
Abstract
Background
Despite extensive efforts devoted to predicting protein-coding genes in genome sequences, many bona fide genes have not been found and many existing gene models are not accurate in all sequenced eukaryote genomes. This situation is partly explained by the fact that gene prediction programs have been developed based on our incomplete understanding of gene feature information such as splicing and promoter characteristics. Additionally, full-length cDNAs of many genes and their isoforms are hard to obtain due to their low level or rare expression. In order to obtain full-length sequences of all protein-coding genes, alternative approaches are required.
Results
In this project, we have developed a method of reconstructing full-length cDNA sequences based on short expressed sequence tags which is called s equence t ag-based a mplification of c DNA e nds (STACE). Expressed tags are used as anchors for retrieving full-length transcripts in two rounds of PCR amplification. We have demonstrated the application of STACE in reconstructing full-length cDNA sequences using expressed tags mined in an array of serial analysis of gene expression (SAGE) of C. elegans cDNA libraries. We have successfully applied STACE to recover sequence information for 12 genes, for two of which we found isoforms. STACE was used to successfully recover full-length cDNA sequences for seven of these genes.
Conclusions
The STACE method can be used to effectively reconstruct full-length cDNA sequences of genes that are under-represented in cDNA sequencing projects and have been missed by existing gene prediction methods, but their existence has been suggested by short sequence tags such as SAGE tags.
Similar content being viewed by others

Background
The nematode Caenorhabditis elegans, which is a well-established model organism for biomedical research [1], is the first metazoan whose genome was subject to whole-genome sequencing [2]. Its gene models were first predicted using the gene prediction program Genefinder (P. Green, unpublished). Over the dozen years since the completion of the C. elegans genome sequencing project [2], the C. elegans gene set has been curated by the C. elegans research community and by WormBase curators [1, 3–5]. However, the C. elegans gene set is still far from complete for the following reasons: First, because Genefinder, like other gene prediction programs, was developed based on an incomplete understanding of gene structures, it suffers from both false positive and false negative predictions; second, many bona fide genes, especially those of unknown character, have been missed. In WormBase http://www.wormbase.org, the official database for the biology and genomics of C. elegans, less than 40% of the annotated gene models are fully confirmed. All others are either partially supported or not supported at all. Additional gene models have been revealed in transcriptome sequencing [6, 7], suggesting many gene models remain to be discovered. This situation is also true for other species [8]. In the human genome, it has been estimated that the most accurate programs only correctly predict 40% of the annotated genes [9].
In this project, we explored how to reconstruct full-length gene models for genes that are not correctly represented in the current gene set, using expressed sequence tags obtained in large-scale gene expression projects. In particular, we attempted to reconstruct novel C. elegans gene models using SAGE (serial analysis of gene expression). The SAGE technique was originally developed for profiling gene expression [10, 11]. The expression profiles created with SAGE have a wide range of applications that include therapeutic target identification in cancerous tissues [12] and others of biological and medical importance [13]. Recently, SAGE was applied to probe gene expression in C. elegans by the C. elegans Gene Expression Consortium http://elegans.bcgsc.bc.ca/home/ge_consortium.html. These SAGE libraries have been fundamental for the success of a variety of research projects [14–19]. While SAGE tags that correspond to existing gene models can be used to evaluate the abundance of gene expression, there are a large number of SAGE tags that do not correspond to existing gene models. These SAGE tags suggest the existence of additional coding exons, splice variants [20], or novel genes.
Results
Tag based reconstruction of full-length cDNA sequence of novel genes
Expressed sequence tags that cannot be aligned to the C. elegans virtual transcriptome (i.e., cDNA sequences of all annotated transcripts) suggest the existence of yet unannotated genes [13, 21]. We have established a protocol, termed as "sequence t ag-based a mplification of c DNA e nds", or STACE, based on the RACE protocol [22], to identify potential novel genes. The method can be used to amplify full-length cDNA transcripts that have been reverse-transcribed from the mRNA sequence of novel genes. STACE uses three primer hybridization sites. The first site (the 5' site) is a sequence located at the extreme 5' end of the target transcript, the second site (the 3' site) is downstream of the polyadenylation sequence, and the third site (the gene-specific site) corresponds to the genomic span where the uncharacterized tag maps. The amplicons are then cloned, sequenced and mapped to the genome. As such, STACE not only confirms the existence of a novel gene, but also defines the full-length transcript sequence of the yet undefined gene.
In this project, in order to get a primer hybridization site at the extreme 5' end of the RNA transcripts, we took advantage of the trans-splice leader 1 (SL1) in C. elegans, and used its sequence as a primer for our 5' site. It is appropriate to design the 5' primer based on the SL1 sequence because SL1 is trans-spliced to the extreme 5' end of nearly 50% of all C. elegans mRNAs [23, 24]. For applications in which the sample transcriptome does not undergo trans-splicing of this nature, a common oligo anchoring sequence can be ligated to the 5'end of each transcript. An oligo sequence was attached to the polyadenylation tracks of mRNA through reverse transcription with a modified oligo d(T) primer that included a 3' common sequence (5' - CCAGACACTATGCTCATACGACGCAGT(16)VN - 3'). This provided us with a cDNA library containing transcripts that had a usable 3' site. Finally, we chose gene-specific sites by bioinformatically identifying SAGE tags. When aligned to the C. elegans genome, qualified SAGE tags do not overlap with existing gene models. For each qualified SAGE tag, a primer was designed and used in conjunction with a primer complementary to the SL1 sequence to amplify the upstream amplicon. A second primer was designed and used in conjunction with the primer complementary to the 3' common sequence (above) to amplify the downstream amplicon. The potential template was amplified, and the amplicon sequences were mapped to the C. elegans genome using BLAT [25], which is available at WormBase http://www.wormbase.org.
Computational selection of SAGE tags that suggest novel genes
SAGE tags used in this study were selected from 33 SAGE libraries, which were sequenced from different tissues and developmental stages of C. eleganshttp://tock.bcgsc.bc.ca/cgi-bin/sage160. There are altogether 220,770 unique SAGE tags in these libraries. Only SAGE tags that did not overlap with annotated protein-coding genes in the WS160 version of the C. elegans genome map were selected for this project.
We obtained four different sets of SAGE tags for testing, one preliminary set and three test sets (Set 1-3) (Table 1). The preliminary set, which was arbitrarily chosen, was used to test the STACE protocol. Set 1 used a longSAGE meta-library as a starting tag set (16,587 SAGE tags). Set 2 was created from the WS160 version of the mixed stage library (14,701), and Set 3 used SAGE libraries derived from Solexa sequencing of the SWN21 and SWN22 embryonic samples (359,457 SAGE tags). Solexa SAGE produced more initial SAGE tags than the previous SAGE libraries because it has much deeper coverage. Note that the Illumina Solexa Genome Analyzer produced a SAGE library that is about 20 times more sensitive than a normal SAGE library [26].
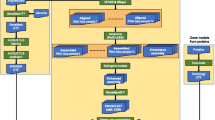
SAGE libraries were filtered to select SAGE tags for finding novel genes (Figure 1). The criteria used included the following: (1) Only SAGE tags that can be aligned to the C. elegans genome were selected; (2) The SAGE tags must not overlap with any annotated coding exon; (3) To avoid tags containing sequencing errors, SAGE tags must have a frequency of at least three for every 100,000 reads; (4) To increase the chances of finding novel genes (rather than novel missing exons), the SAGE tags must not overlap with an intron and have to be at least 500 bp away from an annotated 5' or 3' gene boundary; (5) SAGE tags must have a GC content between 35% and 45%, which is critical for primer design.

SAGE tag primer development. SAGE tag pools are put through various filtrations to produce a source of SAGE tag primers that are used in STACE experiments. 1) SAGE tag libraries are downloaded from the MultiSAGE website. 2) All SAGE tags that do not fully map to the genome as one single uninterrupted alignment are discarded. 3) SAGE tags that map to annotated transcribed sequences are discarded. 4) SAGE tags that have a low level of expression and are possible errors in sequencing are discarded. 5) SAGE tags that are found to overlap with intronic sequences or map near a 5' or 3' boundary are eliminated. 6) Only tags with GC content between 35% and 45% are retained. 7) SAGE tags are edited into primer form. All SAGE tags that are likely to produce secondary structure (i.e. hairpins, homo-dimers, hetero-dimers) are discarded. 8) A final list of SAGE tag primers is procured.
Primers based on SAGE tags were designed to ensure a reduced possibility of formation of secondary structures which would inhibit proper annealing of the primers [27]. For many cases, we trimmed sequences from either end of the SAGE tags to ensure primer quality. SAGE tag sequences that could not be used to guide proper primer design were not used. Primer design was done using the Primer3 program [28].
cDNA libraries
Two different cDNA libraries were created; one from a mixed stage population of C. elegans and another one from embryonic animals. In order to maximize the number of successful experiments, candidate SAGE tags were only screened against the developmental library that corresponded with the time in development that the tags were originally observed.
New transcripts and novel cDNAs
STACE-identified candidates consist of three categories based on the alignments of these candidates to the C. elegans genome (release WS160): (1) novel gene (six candidates), (2) annotation extension (four candidates), and (3) non-protein-coding gene overlap (two candidates) (Figure 2; Table 2). Novel genes are proof of entirely new genes discovered using the STACE method. The exons of these six new genes are all bordered by the canonical GT-AG splice signals, as is the case with most exons [29, 30]. Our identification of six novel genes was based on using the WS160 annotated genome. In the interim four have been annotated in WS200, while the other two are still completely novel. One of these two new gene models was characterized as a full-length gene model with the STACE method, while the other's existence was implied by an upstream amplicon sequence. Four tested SAGE tag primers produced results that suggest an extension to the annotated length of the gene models. These annotated extensions align perfectly with annotated exons, and imply either additional exons are transcribed within the gene, or that the terminal exons are longer than shown by WormBase.

STACE experimental procedure. Template cDNA is used in two separate PCR reactions that utilize primers based on the sequence of a SAGE tag, the SL1 sequence and modified oligo d(T) primer. The PCRs produce an amplicon representative of the sequence upstream of the SAGE tag location (PCR product 1), and another that comes from the downstream sequence (PCR product 2). These products are sequenced, and mapped to the C. elegans genome. Those sequences whose alignments overlap with the SAGE tag used in primer design are considered true positive STACE results.
We found a successful STACE result overlapped with a pseudogene. While this transcript may not be translated, using STACE we have clearly shown that it is processed with introns removed and a polyadenylation track added to the 3'end. We have also found that a STACE result overlapped with an annotated ncRNA gene. The transcript was also processed with a previously unknown intron excised and a polyadenylation track added.
Altogether, we have reconstructed seven full-length, true positive cDNA sequences, corresponding to seven separate gene models (Table 3). All seven cDNAs contain SL1 signals at the 5' ends and polyadenylation at the 3'ends. The remaining seven true positive cDNAs recovered represent the 5'ends of separate gene models, and these too contain full-length 5' SL1 signals. Thus, in this study, we have identified 14 SL1-trans-spliced cDNA sequences. All 14 cDNA sequences have been submitted to GenBank (Table 3).
Discussion
In this project, we have developed an experimental method termed STACE for reconstructing full-length cDNAs of novel genes. The applicability of STACE has been demonstrated by defining novel genes in the well-curated C. elegans genome, using SAGE tags from gene expression studies. We reconstructed seven novel full-length cDNAs and seven partial cDNA sequences that can be merged to existing gene models. Novel genes, annotation extensions, and non-protein-coding gene overlaps are represented by the identified cDNA sequences 3.3 (Figure 3), 3.1 (Figure 4), and P.3 (Figure 5), respectively.

Candidate cDNA 3.3 alignment. Full length gene model reconstructed for candidate cDNA 3.3. This gene model suggests a completely novel gene that is missing from WormBase as of WS200.

Candidate DNA 3.1 alignment. Full length gene model reconstructed for candidate cDNA 3.1. This gene model suggests a 3' UTR extension to the current sox-3 gene model.

Candidate cDNA P.3 alignment. Full length gene model reconstructed for candidate cDNA P.3. This gene model suggests revision of the structure to the tts-2 gene model, and also that this transcript is polyadenylated, a feature that is commonly associated with protein-coding genes.
We compared novel cDNAs with C. elegans gene models predicted using AUGUSTUS [31], mGENE [32], TWINSCAN [33] and FGENESH++ [34], which are available at WormBase. All cDNAs, which were detected using STACE, when aligned to the C. elegans genome overlap to a certain extent with predicted gene models. The novel full-length cDNA 3.3 aligned well with a prediction from TWINSCAN and with a prediction made by FGENESH++. The annotation extension result (full-length cDNA 3.1) was found to overlap with gene predictions from each of the utilized programs. However, a new 3' UTR exon was shown to be part of this gene model, and this exon did not overlap with the predictions made by any of the described programs. Additionally, the P.3 result overlapped with an existing ncRNA gene model. However, the novel intron suggested by this STACE result was not included in the WormBase gene model, although it overlaps with AUGUSTUS prediction.
Conclusions
We have found that the STACE method can be used to recover accurate full-length gene models. This method is useful for reconstructing gene models for genes that have been missed in cDNA sequencing projects and were missed or mispredicted by gene finders. With the wide application of next-generation sequencing methods in the deep sequencing of transcriptomes, more expressed sequence tags, which indicate the presence of novel genes will be uncovered. We expect that these tags will serve as input to the STACE protocol for further novel gene discovery and determination.
Methods
cDNA library production
Two samples of C. elegans were produced that represented both a mixed stage population and an embryonic sample. Tissue samples were put through an RNA extraction using TRIzol (Invitrogen, SKU# 10296-028). The cDNA libraries used in this project were created with the Superscript III reverse transcriptase kit (Invitrogen, SKU# 18080-085), and the primer used to initiate reverse transcription was a modified oligo d(T) primer (5' - CCAGACACTATGCTCATACGACGCAGT(16) VN - 3') (Invitrogen). The protocol accompanying the kit was followed, and the samples were treated with Ribonuclease H (Invitrogen, SKU# 18021-014).
Amplification of tag ends
The reverse complement of each SAGE tag sequence was used to design the SAGE tag primers. These primers were used in conjunction with a primer based on the SL1 sequence (5' - GGTTTAATTACCCAAGTTTGAG - 3') in a PCR. The PCR was initiated with a 94°C melt step for 2 minutes, followed by 32 cycles of a 94°C melt step for 15 seconds, a 60°C annealing step for 45 seconds, and a 72°C extension step for 1 minute. This was followed by a final extension at 72°C for 5 minutes. A Taq polymerase provided by Dr. Harald Hutter was used in all of the PCRs. Amplicons produced by the PCRs were visualized with a 1% gel electrophoresis, and extracted with a QIAquick Gel Extraction kit (Qiagen, ID 28704). These amplicons were then cloned with the InsTAclone kit (Fermentas, #K1214). Cloned amplicons were submitted for sequencing (Macrogen, Seoul, Korea), and returned sequences were mapped back to the C. elegans genome with the BLAT tool [25] on the WormBase website http://www.wormbase.org/. We opted to use BLAT instead of other alignment tools because this program can take spliced mRNA sequences (i.e. STACE cloned sequences) and align them to the genome in a way that reflects intron - exon boundaries [25, 35]. Those amplicons whose sequence alignment indicated a true positive result were then further studied. The returned sequence was used to design an internal primer that would be compatible with the universal primer (5' - CACTATGCTCATACGACGCAGT - 3'). These primers were then used in a PCR with the same parameters described above to produce the downstream amplicons needed for full-length characterization. Internal primers were designed using the Primer3 program [28].
References
Hillier LW, Coulson A, Murray JI, Bao Z, Sulston JE, Waterston RH: Genomics in C. elegans: so many genes, such a little worm. Genome Res. 2005, 15: 1651-1660. 10.1101/gr.3729105
, : Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 1998, 282: 2012-2018. 10.1126/science.282.5396.2012
Chen N, Harris TW, Antoshechkin I, Bastiani C, Bieri T, Blasiar D, Bradnam K, Canaran P, Chan J, Chen CK: WormBase: a comprehensive data resource for Caenorhabditis biology and genomics. Nucleic Acids Res. 2005, 33: D383-389. 10.1093/nar/gki066
Waterston R, Martin C, Craxton M, Huynh C, Coulson A, Hillier L, Durbin R, Green P, Shownkeen R, Halloran N: A survey of expressed genes in Caenorhabditis elegans. Nat Genet. 1992, 1: 114-123. 10.1038/ng0592-114
Reboul J, Vaglio P, Rual JF, Lamesch P, Martinez M, Armstrong CM, Li S, Jacotot L, Bertin N, Janky R: C. elegans ORFeome version 1.1: experimental verification of the genome annotation and resource for proteome-scale protein expression. Nat Genet. 2003, 34: 35-41. 10.1038/ng1140
Hillier LW, Reinke V, Green P, Hirst M, Marra MA, Waterston RH: Massively parallel sequencing of the polyadenylated transcriptome of C. elegans. Genome Res. 2009, 19: 657-666. 10.1101/gr.088112.108
Shin H, Hirst M, Bainbridge MN, Magrini V, Mardis E, Moerman DG, Marra MA, Baillie DL, Jones SJ: Transcriptome analysis for Caenorhabditis elegans based on novel expressed sequence tags. BMC Biol. 2008, 6: 30- 10.1186/1741-7007-6-30
Brent MR: Genome annotation past, present, and future: how to define an ORF at each locus. Genome Res. 2005, 15: 1777-1786. 10.1101/gr.3866105
Guigo R, Flicek P, Abril JF, Reymond A, Lagarde J, Denoeud F, Antonarakis S, Ashburner M, Bajic VB, Birney E: EGASP: the human ENCODE Genome Annotation Assessment Project. Genome Biol. 2006, 7 (Suppl 1): S2 1-31. 10.1186/gb-2006-7-s1-s2. 10.1186/gb-2006-7-s1-s2
Velculescu VE, Zhang L, Vogelstein B, Kinzler KW: Serial analysis of gene expression. Science. 1995, 270: 484-487. 10.1126/science.270.5235.484
Gnatenko DV, Dunn JJ, McCorkle SR, Weissmann D, Perrotta PL, Bahou WF: Transcript profiling of human platelets using microarray and serial analysis of gene expression. Blood. 2003, 101: 2285-2293. 10.1182/blood-2002-09-2797
Porter D, Yao J, Polyak K: SAGE and related approaches for cancer target identification. Drug Discov Today. 2006, 11: 110-118. 10.1016/S1359-6446(05)03694-9
Wang SM: Understanding SAGE data. Trends Genet. 2007, 23: 42-50. 10.1016/j.tig.2006.11.001
Pleasance ED, Marra MA, Jones SJ: Assessment of SAGE in transcript identification. Genome Res. 2003, 13: 1203-1215. 10.1101/gr.873003
Blacque OE, Perens EA, Boroevich KA, Inglis PN, Li C, Warner A, Khattra J, Holt RA, Ou G, Mah AK: Functional genomics of the cilium, a sensory organelle. Curr Biol. 2005, 15: 935-941. 10.1016/j.cub.2005.04.059
Jones SJ, Riddle DL, Pouzyrev AT, Velculescu VE, Hillier L, Eddy SR, Stricklin SL, Baillie DL, Waterston R, Marra MA: Changes in gene expression associated with developmental arrest and longevity in Caenorhabditis elegans. Genome Res. 2001, 11: 1346-1352. 10.1101/gr.184401
McGhee JD, Fukushige T, Krause MW, Minnema SE, Goszczynski B, Gaudet J, Kohara Y, Bossinger O, Zhao Y, Khattra J: ELT-2 is the predominant transcription factor controlling differentiation and function of the C. elegans intestine, from embryo to adult. Dev Biol. 2009, 327: 551-565. 10.1016/j.ydbio.2008.11.034
McGhee JD, Sleumer MC, Bilenky M, Wong K, McKay SJ, Goszczynski B, Tian H, Krich ND, Khattra J, Holt RA: The ELT-2 GATA-factor and the global regulation of transcription in the C. elegans intestine. Dev Biol. 2007, 302: 627-645. 10.1016/j.ydbio.2006.10.024
Wang X, Zhao Y, Wong K, Ehlers P, Kohara Y, Jones SJ, Marra MA, Holt RA, Moerman DG, Hansen D: Identification of genes expressed in the hermaphrodite germ line of C. elegans using SAGE. BMC Genomics. 2009, 10: 213- 10.1186/1471-2164-10-213
Ruzanov P, Jones SJ, Riddle DL: Discovery of novel alternatively spliced C. elegans transcripts by computational analysis of SAGE data. BMC Genomics. 2007, 8: 447- 10.1186/1471-2164-8-447
Chen J, Sun M, Lee S, Zhou G, Rowley JD, Wang SM: Identifying novel transcripts and novel genes in the human genome by using novel SAGE tags. Proc Natl Acad Sci USA. 2002, 99: 12257-12262. 10.1073/pnas.192436499
Schaefer BC: Revolutions in rapid amplification of cDNA ends: new strategies for polymerase chain reaction cloning of full-length cDNA ends. Anal Biochem. 1995, 227: 255-273. 10.1006/abio.1995.1279
Zorio DA, Cheng NN, Blumenthal T, Spieth J: Operons as a common form of chromosomal organization in C. elegans. Nature. 1994, 372: 270-272. 10.1038/372270a0
Blumenthal T: Trans-splicing and operons. WormBook. 2005, 1-9.
Kent WJ: BLAT--the BLAST-like alignment tool. Genome Res. 2002, 12: 656-664.
Bonetta L: Gene expression: an expression of interest. Nature. 2006, 440: 1233-1237. 10.1038/4401233a
Gamper HB, Cimino GD, Hearst JE: Solution hybridization of crosslinkable DNA oligonucleotides to bacteriophage M13 DNA. Effect of secondary structure on hybridization kinetics and equilibria. J Mol Biol. 1987, 197: 349-362. 10.1016/0022-2836(87)90128-8
Rozen S, Skaletsky H: Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000, 132: 365-386.
Breathnach R, Benoist C, O'Hare K, Gannon F, Chambon P: Ovalbumin gene: evidence for a leader sequence in mRNA and DNA sequences at the exon-intron boundaries. Proc Natl Acad Sci USA. 1978, 75: 4853-4857. 10.1073/pnas.75.10.4853
Breathnach R, Chambon P: Organization and expression of eucaryotic split genes coding for proteins. Annu Rev Biochem. 1981, 50: 349-383. 10.1146/annurev.bi.50.070181.002025
Stanke M, Waack S: Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003, 19 (Suppl 2): ii215-225.
Schweikert G, Zien A, Zeller G, Behr J, Dieterich C, Ong CS, Philips P, De Bona F, Hartmann L, Bohlen A: mGene: accurate SVM-based gene finding with an application to nematode genomes. Genome Res. 2009, 19: 2133-2143. 10.1101/gr.090597.108
Korf I, Flicek P, Duan D, Brent MR: Integrating genomic homology into gene structure prediction. Bioinformatics. 2001, 17 (Suppl 1): S140-148.
Solovyev V, Kosarev P, Seledsov I, Vorobyev D: Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 2006, 7 (Suppl 1): S10 11-12. 10.1186/gb-2006-7-s1-s10. 10.1186/gb-2006-7-s1-s10
Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Clawson H, Diekhans M, Furey TS, Harte RA, Hsu F: The UCSC Genome Browser Database: update 2006. Nucleic Acids Res. 2006, 34: D590-598. 10.1093/nar/gkj144
Acknowledgements
We thank Drs. Harald Hutter, David Baillie and Robert Johnsen for their advice, technical assistance, and reagents. We also thank Dr. Johnsen for proofreading the manuscript. We thank members of the Chen, Hutter, and Baillie laboratories for their technical assistance. Lindsay McGhee helped with generating figures. This project is supported by a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada (NSERC) to NC. NC is also a Michael Smith Foundation for Health Research (MSFHR) Scholar and a Canadian Institutes of Health Research (CIHR) New Investigator.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
NC and DGM conceived of the study. MJN conducted the experiments. MJN and NC wrote the manuscript with input from DGM. All authors have read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article
Cite this article
Nesbitt, M.J., Moerman, D.G. & Chen, N. Identifying novel genes in C. elegans using SAGE tags. BMC Molecular Biol 11, 96 (2010). https://doi.org/10.1186/1471-2199-11-96
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2199-11-96