the interaction with the RAD51 recombinase, a protein which, following RPA displacement, rapidly coats ssDNA strands [177, 178]. Although it remains unclear exactly how this interchange occurs, several interactions appear necessary for both the removal of RPA, and the loading of RAD51. Of these, the direct interaction of RPA and RAD51 appears to play a central role. Here, the A-OB-fold of ssDNA-bound RPA interacts directly with the RAD51 N-terminus, potentially representing a means of competitive ssDNA-binding [179]. One mechanism through which this has been suggested to occur is the capture of transiently bound RPA, presumably preventing its re-association with the ssDNA strand [179, 180]. In addition, the RPA-Rad51 exchange seems to be promoted by Rad52, a protein shown to bind the RPA1 and 2 subunits, and to stimulate their displacement [181–183]. This interaction seems to be promoted by phosphorylation of RPA1, an event that also stimulates ssDNA exchange [184].
The displacement of RPA, which is essential for completion of homologous recombination, also appears to be promoted by the OB-fold containing protein, BRAC2 [185]. Structural analysis of this protein has indicated the presence of a DNA-binding region composed of a helical domain, followed by three OB-folds [186]. Interestingly, the central OB-fold also contains a protruding structure composed of five α-helices, three of which form a helix-turn-helix bundle which sits atop the remaining two anti-parallel helices; this structure is referred to as a ‘tower domain’. While the helical domains and three OB-folds may bind ssDNA, the tower domain seems to bind dsDNA [186]. This dual-binding ability presumably allows BRCA2 to interact efficiently with both ssDNA and dsDNA found at the ends of a resected DSB, as has been observed for the Brh2 nuclease, the Ustilago maydis fungal homologue of BRCA2 [187]. In this manner, it seems likely the DNA binding activity of BRCA2 may be involved in modulating RPA-ssDNA-binding, potentially displacing RPA through competitive ssDNA association. In addition to binding DNA, the first and second OB-folds and the tower domain have also been found to interact with deleted in split-hand/split-foot syndrome 1 (DSS1), a protein associated with BRAC2 stability and modulation of DNA binding activity [186, 188, 189]. Although a direct interaction between the RPA complex and the GST-tagged N-terminus of BRCA2 has been suggested [190], this has not been supported by more recent studies using full-length BRCA2 [191].
BRCA2 also interacts directly with RAD51 through a series of eight 35 amino acid repeat motifs, known as the BRC repeats [192–195]. Remarkably, the crystal structure of BRC4 bound to RAD51 identified a conserved BRC fingerprint that mimics the RAD51 homodimer interface; this may potentially support stabilisation of the RAD51 monomeric form [196]. Such stabilisation presumably represents a rationale for interaction of the BRC repeats in RAD51 loading [197, 198]. This has been supported by in vitro data, where BRCA2 was shown to increase RAD51-mediated DNA strand exchange of an RPA-coated ssDNA construct [191]. Additionally, a 7-fold increase in RAD51 binding to RPA-coated ssDNA has been observed in the presence of BRCA2, suggesting a role for BRCA2 in RAD51 nucleofilament formation [199]. Recently the BRC repeats have been further classified into two distinct classes, distinguished by their differing RAD51-binding affinities; while the first series (BRC1-4) bind the free protein with high affinity, the second (BRC5-8) strongly interact only when RAD51 is in a filamentous ssDNA-bound form [200]. The existence of these two classes supports the notion that BRCA2 functions in both loading and stabilisation of RAD51 nucleofilaments.
hSSB1 and RAD51 have also been found to co-localise following ionising radiation exposure, which, on the basis of immunoprecipitation data, could be facilitated through a direct interaction [7]. This suggests that hSSB1 may potentially share an overlapping role with RPA in the modulation of Rad51-mediated strand invasion. Such a suggestion is consistent with in vitro data, where hSSB1 was able to stimulate Rad51-mediated strand exchange to a similar degree as RPA [7]. Further, depletion of the hSSB1 binding partner, INTS3, was shown to suppress RAD51 foci formation, as well as the recruitment of topoisomerase binding protein 1 (TopBP1) and BRCA1 to sites of DNA damage [169]. Interestingly however, using high-resolution microscopy, RPA and hSSB1 were not shown to directly localise at radiation-induced foci, but to form proximal centres [7]. This may suggest that while functional overlap between these proteins exists, their individual roles at each DSB site may be independent, representing different stages of processivity.
Nucleotide excision repair requires the action of SSBs
Nucleotide excision repair is an important pathway in the maintenance of genomic stability, as it represents an essential mechanism through which the cell is able to remove a large number of structurally varied adducts. These adducts may arise through either endogenous or exogenous means and are recognised as a result of the ensuing disruption to base pairing and helical distortion, as opposed to their precise chemical nature [153]. However, adducts caused by ultraviolet (UV) light-induced damage, such as cyclobutane pyrimidine dimers (CPDs) and 6–4 photoproducts, do represent primary targets. This is demonstrated by the conditions Xeroderma pigmentosum (XP), Cockayne syndrome (CS) and Trichothiodystrophy (TTD); these conditions are all associated with diminished NER and severe photosensitivity [201]. In functional cells, NER may occur through two related pathways: global genomic NER (GG-NER), which removes lesions non-specifically throughout the genome, and transcription-coupled NER (TC-NER), which removes lesions from actively transcribed genes.
RPA has a well-established role in NER, where it functions to both stabilise exposed ssDNA and to recruit repair proteins at these sites [202, 203]. Of these, the XP proteins (XPA – XPG) play a direct function both in the recognition and excision of damaged nucleotides [204]. In the earliest stages of NER, damage is recognised by a complex of the XPC protein and hHR23B [205]. This allows unwinding of the lesion, a function further facilitated by the helicase activity of the TFIIH transcription factor [206]. Additionally, this process is stimulated by XPA, which recognises damaged DNA and seems to function in the recruitment of TFIIH [207, 208]. Here, XPA binding of damaged DNA is promoted through the direct interaction with RPA [204, 209, 210]; this interaction is facilitated both by the RPA1 and 2 subunits [211, 212]. Through promotion of TFIIH helicase activity (facilitated by the XPB and XPD components), RPA-XPA expands the damage region [213, 214]. RPA2 also interacts directly with the XPF-ERCC1 exonuclease, while RPA1 interacts with XPG [31, 215–218]. As a result of the differential interaction regions for these proteins, and the binding polarity of RPA, RPA can correctly position XPF-ERCC1 and XPG at sites of damage; XPG is recruited 3′ to the lesion, while XPF-ERCC1 binds on the 5′ side [31, 215, 217]. In addition, by binding the undamaged DNA strand, RPA is able to direct ERCC1-XPF nuclease activity to the site of damage on the opposing strand [31].
In response to UV treatment, RPA2 phosphorylation is mediated through the combined efforts of the ATR and DNA-PK kinases, leading to replication arrest [69, 71, 72, 78, 97, 219]. Interestingly, hSSB1 is also stabilised following exposure to UV, and is localised to sites of the resulting DNA damage [7]. It will therefore be of interest to determine whether hSSB1 plays a role in NER.
SSBs are necessary for cell cycle checkpoint activation following DNA damage
An essential step to prevent the propagation of damaged DNA, either by its replication or division amongst daughter cells, is the damage-induced suppression of cell cycle progression. This is achieved through the activation of cell cycle checkpoints, a cascade of signalling events resulting in the inhibition of cyclin/CDK-mediated checkpoint progression [220, 221]. In mammalian cells, initiation of checkpoint signalling is largely facilitated by two members of the PI3 kinase-related kinases family, ATM and ATR [222–224]. For both ATM and ATR, activation results in the initiation of downstream signalling, facilitating activation of effector kinases, namely Chk1 and Chk2 [225]. Although Chk1 activation is predominantly mediated by ATR, and Chk2 activation predominantly by ATM, considerable crossover is also apparent between these pathways [225–230]. In addition, a temporal contrast exists in the primary initiation of ATM and ATR signalling; while ATM is activated prior to DSB resection as a result of MRN recruitment [221], ATR activity is stimulated by RPA-coating of ssDNA strands [89, 231, 232].
ssDNA-bound RPA partially activates ATR through a direct interaction between the RPA1 N-terminus and the ATR-interacting protein (ATRIP) [89, 161, 232] (Figure 3A). This is supported by suppressed phosphorylation of the CHK1 and Rad17 ATR substrates following RPA1 depletion [89]. Disruption of the RPA-ATRIP interaction is however not sufficient to eliminate localisation of the ATR-ATRIP complex to sites of DNA damage [232], and full RPA-mediated activation requires an additional interaction with the checkpoint complex Rad9/Rad1/Hus1 (9-1-1)/ Rad17-Rfc2-5 [89, 233, 234]. Here, the 9-1-1 component forms a PCNA-like sliding clamp [235], which seems to interact directly with the RPA1 N-terminal OB-fold [94], as well as with the topoisomerase-binding protein 1 (TopBP1) [224, 236, 237]. In turn, recruitment and activation of TopBPI is seen to stimulate the kinase activity of ATR, amplifying downstream signalling [238].

SSBs function in cell cycle checkpoint activation. (A) Schematic of the potential role of RPA in ATR signalling: RPA coated ssDNA stimulates ATR signalling both through direct interaction with its binding partner ATRIP, as well as through the 9-1-1 complex. Activated ATR phosphorylates various cell cycle checkpoint proteins, including p53 and CHK1, allowing for cell cycle arrest. RPA also interacts with RFWD3, an E3 ubiquitin ligase involved in p53 stabilisation. (B) Schematic of the potential role of hSSB1 and Strap in ATM signalling: hSSB1 activation allows for the recruitment of the MRN complex, which activates the ATM kinase. ATM phosphorylation of hSSB1 then facilitates a positive feedback loop, which further activates ATM, as well as many downstream ATM targets. hSSB1 also interacts with and stabilises p53 and p21 (an effector of p53 signalling), as well as the p53-interacting acetyltransferase p300. Strap, a phosphorylation substrate of ATM, interacts with and stabilises p53, and additionally directs it to transcriptional targets.
Recent data has suggested that RPA also mediates p53 activity by interaction with the RING finger and WD repeat domain 3 (RFWD3) E3 ubiquitin ligase [239, 240]. RFWD3 has been identified through proteomic screening as a substrate of ATR kinase [241–243], and is important for CHK1 activation [239]. In response to replication-stress and DNA damage, RFWD3 is rapidly recruited to RPA coated ssDNA stretches [239, 240, 244]. Here it interacts directly with murine double minute 2 (Mdm2), the major E3 ubiquitin ligase involved in p53 poly-ubiquitination [244]. While suppression of p53 poly-ubiquitination immediately following damage appears to be facilitated through various post-translational means [245], competitive ubiquitination by RFWD3 seems to present a method of stabilisation during the later phase (after 2.5 hours) [244]. This may occur through the generation of smaller non-proteasome-targeting ubiquitin chains. In addition to this role, RFWD3 may also have a more direct role in DSB repair, as suggested by disrupted RPA phosphorylation following DNA damage in RFWD3 depleted cells [240]. As yet however, the mechanism through which this occurs remains unclear.
Activation of the ATM kinase is one of the most immediate consequences of MRN recruitment to DSB foci. In addition to a large number of other substrates, this activation allows for the ATM-mediated phosphorylation of hSSB1, facilitating a positive feedback loop that amplifies ATM signalling (Figure 3B). This was demonstrated by mutation of the ATM-mediated hSSB1 phosphorylation site (T117), which led to suppression of ATM auto-phosphorylation [7]. In addition, depletion of hSSB1 from cells demonstrated defective ATM-checkpoint signalling, including loss of G1/S and G2/M checkpoint activation, as well as suppressed NBS1, p53, CHK1 and CHK2 phosphorylation [7]. Interestingly, despite this central role for hSSB1, hSSB2 has been reported as dispensable for ATM activation and checkpoint arrest [1].
hSSB1 has also been seen to interact directly with the cyclin-dependent kinase inhibitor p21 [246], an important effector of p53-mediated G1-S and G2-M checkpoint arrest [247–251]. This is achieved through the inhibition of cyclin E/CDK2 and cyclin B/CDK2 activity, arresting cell cycle progression. In response to DNA damage, hSSB1 was suggested to bind p21 [246], where it inhibits the ubiquitin-mediated degradation associated with the labile protein [252–254]. More recent data has indicated that hSSB1 may also function to promote p53 stabilisation through a direct interaction [255]. Additionally, hSSB1 depletion was found to suppress p300-mediated acetylation of p53 [255], an important modification associated with p53 transcriptional activity. This was supported by the decreased expression of p21 and SULF2, two p53 transcriptional targets [247, 256], following hSSB1 depletion [255]. Together, these data suggest a further upstream function of hSSB1 in the p53 pathway; in addition to amplification of ATM signalling, hSSB1 may regulate the G1/S and G2/M transitions.
Recently, a novel OB-fold was reported in Strap, an important cofactor of p53 [257, 258]. In response to DNA damage, ATM-mediated phosphorylation of Strap allows for nuclear accumulation of the protein, while protein stabilisation is achieved following phosphorylation by CHK2 [259, 260]. This is supported by the observation that in ataxia telangiectasia cells, or cells expressing Strap mutated at the S203 ATM phosphorylation site, Strap is restricted to the cytoplasm [260]. Following damage, localised Strap interacts with other components of the p300 coactivator complex, including p300, the junction mediating and regulatory (JMY) protein, and protein arginine methyltransferase 5 (PRMT5) [258, 261–263]. Such protein interactions are facilitated by both the Strap N-terminus, which contains six tetratricopeptide repeat (TPR) motifs [258], as well as the C-terminus, which largely consists of the OB-fold [257]. Here, JMY is bound by two N-terminal domains [258], identified as TRP motifs 1–3, and 4–5, while TRP6 and the OB-fold interact directly with p300 [257]. As part of the p300 complex, Strap has been shown to promote p53 activity both by promoting stabilisation of p53 through suppression of MDM2-mediated poly-ubiquitination, as well as by stimulating p53 transcription modulating activation [258]. In agreement with the later, in vitro data has indicated the Strap OB-fold is able to bind ssDNA and dsDNA, while chromatin immunoprecipitation has demonstrated localisation of Strap, as well as the TRP and OB-fold domains, at p53 target genes [257].
ssDNA at telomere ends are bound by SSBs
The replication of chromosome ends presents a unique challenge in eukaryotic cells. This arises from the strict requirement of a primed template from which 5′-3′ extension can occur, a constraint preventing the replication of chromosome 3′ terminal regions. To overcome this ‘end-replication problem’, eukaryotic chromosome ends are comprised of protein-nucleic acid structures known as telomeres. These structures contain telomeric DNA arranged in a series of repeats, the sequence of which varies between organisms. In humans, telomeric DNA is composed of precise hexanucleotide (CCCTAA/TTAGGG) repeats constituting 2–50 kb of code (3,000-80,000 repeats) [21, 264, 265]. While telomere repeats are re-synthesised in germ and embryonic cells by the enzyme telomerase, in somatic cells, shortening of chromosomes to a critical length causes the induction of a senescence phase [264].
An additional consequence of incomplete lagging strand replication is the generation of 3′ ssDNA overhangs on each chromosome end. The precise length of these overhangs is fundamentally determined by the positioning of the final RNA primer, generally ranging in human cells from 30–40 nt for leading strand, and 80–120 nt for lagging strand, daughter chromosomes [265, 266]. The presence of these overhangs offers an additional challenge for eukaryotic cells, as to prevent their degradation or deleterious recognition as a site of DNA damage, they must be in some way sequestered [21]. One mechanism through which this occurs is the formation of a T-loop structures, in which the 3′ overhangs invade an upstream telomere duplex region, and by virtue of the telomere hexanucleotide repeats, form stable interactions at the base of the T-loop [267, 268]. However, while this shields the 3′ termini, ssDNA is still exposed in the displaced section of the telomere duplex (the D-loop). To protect these regions, telomeric ssDNA is protein bound, forming a telomere ‘caps’ [24].
An important component of the telomere cap is the shelterin/telosome complex, a protein complex composed of six subunits: TRF1, TRF2, TIN2, TERF2IP, TPP1, and POT1 [269]. Of these, protection of telomeres 1 (POT1) and TPP1 (also known as POT1-interacting protein; PIP1), form a DNA-binding heterodimer that interacts directly with teleromic ssDNA [270, 271]. Although OB-folds have been detected in both proteins, DNA-binding is predominantly facilitated by POT1, which interacts with strong specificity to 5′-(T)TAGGGTTAG-3′ sequences in the T- and D- loops [272–274]. Crystallography data has indicated that POT1 contains tandem OB-folds, both of which bind ssDNA. This is facilitated by the special arrangement of the OB-folds, where the binding grooves of each domain form a continuous channel, with both OB-folds arranged in-line [273]. In addition, the second of these domains is somewhat modified from the archetypal OB-fold, containing a 31-residue insertion between the first and second β-barrels, similar to that observed in the CTC1 DNA binding domain [275]. While suggested not to bind ssDNA directly, a third POT1 OB-fold has also been suggested in the TPP1 interaction region [271]. In addition, TPP1 also contains a putative OB-fold [270], although it does not appear to bind ssDNA directly, and instead TPP1 localisation at telomeric DNA is mediated by POT1 [270, 271]. A protein-interaction function has however recently been described for the TPP1 OB-fold, where the domain was observed to bind and recruit the telomerase reverse transcriptase (TERT) [276–278]. Interestingly, this is in addition to the observation that POT1-TPP1 binding delays primer dissociation [279], suggesting at least two possible mechanisms through which the dimer may function in telomere processivity. Contrasting this idea however is the observation that, in vitro, POT1 binding of 3′ terminal ends sterically inhibits telomerase access [273]. Furthermore, deletion of the POT1 N-terminal OB-fold resulted in an increase in telomere length in vivo[280]. Together, these seemingly conflicting roles may indicate the POT1-TPP1 heterodimer is involved both in the stimulation and suppression of telomerase activity, however as yet the coordination of these activities remains unclear.
In addition to binding DNA by virtue of the POT1-TPP1 heterodimer, the telomeric repeat-binding factors 1 and 2 (TRF1 and 2), as well as TRF2 interacting protein (TERF2IP, also known as RAP1), also allow the shelterin complex to bind dsDNA at sites adjacent to the T-loop [269, 281]. As with POT1, DNA-binding of TRF1 and 2 is highly sequence specific, with both proteins recognising telomeric duplex DNA of sequence 5′-GTTAGGGTTAGGG-3′[282–284]. Such sequence-specificity is however not evident for TERF2IP, where in vitro data has demonstrated similar binding of the protein with telomeric and non-telomeric DNA [281]. Even so, TERF2IP recruitment remains sequence-specific in vivo, based on the requirement of TERF2 interaction for efficient DNA binding [285]. Another important factor of teleromic DNA protection is stabilisation of the shelterin complex, a process facilitated by the scaffolding protein TIN2, which couples TRF1 with TRF2, as well as linking TRF1 to TPP1 [279, 286, 287]. Additionally, this also represents a mechanism through which TRF1 and 2 are able to recruit POT1 by indirect interaction with TPP1 [288, 289].
A central function of the shelterin complex is to prevent induction of a DSB response against exposed telomeric DNA. Recently this has been demonstrated by double knockout of TRF1 and 2 in mouse embryonic fibroblasts, leading to generation of shelterin-free telomeres. Here, both ATM and ATR responses were elicited against the exposed chromosome ends, as indicated by the accumulation of 53BP1 foci, CHK1 and 2 phosphorylation, and increased telomere fusion events [290]. These data were consistent with previous observations in senescent cells [291, 292], as well as cells where TRF2 and/or POT1 was depleted [293–296]. Interestingly, TRF2 and POT1 seem to play independent roles in the suppression of DSB signalling; while ATM suppression seems to occur through TRF2, POT1 is involved in the prevention of ATR activation [293]. Here, POT1 suppression of ATR signalling was suggested to be through the steric inhibition of RPA localisation, a process described in previous sections as essential for such signalling. Interestingly however, RPA is known to bind telomeres during S-phase where it is suggested to promote telomerase activity [297, 298]. Although POT1 is unable to displace RPA on its own, the switch from RPA to POT1 binding seems to be facilitated outside of S-phase by the heterogenous nuclear ribonucleoprotein A1 (hnRNPA1) [299].
An additional protein complex central for the capping of telomeres is composed of the proteins CTC1 (CDC13), STN1 and TEN1 [24, 300–302]. Together, this complex (CST) constitutes a ssDNA-binding unit which interacts independently of POT1 [301]. Furthermore, as each of these subunits contain putative OB-fold domains, structural and functional similarities have been drawn with the RPA heterotrimer [302, 303]. An exception to this is the presence of an additional C-terminal winged helix-turn helix (wHTH) motif on the STN1 subunit, which confers a telomere-specific binding function [24]. CTC1 appears to contain 3 OB-folds, in contrast to the 4 of RPA1, two of which have been further confirmed by structural analysis [275, 304–306]; here, ssDNA-binding is largely facilitated by the CTC1 C-terminal OB-fold [307, 308]. Currently the end-protection role of CST remains unclear, and indeed while some authors have reported de-protection of telomeres following CST component depletion [301, 309], others have suggested only a minimal effect [302, 310]. Furthermore, while it seems uncontested that CST functions in telomere length control, both telomere lengthening [302], and shortening [310] has been reported in cells deficient of CST. As the N-terminal OB-fold of CTC1 is known to interact with telomerase [304, 308], these length control functions are likely due to regulation of telomerase activity. As yet the coordination of this function, as with the POT1-TPP1 dimer, remains unclear.
Summary
SSBs from the OB domain family play an essential role in the maintenance of genome stability, functioning in DNA replication, the repair of damaged DNA, the activation of cell cycle checkpoints, and in telomere maintenance. The importance of SSBs in these processes is highlighted by their ubiquitous nature in all kingdoms of life [1]. Here, in addition to genome stability maintenance, SSBs function in all known processes involving the exposure of ssDNA, such as transcriptional activation [311]. In humans, RPA has long been known to play an important role in the processing of ssDNA, however the recent identification of hSSB1 and 2 has raised several questions regarding the coordination of these processes. Additionally, the diversity of OB-fold primary sequences has made it difficult to detect these domains by non-biophysical means, allowing for the continued identification of OB-folds in previously identified proteins. This is highlighted by the recent identification of Strap and the RMI dimer, and suggests that further SSBs are likely to be identified.
References
Richard DJ, Bolderson E, Khanna KK: Multiple human single-stranded DNA binding proteins function in genome maintenance: structural, biochemical and functional analysis. Crit Rev Biochem Mol Biol. 2009, 44 (2–3): 98-116.
Broderick S, Rehmet K, Concannon C, Nasheuer HP: Eukaryotic single-stranded DNA binding proteins: central factors in genome stability. Subcell Biochem. 2010, 50: 143-163.
Flynn RL, Zou L: Oligonucleotide/oligosaccharide-binding fold proteins: a growing family of genome guardians. Crit Rev Biochem Mol Biol. 2010, 45 (4): 266-275.
Horvath MP: Structural anatomy of telomere OB proteins. Crit Rev Biochem Mol Biol. 2011, 46 (5): 409-435.
Oliveira MT, Kaguni LS: Functional roles of the N- and C-terminal regions of the human mitochondrial single-stranded DNA-binding protein. PLoS One. 2010, 5 (10): e15379-
Datta PK, Moses HL: STRAP and Smad7 synergize in the inhibition of transforming growth factor beta signaling. Mol Cell Biol. 2000, 20 (9): 3157-3167.
Richard DJ, Bolderson E, Cubeddu L, Wadsworth RI, Savage K, Sharma GG, Nicolette ML, Tsvetanov S, McIlwraith MJ, Pandita RK: Single-stranded DNA-binding protein hSSB1 is critical for genomic stability. Nature. 2008, 453 (7195): 677-681.
Murzin AG: OB(oligonucleotide/oligosaccharide binding)-fold: common structural and functional solution for non-homologous sequences. EMBO J. 1993, 12 (3): 861-867.
Merrill BM, Williams KR, Chase JW, Konigsberg WH: Photochemical cross-linking of the Escherichia coli single-stranded DNA-binding protein to oligodeoxynucleotides. Identification of phenylalanine 60 as the site of cross-linking. J Biol Chem. 1984, 259 (17): 10850-10856.
Casas-Finet JR, Khamis MI, Maki AH, Chase JW: Tryptophan 54 and phenylalanine 60 are involved synergistically in the binding of E. coli SSB protein to single-stranded polynucleotides. FEBS Lett. 1987, 220 (2): 347-352.
Casas-Finet JR, Khamis MI, Maki AH, Ruvolo PP, Chase JW: Optically detected magnetic resonance of tryptophan residues in Escherichia coli ssb gene product and E. coli plasmid-encoded single-stranded DNA-binding proteins and their complexes with poly(deoxythymidylic) acid. J Biol Chem. 1987, 262 (18): 8574-8583.
Khamis MI, Casas-Finet JR, Maki AH: Stacking interactions of tryptophan residues and nucleotide bases in complexes formed between Escherichia coli single-stranded DNA binding protein and heavy atom-modified poly(uridylic) acid. A study by optically detected magnetic resonance spectroscopy. J Biol Chem. 1987, 262 (4): 1725-1733.
Khamis MI, Casas-Finet JR, Maki AH, Murphy JB, Chase JW: Role of tryptophan 54 in the binding of E. coli single-stranded DNA-binding protein to single-stranded polynucleotides. FEBS Lett. 1987, 211 (2): 155-159.
Bujalowski W, Overman LB, Lohman TM: Binding mode transitions of Escherichia coli single strand binding protein-single-stranded DNA complexes. Cation, anion, pH, and binding density effects. J Biol Chem. 1988, 263 (10): 4629-4640.
Lohman TM, Bujalowski W, Overman LB, Wei TF: Interactions of the E. coli single strand binding (SSB) protein with ss nucleic acids. Binding mode transitions and equilibrium binding studies. Biochem Pharmacol. 1988, 37 (9): 1781-1782.
Overman LB, Lohman TM: Linkage of pH, anion and cation effects in protein-nucleic acid equilibria. Escherichia coli SSB protein-single stranded nucleic acid interactions. J Mol Biol. 1994, 236 (1): 165-178.
Overman LB, Bujalowski W, Lohman TM: Equilibrium binding of Escherichia coli single-strand binding protein to single-stranded nucleic acids in the (SSB)65 binding mode. Cation and anion effects and polynucleotide specificity. Biochemistry (Mosc). 1988, 27 (1): 456-471.
Curth U, Greipel J, Urbanke C, Maass G: Multiple binding modes of the single-stranded DNA binding protein from Escherichia coli as detected by tryptophan fluorescence and site-directed mutagenesis. Biochemistry (Mosc). 1993, 32 (10): 2585-2591.
Raghunathan S, Kozlov AG, Lohman TM, Waksman G: Structure of the DNA binding domain of E. coli SSB bound to ssDNA. Nat Struct Biol. 2000, 7 (8): 648-652.
Savvides SN, Raghunathan S, Futterer K, Kozlov AG, Lohman TM, Waksman G: The C-terminal domain of full-length E. coli SSB is disordered even when bound to DNA. Protein Sci. 2004, 13 (7): 1942-1947.
de Lange T: T-loops and the origin of telomeres. Nat Rev Mol Cell Biol. 2004, 5 (4): 323-329.
Ferrari ME, Lohman TM: Apparent heat capacity change accompanying a nonspecific protein-DNA interaction. Escherichia coli SSB tetramer binding to oligodeoxyadenylates. Biochemistry (Mosc). 1994, 33 (43): 12896-12910.
Kozlov AG, Lohman TM: Calorimetric studies of E. coli SSB protein-single-stranded DNA interactions. Effects of monovalent salts on binding enthalpy. J Mol Biol. 1998, 278 (5): 999-1014.
Gelinas AD, Paschini M, Reyes FE, Heroux A, Batey RT, Lundblad V, Wuttke DS: Telomere capping proteins are structurally related to RPA with an additional telomere-specific domain. Proc Natl Acad Sci USA. 2009, 106 (46): 19298-19303.
Fan J, Pavletich NP: Structure and conformational change of a replication protein A heterotrimer bound to ssDNA. Genes Dev. 2012, 26 (20): 2337-2347.
Bochkareva E, Korolev S, Lees-Miller SP, Bochkarev A: Structure of the RPA trimerization core and its role in the multistep DNA-binding mechanism of RPA. EMBO J. 2002, 21 (7): 1855-1863.
Iftode C, Daniely Y, Borowiec JA: Replication protein A (RPA): the eukaryotic SSB. Crit Rev Biochem Mol Biol. 1999, 34 (3): 141-180.
Arunkumar AI, Stauffer ME, Bochkareva E, Bochkarev A, Chazin WJ: Independent and coordinated functions of replication protein A tandem high affinity single-stranded DNA binding domains. J Biol Chem. 2003, 278 (42): 41077-41082.
Blackwell LJ, Borowiec JA: Human replication protein A binds single-stranded DNA in two distinct complexes. Mol Cell Biol. 1994, 14 (6): 3993-4001.
Jiang X, Klimovich V, Arunkumar AI, Hysinger EB, Wang Y, Ott RD, Guler GD, Weiner B, Chazin WJ, Fanning E: Structural mechanism of RPA loading on DNA during activation of a simple pre-replication complex. EMBO J. 2006, 25 (23): 5516-5526.
de Laat WL, Appeldoorn E, Sugasawa K, Weterings E, Jaspers NG, Hoeijmakers JH: DNA-binding polarity of human replication protein A positions nucleases in nucleotide excision repair. Genes Dev. 1998, 12 (16): 2598-2609.
Iftode C, Borowiec JA: 5′ –> 3′ molecular polarity of human replication protein A (hRPA) binding to pseudo-origin DNA substrates. Biochemistry (Mosc). 2000, 39 (39): 11970-11981.
Kolpashchikov DM, Khodyreva SN, Khlimankov DY, Wold MS, Favre A, Lavrik OI: Polarity of human replication protein A binding to DNA. Nucleic Acids Res. 2001, 29 (2): 373-379.
Richard DJ, Cubeddu L, Urquhart AJ, Bain A, Bolderson E, Menon D, White MF, Khanna KK: hSSB1 interacts directly with the MRN complex stimulating its recruitment to DNA double-strand breaks and its endo-nuclease activity. Nucleic Acids Res. 2011, 39 (9): 3643-3651.
Skaar JR, Richard DJ, Saraf A, Toschi A, Bolderson E, Florens L, Washburn MP, Khanna KK, Pagano M: INTS3 controls the hSSB1-mediated DNA damage response. J Cell Biol. 2009, 187 (1): 25-32.
Yang SH, Zhou R, Campbell J, Chen J, Ha T, Paull TT: The SOSS1 single-stranded DNA binding complex promotes DNA end resection in concert with Exo1. EMBO J. 2013, 32 (1): 126-139.
Richard DJ, Savage K, Bolderson E, Cubeddu L, So S, Ghita M, Chen DJ, White MF, Richard K, Prise KM: hSSB1 rapidly binds at the sites of DNA double-strand breaks and is required for the efficient recruitment of the MRN complex. Nucleic Acids Res. 2011, 39 (5): 1692-1702.
Cubeddu L, White MF: DNA damage detection by an archaeal single-stranded DNA-binding protein. J Mol Biol. 2005, 353 (3): 507-516.
Lohman TM, Ferrari ME: Escherichia coli single-stranded DNA-binding protein: multiple DNA-binding modes and cooperativities. Annu Rev Biochem. 1994, 63: 527-570.
Blackwell LJ, Borowiec JA, Mastrangelo IA: Single-stranded-DNA binding alters human replication protein A structure and facilitates interaction with DNA-dependent protein kinase. Mol Cell Biol. 1996, 16 (9): 4798-4807.
Bell SP, Dutta A: DNA replication in eukaryotic cells. Annu Rev Biochem. 2002, 71: 333-374.
Yuzhakov A, Kelman Z, Hurwitz J, O’Donnell M: Multiple competition reactions for RPA order the assembly of the DNA polymerase delta holoenzyme. EMBO J. 1999, 18 (21): 6189-6199.
Dornreiter I, Erdile LF, Gilbert IU, von Winkler D, Kelly TJ, Fanning E: Interaction of DNA polymerase alpha-primase with cellular replication protein A and SV40 T antigen. EMBO J. 1992, 11 (2): 769-776.
Burgers PM: Polymerase dynamics at the eukaryotic DNA replication fork. J Biol Chem. 2009, 284 (7): 4041-4045.
Garg P, Burgers PM: DNA polymerases that propagate the eukaryotic DNA replication fork. Crit Rev Biochem Mol Biol. 2005, 40 (2): 115-128.
Pellegrini L: The Pol alpha-Primase Complex. Subcell Biochem. 2012, 62: 157-169.
Nunez-Ramirez R, Klinge S, Sauguet L, Melero R, Recuero-Checa MA, Kilkenny M, Perera RL, Garcia-Alvarez B, Hall RJ, Nogales E: Flexible tethering of primase and DNA Pol alpha in the eukaryotic primosome. Nucleic Acids Res. 2011, 39 (18): 8187-8199.
Braun KA, Lao Y, He Z, Ingles CJ, Wold MS: Role of protein-protein interactions in the function of replication protein A (RPA): RPA modulates the activity of DNA polymerase alpha by multiple mechanisms. Biochemistry (Mosc). 1997, 36 (28): 8443-8454.
Waga S, Stillman B: The DNA replication fork in eukaryotic cells. Annu Rev Biochem. 1998, 67: 721-751.
Burgers PM: Mammalian cyclin/PCNA (DNA polymerase delta auxiliary protein) stimulates processive DNA synthesis by yeast DNA polymerase III. Nucleic Acids Res. 1988, 16 (14A): 6297-6307.
Krishna TS, Kong XP, Gary S, Burgers PM, Kuriyan J: Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell. 1994, 79 (7): 1233-1243.
Schurtenberger P, Egelhaaf SU, Hindges R, Maga G, Jonsson ZO, May RP, Glatter O, Hubscher U: The solution structure of functionally active human proliferating cell nuclear antigen determined by small-angle neutron scattering. J Mol Biol. 1998, 275 (1): 123-132.
Adachi Y, Laemmli UK: Identification of nuclear pre-replication centers poised for DNA synthesis in Xenopus egg extracts: immunolocalization study of replication protein A. J Cell Biol. 1992, 119 (1): 1-15.
Dimitrova DS, Todorov IT, Melendy T, Gilbert DM: Mcm2, but not RPA, is a component of the mammalian early G1-phase prereplication complex. J Cell Biol. 1999, 146 (4): 709-722.
Henry RA, Balakrishnan L, Ying-Lin ST, Campbell JL, Bambara RA: Components of the secondary pathway stimulate the primary pathway of eukaryotic Okazaki fragment processing. J Biol Chem. 2010, 285 (37): 28496-28505.
Ayyagari R, Gomes XV, Gordenin DA, PM B: Okazaki fragment maturation in yeast. I. Distribution of functions between FEN1 AND DNA2. J Biol Chem. 2003, 278 (3): 1618-1625.
Garg P, Stith CM, Sabouri N, Johansson E, Burgers PM: Idling by DNA polymerase delta maintains a ligatable nick during lagging-strand DNA replication. Genes Dev. 2004, 18 (22): 2764-2773.
Jin YH, Ayyagari R, Resnick MA, Gordenin DA, Burgers PM: Okazaki fragment maturation in yeast. II. Cooperation between the polymerase and 3′-5′-exonuclease activities of Pol delta in the creation of a ligatable nick. J Biol Chem. 2003, 278 (3): 1626-1633.
Rossi ML, Bambara RA: Reconstituted Okazaki fragment processing indicates two pathways of primer removal. J Biol Chem. 2006, 281 (36): 26051-26061.
Rossi ML, Pike JE, Wang W, Burgers PM, Campbell JL, Bambara RA: Pif1 helicase directs eukaryotic Okazaki fragments toward the two-nuclease cleavage pathway for primer removal. J Biol Chem. 2008, 283 (41): 27483-27493.
Bae SH, Bae KH, Kim JA, Seo YS: RPA governs endonuclease switching during processing of Okazaki fragments in eukaryotes. Nature. 2001, 412 (6845): 456-461.
Bae SH, Seo YS: Characterization of the enzymatic properties of the yeast dna2 Helicase/endonuclease suggests a new model for Okazaki fragment processing. J Biol Chem. 2000, 275 (48): 38022-38031.
Dutta A, Stillman B: cdc2 family kinases phosphorylate a human cell DNA replication factor, RPA, and activate DNA replication. EMBO J. 1992, 11 (6): 2189-2199.
Henricksen LA, Wold MS: Replication protein A mutants lacking phosphorylation sites for p34cdc2 kinase support DNA replication. J Biol Chem. 1994, 269 (39): 24203-24208.
Lee SH, Kim DK: The role of the 34-kDa subunit of human replication protein A in simian virus 40 DNA replication in vitro. J Biol Chem. 1995, 270 (21): 12801-12807.
Fang F, Newport JW: Distinct roles of cdk2 and cdc2 in RP-A phosphorylation during the cell cycle. J Cell Sci. 1993, 106 (Pt 3): 983-994.
Oakley GG, Patrick SM, Yao J, Carty MP, Turchi JJ, Dixon K: RPA phosphorylation in mitosis alters DNA binding and protein-protein interactions. Biochemistry (Mosc). 2003, 42 (11): 3255-3264.
Cuvier O, Lutzmann M, Mechali M: ORC is necessary at the interphase-to-mitosis transition to recruit cdc2 kinase and disassemble RPA foci. Curr Biol. 2006, 16 (5): 516-523.
Park JS, Park SJ, Peng X, Wang M, Yu MA, Lee SH: Involvement of DNA-dependent protein kinase in UV-induced replication arrest. J Biol Chem. 1999, 274 (45): 32520-32527.
Vassin VM, Wold MS, Borowiec JA: Replication protein A (RPA) phosphorylation prevents RPA association with replication centers. Mol Cell Biol. 2004, 24 (5): 1930-1943.
Carty MP, Zernik-Kobak M, McGrath S, Dixon K: UV light-induced DNA synthesis arrest in HeLa cells is associated with changes in phosphorylation of human single-stranded DNA-binding protein. EMBO J. 1994, 13 (9): 2114-2123.
Shao RG, Cao CX, Zhang H, Kohn KW, Wold MS, Pommier Y: Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. EMBO J. 1999, 18 (5): 1397-1406.
Liu VF, Weaver DT: The ionizing radiation-induced replication protein A phosphorylation response differs between ataxia telangiectasia and normal human cells. Mol Cell Biol. 1993, 13 (12): 7222-7231.
Vassin VM, Anantha RW, Sokolova E, Kanner S, Borowiec JA: Human RPA phosphorylation by ATR stimulates DNA synthesis and prevents ssDNA accumulation during DNA-replication stress. J Cell Sci. 2009, 122 (Pt 22): 4070-4080.
Brush GS, Anderson CW, Kelly TJ: The DNA-activated protein kinase is required for the phosphorylation of replication protein A during simian virus 40 DNA replication. Proc Natl Acad Sci USA. 1994, 91 (26): 12520-12524.
Niu H, Erdjument-Bromage H, Pan ZQ, Lee SH, Tempst P, Hurwitz J: Mapping of amino acid residues in the p34 subunit of human single-stranded DNA-binding protein phosphorylated by DNA-dependent protein kinase and Cdc2 kinase in vitro. J Biol Chem. 1997, 272 (19): 12634-12641.
Pan ZQ, Amin AA, Gibbs E, Niu H, Hurwitz J: Phosphorylation of the p34 subunit of human single-stranded-DNA-binding protein in cyclin A-activated G1 extracts is catalyzed by cdk-cyclin A complex and DNA-dependent protein kinase. Proc Natl Acad Sci USA. 1994, 91 (18): 8343-8347.
Zernik-Kobak M, Vasunia K, Connelly M, Anderson CW, Dixon K: Sites of UV-induced phosphorylation of the p34 subunit of replication protein A from HeLa cells. J Biol Chem. 1997, 272 (38): 23896-23904.
Oakley GG, Loberg LI, Yao J, Risinger MA, Yunker RL, Zernik-Kobak M, Khanna KK, Lavin MF, Carty MP, Dixon K: UV-induced hyperphosphorylation of replication protein a depends on DNA replication and expression of ATM protein. Mol Biol Cell. 2001, 12 (5): 1199-1213.
Gately DP, Hittle JC, Chan GK, Yen TJ: Characterization of ATM expression, localization, and associated DNA-dependent protein kinase activity. Mol Biol Cell. 1998, 9 (9): 2361-2374.
Chan DW, Son SC, Block W, Ye R, Khanna KK, Wold MS, Douglas P, Goodarzi AA, Pelley J, Taya Y: Purification and characterization of ATM from human placenta. A manganese-dependent, wortmannin-sensitive serine/threonine protein kinase. J Biol Chem. 2000, 275 (11): 7803-7810.
Wang H, Guan J, Perrault AR, Wang Y, Iliakis G: Replication protein A2 phosphorylation after DNA damage by the coordinated action of ataxia telangiectasia-mutated and DNA-dependent protein kinase. Cancer Res. 2001, 61 (23): 8554-8563.
Stephan H, Concannon C, Kremmer E, Carty MP, Nasheuer HP: Ionizing radiation-dependent and independent phosphorylation of the 32-kDa subunit of replication protein A during mitosis. Nucleic Acids Res. 2009, 37 (18): 6028-6041.
Francon P, Lemaitre JM, Dreyer C, Maiorano D, Cuvier O, Mechali M: A hypophosphorylated form of RPA34 is a specific component of pre-replication centers. J Cell Sci. 2004, 117 (Pt 21): 4909-4920.
Feng J, Wakeman T, Yong S, Wu X, Kornbluth S, Wang XF: Protein phosphatase 2A-dependent dephosphorylation of replication protein A is required for the repair of DNA breaks induced by replication stress. Mol Cell Biol. 2009, 29 (21): 5696-5709.
Petermann E, Helleday T: Pathways of mammalian replication fork restart. Nat Rev Mol Cell Biol. 2010, 11 (10): 683-687.
Schwartz M, Zlotorynski E, Kerem B: The molecular basis of common and rare fragile sites. Cancer Lett. 2006, 232 (1): 13-26.
Friedberg EC: Suffering in silence: the tolerance of DNA damage. Nat Rev Mol Cell Biol. 2005, 6 (12): 943-953.
Zou L, Elledge SJ: Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003, 300 (5625): 1542-1548.
Robison JG, Elliott J, Dixon K, Oakley GG: Replication protein A and the Mre11.Rad50.Nbs1 complex co-localize and interact at sites of stalled replication forks. J Biol Chem. 2004, 279 (33): 34802-34810.
Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA: Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005, 19 (9): 1040-1052.
Recolin B, Van der Laan S, Maiorano D: Role of replication protein A as sensor in activation of the S-phase checkpoint in Xenopus egg extracts. Nucleic Acids Res. 2012, 40 (8): 3431-3442.
Manthey KC, Opiyo S, Glanzer JG, Dimitrova D, Elliott J, Oakley GG: NBS1 mediates ATR-dependent RPA hyperphosphorylation following replication-fork stall and collapse. J Cell Sci. 2007, 120 (Pt 23): 4221-4229.
Xu X, Vaithiyalingam S, Glick GG, Mordes DA, Chazin WJ, Cortez D: The basic cleft of RPA70N binds multiple checkpoint proteins, including RAD9, to regulate ATR signaling. Mol Cell Biol. 2008, 28 (24): 7345-7353.
Olson E, Nievera CJ, Liu E, Lee AY, Chen L, Wu X: The Mre11 complex mediates the S-phase checkpoint through an interaction with replication protein A. Mol Cell Biol. 2007, 27 (17): 6053-6067.
Falck J, Petrini JH, Williams BR, Lukas J, Bartek J: The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat Genet. 2002, 30 (3): 290-294.
Olson E, Nievera CJ, Klimovich V, Fanning E, Wu X: RPA2 is a direct downstream target for ATR to regulate the S-phase checkpoint. J Biol Chem. 2006, 281 (51): 39517-39533.
Block WD, Yu Y, Lees-Miller SP: Phosphatidyl inositol 3-kinase-like serine/threonine protein kinases (PIKKs) are required for DNA damage-induced phosphorylation of the 32 kDa subunit of replication protein A at threonine 21. Nucleic Acids Res. 2004, 32 (3): 997-1005.
Shi W, Feng Z, Zhang J, Gonzalez-Suarez I, Vanderwaal RP, Wu X, Powell SN, Roti Roti JL, Gonzalo S: The role of RPA2 phosphorylation in homologous recombination in response to replication arrest. Carcinogenesis. 2010, 31 (6): 994-1002.
Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M: Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011, 145 (4): 529-542.
Unsal-Kacmaz K, Chastain PD, Qu PP, Minoo P, Cordeiro-Stone M, Sancar A, Kaufmann WK: The human Tim/Tipin complex coordinates an Intra-S checkpoint response to UV that slows replication fork displacement. Mol Cell Biol. 2007, 27 (8): 3131-3142.
Kemp MG, Akan Z, Yilmaz S, Grillo M, Smith-Roe SL, Kang TH, Cordeiro-Stone M, Kaufmann WK, Abraham RT, Sancar A: Tipin-replication protein A interaction mediates Chk1 phosphorylation by ATR in response to genotoxic stress. J Biol Chem. 2010, 285 (22): 16562-16571.
Errico A, Costanzo V: Mechanisms of replication fork protection: a safeguard for genome stability. Crit Rev Biochem Mol Biol. 2012, 47 (3): 222-235.
Katou Y, Kanoh Y, Bando M, Noguchi H, Tanaka H, Ashikari T, Sugimoto K, Shirahige K: S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature. 2003, 424 (6952): 1078-1083.
Noguchi E, Noguchi C, Du LL, Russell P: Swi1 prevents replication fork collapse and controls checkpoint kinase Cds1. Mol Cell Biol. 2003, 23 (21): 7861-7874.
Noguchi E, Noguchi C, McDonald WH, Yates JR, Russell P: Swi1 and Swi3 are components of a replication fork protection complex in fission yeast. Mol Cell Biol. 2004, 24 (19): 8342-8355.
Krings G, Bastia D: swi1- and swi3-dependent and independent replication fork arrest at the ribosomal DNA of Schizosaccharomyces pombe. Proc Natl Acad Sci USA. 2004, 101 (39): 14085-14090.
Calzada A, Hodgson B, Kanemaki M, Bueno A, Labib K: Molecular anatomy and regulation of a stable replisome at a paused eukaryotic DNA replication fork. Genes Dev. 2005, 19 (16): 1905-1919.
Mohanty BK, Bairwa NK, Bastia D: The Tof1p-Csm3p protein complex counteracts the Rrm3p helicase to control replication termination of Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2006, 103 (4): 897-902.
Hodgson B, Calzada A, Labib K: Mrc1 and Tof1 regulate DNA replication forks in different ways during normal S phase. Mol Biol Cell. 2007, 18 (10): 3894-3902.
Chou DM, Elledge SJ: Tipin and Timeless form a mutually protective complex required for genotoxic stress resistance and checkpoint function. Proc Natl Acad Sci USA. 2006, 103 (48): 18143-18147.
Gotter AL, Suppa C, Emanuel BS: Mammalian TIMELESS and Tipin are evolutionarily conserved replication fork-associated factors. J Mol Biol. 2007, 366 (1): 36-52.
Unsal-Kacmaz K, Mullen TE, Kaufmann WK, Sancar A: Coupling of human circadian and cell cycles by the timeless protein. Mol Cell Biol. 2005, 25 (8): 3109-3116.
Yoshizawa-Sugata N, Masai H: Human Tim/Timeless-interacting protein, Tipin, is required for efficient progression of S phase and DNA replication checkpoint. J Biol Chem. 2007, 282 (4): 2729-2740.
Constantinou A, Tarsounas M, Karow JK, Brosh RM, Bohr VA, Hickson ID, West SC: Werner’s syndrome protein (WRN) migrates Holliday junctions and co-localizes with RPA upon replication arrest. EMBO Rep. 2000, 1 (1): 80-84.
Sanz MM, Proytcheva M, Ellis NA, Holloman WK, German J: BLM, the Bloom’s syndrome protein, varies during the cell cycle in its amount, distribution, and co-localization with other nuclear proteins. Cytogenet Cell Genet. 2000, 91 (1–4): 217-223.
Sakamoto S, Nishikawa K, Heo SJ, Goto M, Furuichi Y, Shimamoto A: Werner helicase relocates into nuclear foci in response to DNA damaging agents and co-localizes with RPA and Rad51. Genes Cells. 2001, 6 (5): 421-430.
Brosh RM, Li JL, Kenny MK, Karow JK, Cooper MP, Kureekattil RP, Hickson ID, Bohr VA: Replication protein A physically interacts with the Bloom’s syndrome protein and stimulates its helicase activity. J Biol Chem. 2000, 275 (31): 23500-23508.
Bugreev DV, Yu X, Egelman EH, Mazin AV: Novel pro- and anti-recombination activities of the Bloom’s syndrome helicase. Genes Dev. 2007, 21 (23): 3085-3094.
Wu L: Wrestling off RAD51: a novel role for RecQ helicases. BioEssays. 2008, 30 (4): 291-295.
Doherty KM, Sommers JA, Gray MD, Lee JW, von Kobbe C, Thoma NH, Kureekattil RP, Kenny MK, Brosh RM: Physical and functional mapping of the replication protein a interaction domain of the werner and bloom syndrome helicases. J Biol Chem. 2005, 280 (33): 29494-29505.
Brosh RM, Orren DK, Nehlin JO, Ravn PH, Kenny MK, Machwe A, Bohr VA: Functional and physical interaction between WRN helicase and human replication protein A. J Biol Chem. 1999, 274 (26): 18341-18350.
Yodh JG, Stevens BC, Kanagaraj R, Janscak P, Ha T: BLM helicase measures DNA unwound before switching strands and hRPA promotes unwinding reinitiation. EMBO J. 2009, 28 (4): 405-416.
Xu D, Guo R, Sobeck A, Bachrati CZ, Yang J, Enomoto T, Brown GW, Hoatlin ME, Hickson ID, Wang W: RMI, a new OB-fold complex essential for Bloom syndrome protein to maintain genome stability. Genes Dev. 2008, 22 (20): 2843-2855.
Yin J, Sobeck A, Xu C, Meetei AR, Hoatlin M, Li L, Wang W: BLAP75, an essential component of Bloom’s syndrome protein complexes that maintain genome integrity. EMBO J. 2005, 24 (7): 1465-1476.
Singh TR, Ali AM, Busygina V, Raynard S, Fan Q, Du CH, Andreassen PR, Sung P, Meetei AR: BLAP18/RMI2, a novel OB-fold-containing protein, is an essential component of the Bloom helicase-double Holliday junction dissolvasome. Genes Dev. 2008, 22 (20): 2856-2868.
Gupta R, Sharma S, Sommers JA, Kenny MK, Cantor SB, Brosh RM: FANCJ (BACH1) helicase forms DNA damage inducible foci with replication protein A and interacts physically and functionally with the single-stranded DNA-binding protein. Blood. 2007, 110 (7): 2390-2398.
Suhasini AN, Brosh RM: Fanconi anemia and Bloom’s syndrome crosstalk through FANCJ-BLM helicase interaction. Trends Genet. 2012, 28 (1): 7-13.
Cybulski KE, Howlett NG: FANCP/SLX4: a Swiss army knife of DNA interstrand crosslink repair. Cell Cycle. 2011, 10 (11): 1757-1763.
Kennedy RD, D’Andrea AD: The Fanconi Anemia/BRCA pathway: new faces in the crowd. Genes Dev. 2005, 19 (24): 2925-2940.
Grompe M, D’Andrea A: Fanconi anemia and DNA repair. Hum Mol Genet. 2001, 10 (20): 2253-2259.
Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, Wahrer DC, Sgroi DC, Lane WS, Haber DA: BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell. 2001, 105 (1): 149-160.
Suhasini AN, Rawtani NA, Wu Y, Sommers JA, Sharma S, Mosedale G, North PS, Cantor SB, Hickson ID, Brosh RM: Interaction between the helicases genetically linked to Fanconi anemia group J and Bloom’s syndrome. EMBO J. 2011, 30 (4): 692-705.
Gong Z, Kim JE, Leung CC, Glover JN, Chen J: BACH1/FANCJ acts with TopBP1 and participates early in DNA replication checkpoint control. Mol Cell. 2010, 37 (3): 438-446.
Leung CC, Gong Z, Chen J, Glover JN: Molecular basis of BACH1/FANCJ recognition by TopBP1 in DNA replication checkpoint control. J Biol Chem. 2011, 286 (6): 4292-4301.
Betous R, Mason AC, Rambo RP, Bansbach CE, Badu-Nkansah A, Sirbu BM, Eichman BF, Cortez D: SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012, 26 (2): 151-162.
Bansbach CE, Betous R, Lovejoy CA, Glick GG, Cortez D: The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 2009, 23 (20): 2405-2414.
Ciccia A, Bredemeyer AL, Sowa ME, Terret ME, Jallepalli PV, Harper JW, Elledge SJ: The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev. 2009, 23 (20): 2415-2425.
Yuan J, Ghosal G, Chen J: The annealing helicase HARP protects stalled replication forks. Genes Dev. 2009, 23 (20): 2394-2399.
Yusufzai T, Kadonaga JT: HARP is an ATP-driven annealing helicase. Science. 2008, 322 (5902): 748-750.
Feldhahn N, Ferretti E, Robbiani DF, Callen E, Deroubaix S, Sellero L, Nussenzweig A, Nussenzweig MC: The hSSB1 orthologue Obfc2b is essential for skeletogenesis but dispensable for the DNA damage response in vivo. EMBO J. 2012, 31 (20): 4045-4056.
Shi W, Bain AL, Schwer B, Al-Ejeh F, Smith C, Wong L, Chai H, Miranda MS, Ho U, Kawaguchi M: Essential Developmental, Genomic Stability, and Tumour Suppressor Functions of the Mouse Orthologue of hSSB1/NABP2. PLoS Genet. 2013, 9 (2): e1003298-
Kannouche PL, Wing J, Lehmann AR: Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell. 2004, 14 (4): 491-500.
Watanabe K, Tateishi S, Kawasuji M, Tsurimoto T, Inoue H, Yamaizumi M: Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004, 23 (19): 3886-3896.
Bienko M, Green CM, Crosetto N, Rudolf F, Zapart G, Coull B, Kannouche P, Wider G, Peter M, Lehmann AR: Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science. 2005, 310 (5755): 1821-1824.
Acharya N, Yoon JH, Gali H, Unk I, Haracska L, Johnson RE, Hurwitz J, Prakash L, Prakash S: Roles of PCNA-binding and ubiquitin-binding domains in human DNA polymerase eta in translesion DNA synthesis. PLoS Genet. 2008, 105 (46): 17724-17729.
Acharya N, Brahma A, Haracska L, Prakash L, Prakash S: Mutations in the ubiquitin binding UBZ motif of DNA polymerase eta do not impair its function in translesion synthesis during replication. Mol Cell Biol. 2007, 27 (20): 7266-7272.
Hendel A, Krijger PH, Diamant N, Goren Z, Langerak P, Kim J, Reissner T, Lee KY, Geacintov NE, Carell T: PCNA ubiquitination is important, but not essential for translesion DNA synthesis in mammalian cells. PLoS Genet. 2011, 7 (9): e1002262-
Andersen PL, Xu F, Xiao W: Eukaryotic DNA damage tolerance and translesion synthesis through covalent modifications of PCNA. Cell Res. 2008, 18 (1): 162-173.
Huttner D, Ulrich HD: Cooperation of replication protein A with the ubiquitin ligase Rad18 in DNA damage bypass. Cell Cycle. 2008, 7 (23): 3629-3633.
Maga G, Villani G, Crespan E, Wimmer U, Ferrari E, Bertocci B, Hubscher U: 8-oxo-guanine bypass by human DNA polymerases in the presence of auxiliary proteins. Nature. 2007, 447 (7144): 606-608.
Polo SE, Jackson SP: Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011, 25 (5): 409-433.
Hoeijmakers JH: Genome maintenance mechanisms for preventing cancer. Nature. 2001, 411 (6835): 366-374.
Lord CJ, Ashworth A: The DNA damage response and cancer therapy. Nature. 2012, 481 (7381): 287-294.
Bekker-Jensen S, Mailand N: Assembly and function of DNA double-strand break repair foci in mammalian cells. DNA Repair (Amst). 2010, 9 (12): 1219-1228.
Shiotani B, Zou L: Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol Cell. 2009, 33 (5): 547-558.
Lee JH, Paull TT: ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005, 308 (5721): 551-554.
Bolderson E, Tomimatsu N, Richard DJ, Boucher D, Kumar R, Pandita TK, Burma S, Khanna KK: Phosphorylation of Exo1 modulates homologous recombination repair of DNA double-strand breaks. Nucleic Acids Res. 2010, 38 (6): 1821-1831.
Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y: Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22 (20): 5612-5621.
Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD: The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 2003, 22 (24): 6610-6620.
Falck J, Coates J, Jackson SP: Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005, 434 (7033): 605-611.
Schiller CB, Lammens K, Guerini I, Coordes B, Feldmann H, Schlauderer F, Mockel C, Schele A, Strasser K, Jackson SP: Structure of Mre11-Nbs1 complex yields insights into ataxia-telangiectasia-like disease mutations and DNA damage signaling. Nat Struct Mol Biol. 2012, 19 (7): 693-700.
Lammens K, Bemeleit DJ, Mockel C, Clausing E, Schele A, Hartung S, Schiller CB, Lucas M, Angermuller C, Soding J: The Mre11:Rad50 structure shows an ATP-dependent molecular clamp in DNA double-strand break repair. Cell. 2011, 145 (1): 54-66.
Hopfner KP, Karcher A, Craig L, Woo TT, Carney JP, Tainer JA: Structural biochemistry and interaction architecture of the DNA double-strand break repair Mre11 nuclease and Rad50-ATPase. Cell. 2001, 105 (4): 473-485.
Park YB, Chae J, Kim YC, Cho Y: Crystal structure of human Mre11: understanding tumorigenic mutations. Structure. 2011, 19 (11): 1591-1602.
de Jager M, van Noort J, van Gent DC, Dekker C, Kanaar R, Wyman C: Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol Cell. 2001, 8 (5): 1129-1135.
Li Y, Bolderson E, Kumar R, Muniandy PA, Xue Y, Richard DJ, Seidman M, Pandita TK, Khanna KK, Wang W: hSSB1 and hSSB2 form similar multiprotein complexes that participate in DNA damage response. J Biol Chem. 2009, 284 (35): 23525-23531.
Huang J, Gong Z, Ghosal G, Chen J: SOSS complexes participate in the maintenance of genomic stability. Mol Cell. 2009, 35 (3): 384-393.
Zhang F, Wu J, Yu X: Integrator3, a partner of single-stranded DNA-binding protein 1, participates in the DNA damage response. J Biol Chem. 2009, 284 (44): 30408-30415.
Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, Groocock LM, Cahill D, Hitomi C, Guenther G: Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell. 2008, 135 (1): 97-109.
Garcia V, Phelps SE, Gray S, Neale MJ: Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature. 2011, 479 (7372): 241-244.
Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP: Human CtIP promotes DNA end resection. Nature. 2007, 450 (7169): 509-514.
Chen L, Nievera CJ, Lee AY, Wu X: Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J Biol Chem. 2008, 283 (12): 7713-7720.
Yun MH, Hiom K: CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature. 2009, 459 (7245): 460-463.
Huertas P, Jackson SP: Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem. 2009, 284 (14): 9558-9565.
Binz SK, Lao Y, Lowry DF, Wold MS: The phosphorylation domain of the 32-kDa subunit of replication protein A (RPA) modulates RPA-DNA interactions. Evidence for an intersubunit interaction. J Biol Chem. 2003, 278 (37): 35584-35591.
Schild D, Wiese C: Overexpression of RAD51 suppresses recombination defects: a possible mechanism to reverse genomic instability. Nucleic Acids Res. 2010, 38 (4): 1061-1070.
Sung P, Krejci L, Van Komen S, Sehorn MG: Rad51 recombinase and recombination mediators. J Biol Chem. 2003, 278 (44): 42729-42732.
Stauffer ME, Chazin WJ: Physical interaction between replication protein A and Rad51 promotes exchange on single-stranded DNA. J Biol Chem. 2004, 279 (24): 25638-25645.
Fanning E, Klimovich V, Nager AR: A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006, 34 (15): 4126-4137.
Davis AP, Symington LS: The Rad52-Rad59 complex interacts with Rad51 and replication protein A. DNA Repair (Amst). 2003, 2 (10): 1127-1134.
Jackson D, Dhar K, Wahl JK, Wold MS, Borgstahl GE: Analysis of the human replication protein A:Rad52 complex: evidence for crosstalk between RPA32, RPA70, Rad52 and DNA. J Mol Biol. 2002, 321 (1): 133-148.
Mer G, Bochkarev A, Gupta R, Bochkareva E, Frappier L, Ingles CJ, Edwards AM, Chazin WJ: Structural basis for the recognition of DNA repair proteins UNG2, XPA, and RAD52 by replication factor RPA. Cell. 2000, 103 (3): 449-456.
Deng X, Prakash A, Dhar K, Baia GS, Kolar C, Oakley GG, Borgstahl GE: Human replication protein A-Rad52-single-stranded DNA complex: stoichiometry and evidence for strand transfer regulation by phosphorylation. Biochemistry (Mosc). 2009, 48 (28): 6633-6643.
Roy R, Chun J, Powell SN: BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2012, 12 (1): 68-78.
Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH, Zheng N, Chen PL, Lee WH, Pavletich NP: BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science. 2002, 297 (5588): 1837-1848.
Yang H, Li Q, Fan J, Holloman WK, Pavletich NP: The BRCA2 homologue Brh2 nucleates RAD51 filament formation at a dsDNA-ssDNA junction. Nature. 2005, 433 (7026): 653-657.
Li J, Zou C, Bai Y, Wazer DE, Band V, Gao Q: DSS1 is required for the stability of BRCA2. Oncogene. 2006, 25 (8): 1186-1194.
Kojic M, Yang H, Kostrub CF, Pavletich NP, Holloman WK: The BRCA2-interacting protein DSS1 is vital for DNA repair, recombination, and genome stability in Ustilago maydis. Mol Cell. 2003, 12 (4): 1043-1049.
Wong JM, Ionescu D, Ingles CJ: Interaction between BRCA2 and replication protein A is compromised by a cancer-predisposing mutation in BRCA2. Oncogene. 2003, 22 (1): 28-33.
Jensen RB, Carreira A, Kowalczykowski SC: Purified human BRCA2 stimulates RAD51-mediated recombination. Nature. 2010, 467 (7316): 678-683.
Bork P, Blomberg N, Nilges M: Internal repeats in the BRCA2 protein sequence. Nat Genet. 1996, 13 (1): 22-23.
Bignell G, Micklem G, Stratton MR, Ashworth A, Wooster R: The BRC repeats are conserved in mammalian BRCA2 proteins. Hum Mol Genet. 1997, 6 (1): 53-58.
Wong AK, Pero R, Ormonde PA, Tavtigian SV, Bartel PL: RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J Biol Chem. 1997, 272 (51): 31941-31944.
Galkin VE, Esashi F, Yu X, Yang S, West SC, Egelman EH: BRCA2 BRC motifs bind RAD51-DNA filaments. Proc Natl Acad Sci USA. 2005, 102 (24): 8537-8542.
Pellegrini L, Yu DS, Lo T, Anand S, Lee M, Blundell TL, Venkitaraman AR: Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature. 2002, 420 (6913): 287-293.
Shivji MK, Davies OR, Savill JM, Bates DL, Pellegrini L, Venkitaraman AR: A region of human BRCA2 containing multiple BRC repeats promotes RAD51-mediated strand exchange. Nucleic Acids Res. 2006, 34 (14): 4000-4011.
Carreira A, Hilario J, Amitani I, Baskin RJ, Shivji MK, Venkitaraman AR, Kowalczykowski SC: The BRC repeats of BRCA2 modulate the DNA-binding selectivity of RAD51. Cell. 2009, 136 (6): 1032-1043.
Liu J, Doty T, Gibson B, Heyer WD: Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat Struct Mol Biol. 2010, 17 (10): 1260-1262.
Carreira A, Kowalczykowski SC: Two classes of BRC repeats in BRCA2 promote RAD51 nucleoprotein filament function by distinct mechanisms. Proc Natl Acad Sci USA. 2011, 108 (26): 10448-10453.
van Vuuren AJ, Appeldoorn E, Odijk H, Yasui A, Jaspers NG, Bootsma D, Hoeijmakers JH: Evidence for a repair enzyme complex involving ERCC1 and complementing activities of ERCC4, ERCC11 and xeroderma pigmentosum group F. EMBO J. 1993, 12 (9): 3693-3701.
Oakley GG, Patrick SM: Replication protein A: directing traffic at the intersection of replication and repair. Front Biosci. 2010, 15: 883-900.
Coverley D, Kenny MK, Lane DP, Wood RD: A role for the human single-stranded DNA binding protein HSSB/RPA in an early stage of nucleotide excision repair. Nucleic Acids Res. 1992, 20 (15): 3873-3880.
Saijo M, Takedachi A, Tanaka K: Nucleotide excision repair by mutant xeroderma pigmentosum group A (XPA) proteins with deficiency in interaction with RPA. J Biol Chem. 2011, 286 (7): 5476-5483.
Sugasawa K, Ng JM, Masutani C, Iwai S, van der Spek PJ, Eker AP, Hanaoka F, Bootsma D, Hoeijmakers JH: Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol Cell. 1998, 2 (2): 223-232.
Tirode F, Busso D, Coin F, Egly JM: Reconstitution of the transcription factor TFIIH: assignment of functions for the three enzymatic subunits, XPB, XPD, and cdk7. Mol Cell. 1999, 3 (1): 87-95.
Park CH, Mu D, Reardon JT, Sancar A: The general transcription-repair factor TFIIH is recruited to the excision repair complex by the XPA protein independent of the TFIIE transcription factor. J Biol Chem. 1995, 270 (9): 4896-4902.
Nocentini S, Coin F, Saijo M, Tanaka K, Egly JM: DNA damage recognition by XPA protein promotes efficient recruitment of transcription factor II H. J Biol Chem. 1997, 272 (37): 22991-22994.
Patrick SM, Turchi JJ: Xeroderma pigmentosum complementation group A protein (XPA) modulates RPA-DNA interactions via enhanced complex stability and inhibition of strand separation activity. J Biol Chem. 2002, 277 (18): 16096-16101.
Missura M, Buterin T, Hindges R, Hubscher U, Kasparkova J, Brabec V, Naegeli H: Double-check probing of DNA bending and unwinding by XPA-RPA: an architectural function in DNA repair. EMBO J. 2001, 20 (13): 3554-3564.
Saijo M, Kuraoka I, Masutani C, Hanaoka F, Tanaka K: Sequential binding of DNA repair proteins RPA and ERCC1 to XPA in vitro. Nucleic Acids Res. 1996, 24 (23): 4719-4724.
Li L, Lu X, Peterson CA, Legerski RJ: An interaction between the DNA repair factor XPA and replication protein A appears essential for nucleotide excision repair. Mol Cell Biol. 1995, 15 (10): 5396-5402.
Evans E, Moggs JG, Hwang JR, Egly JM, Wood RD: Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J. 1997, 16 (21): 6559-6573.
Mu D, Wakasugi M, Hsu DS, Sancar A: Characterization of reaction intermediates of human excision repair nuclease. J Biol Chem. 1997, 272 (46): 28971-28979.
He Z, Henricksen LA, Wold MS, Ingles CJ: RPA involvement in the damage-recognition and incision steps of nucleotide excision repair. Nature. 1995, 374 (6522): 566-569.
Stigger E, Drissi R, Lee SH: Functional analysis of human replication protein A in nucleotide excision repair. J Biol Chem. 1998, 273 (15): 9337-9343.
Bessho T, Sancar A, Thompson LH, Thelen MP: Reconstitution of human excision nuclease with recombinant XPF-ERCC1 complex. J Biol Chem. 1997, 272 (6): 3833-3837.
Matsunaga T, Park CH, Bessho T, Mu D, Sancar A: Replication protein A confers structure-specific endonuclease activities to the XPF-ERCC1 and XPG subunits of human DNA repair excision nuclease. J Biol Chem. 1996, 271 (19): 11047-11050.
Cruet-Hennequart S, Coyne S, Glynn MT, Oakley GG, Carty MP: UV-induced RPA phosphorylation is increased in the absence of DNA polymerase eta and requires DNA-PK. DNA Repair (Amst). 2006, 5 (4): 491-504.
Zhou BB, Elledge SJ: The DNA damage response: putting checkpoints in perspective. Nature. 2000, 408 (6811): 433-439.
Finn K, Lowndes NF, Grenon M: Eukaryotic DNA damage checkpoint activation in response to double-strand breaks. Cell Mol Life Sci. 2012, 69 (9): 1447-1473.
Abraham RT: Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15 (17): 2177-2196.
Khanna KK, Jackson SP: DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001, 27 (3): 247-254.
Cimprich KA, Cortez D: ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008, 9 (8): 616-627.
Stracker TH, Usui T, Petrini JH: Taking the time to make important decisions: the checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair (Amst). 2009, 8 (9): 1047-1054.
Gatei M, Sloper K, Sorensen C, Syljuasen R, Falck J, Hobson K, Savage K, Lukas J, Zhou BB, Bartek J: Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation. J Biol Chem. 2003, 278 (17): 14806-14811.
Garcia-Muse T, Boulton SJ: Distinct modes of ATR activation after replication stress and DNA double-strand breaks in Caenorhabditis elegans. EMBO J. 2005, 24 (24): 4345-4355.
Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP: ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006, 8 (1): 37-45.
Hirao A, Cheung A, Duncan G, Girard PM, Elia AJ, Wakeham A, Okada H, Sarkissian T, Wong JA, Sakai T: Chk2 is a tumor suppressor that regulates apoptosis in both an ataxia telangiectasia mutated (ATM)-dependent and an ATM-independent manner. Mol Cell Biol. 2002, 22 (18): 6521-6532.
Li J, Stern DF: Regulation of CHK2 by DNA-dependent protein kinase. J Biol Chem. 2005, 280 (12): 12041-12050.
Dart DA, Adams KE, Akerman I, Lakin ND: Recruitment of the cell cycle checkpoint kinase ATR to chromatin during S-phase. J Biol Chem. 2004, 279 (16): 16433-16440.
Ball HL, Myers JS, Cortez D: ATRIP binding to replication protein A-single-stranded DNA promotes ATR-ATRIP localization but is dispensable for Chk1 phosphorylation. Mol Biol Cell. 2005, 16 (5): 2372-2381.
Wu X, Shell SM, Zou Y: Interaction and colocalization of Rad9/Rad1/Hus1 checkpoint complex with replication protein A in human cells. Oncogene. 2005, 24 (29): 4728-4735.
Zou L, Liu D, Elledge SJ: Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc Natl Acad Sci USA. 2003, 100 (24): 13827-13832.
Parrilla-Castellar ER, Arlander SJ, Karnitz L: Dial 9-1-1 for DNA damage: the Rad9-Hus1-Rad1 (9-1-1) clamp complex. DNA Repair (Amst). 2004, 3 (8–9): 1009-1014.
Lee J, Kumagai A, Dunphy WG: The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J Biol Chem. 2007, 282 (38): 28036-28044.
Delacroix S, Wagner JM, Kobayashi M, Yamamoto K, Karnitz LM: The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 2007, 21 (12): 1472-1477.
Kumagai A, Lee J, Yoo HY, Dunphy WG: TopBP1 activates the ATR-ATRIP complex. Cell. 2006, 124 (5): 943-955.
Gong Z, Chen J: E3 ligase RFWD3 participates in replication checkpoint control. J Biol Chem. 2011, 286 (25): 22308-22313.
Liu S, Chu J, Yucer N, Leng M, Wang SY, Chen BP, Hittelman WN, Wang Y: RING finger and WD repeat domain 3 (RFWD3) associates with replication protein A (RPA) and facilitates RPA-mediated DNA damage response. J Biol Chem. 2011, 286 (25): 22314-22322.
Mu JJ, Wang Y, Luo H, Leng M, Zhang J, Yang T, Besusso D, Jung SY, Qin J: A proteomic analysis of ataxia telangiectasia-mutated (ATM)/ATM-Rad3-related (ATR) substrates identifies the ubiquitin-proteasome system as a regulator for DNA damage checkpoints. J Biol Chem. 2007, 282 (24): 17330-17334.
Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y: ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007, 316 (5828): 1160-1166.
Smolka MB, Albuquerque CP, Chen SH, Zhou H: Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc Natl Acad Sci USA. 2007, 104 (25): 10364-10369.
Fu X, Yucer N, Liu S, Li M, Yi P, Mu JJ, Yang T, Chu J, Jung SY, O’Malley BW: RFWD3-Mdm2 ubiquitin ligase complex positively regulates p53 stability in response to DNA damage. Proc Natl Acad Sci USA. 2010, 107 (10): 4579-4584.
Kruse JP, Gu W: Modes of p53 regulation. Cell. 2009, 137 (4): 609-622.
Xu S, Feng Z, Zhang M, Wu Y, Sang Y, Xu H, Lv X, Hu K, Cao J, Zhang R: hSSB1 binds and protects p21 from ubiquitin-mediated degradation and positively correlates with p21 in human hepatocellular carcinomas. Oncogene. 2011, 30 (19): 2219-2229.
El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B: WAF1, a potential mediator of p53 tumor suppression. Cell. 1993, 75 (4): 817-825.
El-Deiry WS, Harper JW, O’Connor PM, Velculescu VE, Canman CE, Jackman J, Pietenpol JA, Burrell M, Hill DE, Wang Y: WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994, 54 (5): 1169-1174.
Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ: The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993, 75 (4): 805-816.
Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D: p21 is a universal inhibitor of cyclin kinases. Nature. 1993, 366 (6456): 701-704.
Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B: Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998, 282 (5393): 1497-1501.
Blagosklonny MV, Wu GS, Omura S, el-Deiry WS: Proteasome-dependent regulation of p21WAF1/CIP1 expression. Biochem Biophys Res Commun. 1996, 227 (2): 564-569.
Maki CG, Howley PM: Ubiquitination of p53 and p21 is differentially affected by ionizing and UV radiation. Mol Cell Biol. 1997, 17 (1): 355-363.
Fukuchi K, Maruyama H, Takagi Y, Gomi K: Direct proteasome inhibition by clasto-lactacystin beta-lactone permits the detection of ubiquitinated p21(waf1) in ML-1 cells. Biochim Biophys Acta. 1999, 1451 (1): 206-210.
Xu S, Wu Y, Chen Q, Cao J, Hu K, Tang J, Sang Y, Lai F, Wang L, Zhang R: hSSB1 regulates both the stability and the transcriptional activity of p53. Cell Res. 2013, 23 (3): 423-435.
Chau BN, Diaz RL, Saunders MA, Cheng C, Chang AN, Warrener P, Bradshaw J, Linsley PS, Cleary MA: Identification of SULF2 as a novel transcriptional target of p53 by use of integrated genomic analyses. Cancer Res. 2009, 69 (4): 1368-1374.
Adams CJ, Pike AC, Maniam S, Sharpe TD, Coutts AS, Knapp S, La Thangue NB, Bullock AN: The p53 cofactor Strap exhibits an unexpected TPR motif and oligonucleotide-binding (OB)-fold structure. Proc Natl Acad Sci USA. 2012, 109 (10): 3778-3783.
Demonacos C, Krstic-Demonacos M, La Thangue NB: A TPR motif cofactor contributes to p300 activity in the p53 response. Mol Cell. 2001, 8 (1): 71-84.
Adams CJ, Graham AL, Jansson M, Coutts AS, Edelmann M, Smith L, Kessler B, La Thangue NB: ATM and Chk2 kinase target the p53 cofactor Strap. EMBO Rep. 2008, 9 (12): 1222-1229.
Demonacos C, Krstic-Demonacos M, Smith L, Xu D, O’Connor DP, Jansson M, La Thangue NB: A new effector pathway links ATM kinase with the DNA damage response. Nat Cell Biol. 2004, 6 (10): 968-976.
Jansson M, Durant ST, Cho EC, Sheahan S, Edelmann M, Kessler B, La Thangue NB: Arginine methylation regulates the p53 response. Nat Cell Biol. 2008, 10 (12): 1431-1439.
Coutts AS, Weston L, La Thangue NB: A transcription co-factor integrates cell adhesion and motility with the p53 response. Proc Natl Acad Sci USA. 2009, 106 (47): 19872-19877.
Shikama N, Lee CW, France S, Delavaine L, Lyon J, Krstic-Demonacos M, La Thangue NB: A novel cofactor for p300 that regulates the p53 response. Mol Cell. 1999, 4 (3): 365-376.
Vega LR, Mateyak MK, Zakian VA: Getting to the end: telomerase access in yeast and humans. Nat Rev Mol Cell Biol. 2003, 4 (12): 948-959.
Chow TT, Zhao Y, Mak SS, Shay JW, Wright WE: Early and late steps in telomere overhang processing in normal human cells: the position of the final RNA primer drives telomere shortening. Genes Dev. 2012, 26 (11): 1167-1178.
Zhao Y, Hoshiyama H, Shay JW, Wright WE: Quantitative telomeric overhang determination using a double-strand specific nuclease. Nucleic Acids Res. 2008, 36 (3): e14-
Zhu XD, Kuster B, Mann M, Petrini JH, de Lange T: Cell-cycle-regulated association of RAD50/MRE11/NBS1 with TRF2 and human telomeres. Nat Genet. 2000, 25 (3): 347-352.
Paeschke K, McDonald KR, Zakian VA: Telomeres: structures in need of unwinding. FEBS Lett. 2010, 584 (17): 3760-3772.
de Lange T: Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19 (18): 2100-2110.
Wang F, Podell ER, Zaug AJ, Yang Y, Baciu P, Cech TR, Lei M: The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature. 2007, 445 (7127): 506-510.
Xin H, Liu D, Wan M, Safari A, Kim H, Sun W, O’Connor MS, Songyang Z: TPP1 is a homologue of ciliate TEBP-beta and interacts with POT1 to recruit telomerase. Nature. 2007, 445 (7127): 559-562.
Loayza D, Parsons H, Donigian J, Hoke K, de Lange T: DNA binding features of human POT1: a nonamer 5′-TAGGGTTAG-3′ minimal binding site, sequence specificity, and internal binding to multimeric sites. J Biol Chem. 2004, 279 (13): 13241-13248.
Lei M, Podell ER, Cech TR: Structure of human POT1 bound to telomeric single-stranded DNA provides a model for chromosome end-protection. Nat Struct Mol Biol. 2004, 11 (12): 1223-1229.
Taylor DJ, Podell ER, Taatjes DJ, Cech TR: Multiple POT1-TPP1 proteins coat and compact long telomeric single-stranded DNA. J Mol Biol. 2011, 410 (1): 10-17.
Mitton-Fry RM, Anderson EM, Theobald DL, Glustrom LW, Wuttke DS: Structural basis for telomeric single-stranded DNA recognition by yeast Cdc13. J Mol Biol. 2004, 338 (2): 241-255.
Zhong FL, Batista LF, Freund A, Pech MF, Venteicher AS, Artandi SE: TPP1 OB-fold domain controls telomere maintenance by recruiting telomerase to chromosome ends. Cell. 2012, 150 (3): 481-494.
Nandakumar J, Bell CF, Weidenfeld I, Zaug AJ, Leinwand LA, Cech TR: The TEL patch of telomere protein TPP1 mediates telomerase recruitment and processivity. Nature. 2012, 492 (7428): 285-289.
Zaug AJ, Podell ER, Nandakumar J, Cech TR: Functional interaction between telomere protein TPP1 and telomerase. Genes Dev. 2010, 24 (6): 613-622.
Latrick CM, Cech TR: POT1-TPP1 enhances telomerase processivity by slowing primer dissociation and aiding translocation. EMBO J. 2010, 29 (5): 924-933.
Loayza D, De Lange T: POT1 as a terminal transducer of TRF1 telomere length control. Nature. 2003, 423 (6943): 1013-1018.
Arat NO, Griffith JD: Human Rap1 interacts directly with telomeric DNA and regulates TRF2 localization at the telomere. J Biol Chem. 2012, 287 (50): 41583-41594.
Hanaoka S, Nagadoi A, Nishimura Y: Comparison between TRF2 and TRF1 of their telomeric DNA-bound structures and DNA-binding activities. Protein Sci. 2005, 14 (1): 119-130.
Court R, Chapman L, Fairall L, Rhodes D: How the human telomeric proteins TRF1 and TRF2 recognize telomeric DNA: a view from high-resolution crystal structures. EMBO Rep. 2005, 6 (1): 39-45.
Bianchi A, Stansel RM, Fairall L, Griffith JD, Rhodes D, de Lange T: TRF1 binds a bipartite telomeric site with extreme spatial flexibility. EMBO J. 1999, 18 (20): 5735-5744.
Li B, Oestreich S, de Lange T: Identification of human Rap1: implications for telomere evolution. Cell. 2000, 101 (5): 471-483.
Liu D, O’Connor MS, Qin J, Songyang Z: Telosome, a mammalian telomere-associated complex formed by multiple telomeric proteins. J Biol Chem. 2004, 279 (49): 51338-51342.
Ye JZ, Donigian JR, van Overbeek M, Loayza D, Luo Y, Krutchinsky AN, Chait BT, de Lange T: TIN2 binds TRF1 and TRF2 simultaneously and stabilizes the TRF2 complex on telomeres. J Biol Chem. 2004, 279 (45): 47264-47271.
Liu D, Safari A, O’Connor MS, Chan DW, Laegeler A, Qin J, Songyang Z: PTOP interacts with POT1 and regulates its localization to telomeres. Nat Cell Biol. 2004, 6 (7): 673-680.
Ye JZ, Hockemeyer D, Krutchinsky AN, Loayza D, Hooper SM, Chait BT, de Lange T: POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 2004, 18 (14): 1649-1654.
Sfeir A, de Lange T: Removal of shelterin reveals the telomere end-protection problem. Science. 2012, 336 (6081): 593-597.
d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP: A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003, 426 (6963): 194-198.
Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM: Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol Cell. 2004, 14 (4): 501-513.
Denchi EL, de Lange T: Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 2007, 448 (7157): 1068-1071.
Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T: p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science. 1999, 283 (5406): 1321-1325.
Martinez P, Flores JM, Blasco MA: 53BP1 deficiency combined with telomere dysfunction activates ATR-dependent DNA damage response. J Cell Biol. 2012, 197 (2): 283-300.
Takai H, Smogorzewska A, de Lange T: DNA damage foci at dysfunctional telomeres. Curr Biol. 2003, 13 (17): 1549-1556.
Schramke V, Luciano P, Brevet V, Guillot S, Corda Y, Longhese MP, Gilson E, Geli V: RPA regulates telomerase action by providing Est1p access to chromosome ends. Nat Genet. 2004, 36 (1): 46-54.
Verdun RE, Karlseder J: The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. Cell. 2006, 127 (4): 709-720.
Flynn RL, Centore RC, O’Sullivan RJ, Rai R, Tse A, Songyang Z, Chang S, Karlseder J, Zou L: TERRA and hnRNPA1 orchestrate an RPA-to-POT1 switch on telomeric single-stranded DNA. Nature. 2011, 471 (7339): 532-536.
Thanasoula M, Escandell JM, Suwaki N, Tarsounas M: ATM/ATR checkpoint activation downregulates CDC25C to prevent mitotic entry with uncapped telomeres. EMBO J. 2012, 31 (16): 3398-3410.
Miyake Y, Nakamura M, Nabetani A, Shimamura S, Tamura M, Yonehara S, Saito M, Ishikawa F: RPA-like mammalian Ctc1-Stn1-Ten1 complex binds to single-stranded DNA and protects telomeres independently of the Pot1 pathway. Mol Cell. 2009, 36 (2): 193-206.
Chen LY, Redon S, Lingner J: The human CST complex is a terminator of telomerase activity. Nature. 2012, 488 (7412): 540-544.
Gao H, Cervantes RB, Mandell EK, Otero JH, Lundblad V: RPA-like proteins mediate yeast telomere function. Nat Struct Mol Biol. 2007, 14 (3): 208-214.
Mitchell MT, Smith JS, Mason M, Harper S, Speicher DW, Johnson FB, Skordalakes E: Cdc13 N-terminal dimerization, DNA binding, and telomere length regulation. Mol Cell Biol. 2010, 30 (22): 5325-5334.
Sun J, Yang Y, Wan K, Mao N, Yu TY, Lin YC, DeZwaan DC, Freeman BC, Lin JJ, Lue NF: Structural bases of dimerization of yeast telomere protein Cdc13 and its interaction with the catalytic subunit of DNA polymerase alpha. Cell Res. 2011, 21 (2): 258-274.
Theobald DL, Wuttke DS: Divergent evolution within protein superfolds inferred from profile-based phylogenetics. J Mol Biol. 2005, 354 (3): 722-737.
Anderson EM, Halsey WA, Wuttke DS: Delineation of the high-affinity single-stranded telomeric DNA-binding domain of Saccharomyces cerevisiae Cdc13. Nucleic Acids Res. 2002, 30 (19): 4305-4313.
Hughes TR, Weilbaecher RG, Walterscheid M, Lundblad V: Identification of the single-strand telomeric DNA binding domain of the Saccharomyces cerevisiae Cdc13 protein. Proc Natl Acad Sci USA. 2000, 97 (12): 6457-6462.
Surovtseva YV, Churikov D, Boltz KA, Song X, Lamb JC, Warrington R, Leehy K, Heacock M, Price CM, Shippen DE: Conserved telomere maintenance component 1 interacts with STN1 and maintains chromosome ends in higher eukaryotes. Mol Cell. 2009, 36 (2): 207-218.
Gu P, Min JN, Wang Y, Huang C, Peng T, Chai W, Chang S: CTC1 deletion results in defective telomere replication, leading to catastrophic telomere loss and stem cell exhaustion. EMBO J. 2012, 31 (10): 2309-2321.
Richard DJ, Bell SD, White MF: Physical and functional interaction of the archaeal single-stranded DNA-binding protein SSB with RNA polymerase. Nucleic Acids Res. 2004, 32 (3): 1065-1074.
Acknowledgments
We would like to thank Mr Joshua Burgess, Dr Aaron Urquhart and Dr Nicolas Paquet for critical reading of the manuscript. Research by the Genome Stability Laboratory is funded by the Australian Research Council (ARC). NWA is supported by a scholarship awarded by Cancer Council Queensland. LC acknowledges funding from ARC and the National Health and Medical Research Council (NHMRC).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NWA, EB, and LC contributed significant intellectual input to both writing and editing. DJR and KJO contributed significant intellectual input to content, structure and editing. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ashton, N.W., Bolderson, E., Cubeddu, L. et al. Human single-stranded DNA binding proteins are essential for maintaining genomic stability. BMC Molecular Biol 14, 9 (2013). https://doi.org/10.1186/1471-2199-14-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2199-14-9