
Abstract
Background
Staphylococcus aureus is an important pathogen that forms biofilms. The global regulator sarA is essential for biofilm formation. Since the modulator of sarA (msa) is required for full expression of sarA and regulates several virulence factors, we examined the capacity of the msa mutant to form biofilm.
Results
We found that mutation of msa results in reduced expression of sarA in biofilm and that the msa mutant formed a weak and unstable biofilm. The msa mutant is able to adhere to surfaces and begins to form biofilm but fails to mature indicating that the defect of the msa mutant biofilm is in the accumulation stage but not in primary adhesion.
Conclusion
The msa gene plays an important role in biofilm development which is likely due to its role in modulating the expression of sarA. This finding is significant because it identifies a new gene that plays a role in the development of biofilm.
Similar content being viewed by others


Background
Staphylococcus aureus is a gram-positive pathogen that causes potentially life threatening nosocomial- and community-acquired infections, such as osteomyelitis and endocarditis. An important characteristic of S. aureus is its ability to form a biofilm, a characteristic associated with several diseases [1]. Bacteria in biofilms are encased in a polysaccharide glycocalyx [2], which provides them with protection against host defenses and antimicrobial drugs [2]. Staphylococcal biofilms form in two distinct stages: (1) primary adhesion to surfaces by means of adhesins or cell wall components, and (2) accumulation of multilayered clusters of cells via the production of a polysaccharide [3]. Cells are also able to detach from the biofilm and disperse to distant sites for colonization or infection.
The staphylococcal accessory regulator sarA is a major global regulator that is essential for biofilm formation both in vitro and in vivo [4–6]. Additionally, O'Neill et al.[7] showed that sarA is essential for biofilm formation in both MRSA and MSSA. However the mechanism of sarA regulation of biofilm is not yet understood. Previously we identified the msa gene as a positive modulator of sarA [8]. We also showed that mutation of msa resulted in differential expression of several virulence factors [8]. These findings prompted the present study, focused on the role of msa in biofilm formation. We show here that msa indeed modulates the accumulation of biofilm in S. aureus.
Results and discussion
The msa mutant has a weak biofilm defect
Prior studies have shown that the modulator of SarA gene (msa) is required for full expression of SarA, which in turn is essential for biofilm formation [4, 6, 8]. To examine the role of msa in biofilm formation, we generated an msa mutant in the methicillin resistant S. aureus (MRSA) strain COL. S. aureus COL was chosen for study because it forms a biofilm in vitro and is virulent in animal models of endocarditis [9, 10]. We confirmed the msa mutation, and its effect on sarA, by measuring transcription levels of msa and sarA in the wild-type COL strain, the msa mutant, and the complemented msa mutant by real-time quantitative PCR (RT-qPCR). As expected, the msa mutant showed no detectable expression of msa in planktonic cultures or biofilm (Table 1). Additionally, transcription of sarA was reduced at least five-fold in these cultures in both planktonic cultures and biofilm (Table 1). These results are consistent with finding from our previous study [8] and show that msa is a positive modulator of sarA during planktonic growth as well as biofilm in strain COL.
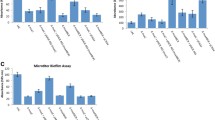
The biofilm forming capacity of the wild-type COL strain, the msa mutant, and the complemented msa mutant were examined by microtiter plate assay. Biofilm formation was observed in at 6, 12, and 24 h post-inoculation in the wild-type COL strain and the complemented msa mutant microtiter plates (Fig. 1). Conversely, there was no evidence of biofilm formation for the msa mutant at 6 or 12 h post-inoculation. At 24 hours post-inoculation, the msa mutant formed a biofilm that appeared similar to that of the wild-type COL strain (Fig. 1). These results were reproduced at least three times and were confirmed in subsequent experiments, suggesting that the msa mutant has a defect in biofilm growth under steady-state conditions.

Biofilm formation in the msa mutant in microtiter plates. The wild type strain COL, the msa mutant and the complemented msa mutant were grown in TSB supplemented with NaCl and glucose. Cultures were incubated for 6, 12 and 24 hours in the wells of microtiter plates with pre-coating with plasma proteins. Biofilm was quantitated by staining with crystal violet and elution with ethanol as described in text. All values have been normalized to wild type levels which were arbitrarily set as 100%.
To further examine this phenotype, we used flow cells to test the ability of the msa mutant to form biofilm under shear forces. Three flow cells coated with human plasma were each inoculated with the wild-type COL strain, the msa mutant, and the complemented msa mutant, and then monitored continuously for biofilm formation while media flowed through the chamber. Wild-type COL strain and the complemented msa mutant formed robust and mature biofilms by 12 hours post-inoculation (Fig. 2). The msa mutant, however, failed to form a robust and uniform biofilm within the flow cell in the first 12 hours (Fig. 2). The gross morphology of the partial biofilm formed by the msa mutant was similar to those formed by the wild type strain and the complemented mutant (Fig. 2). However, while the biofilms formed by the wild-type and the complemented msa mutant persisted for up to 36 h before sloughing off, the biofilm formed by the msa mutant rapidly disintegrated (Fig. 2). These results were confirmed in three independent experiments, indicating that the msa mutant is defective in its ability to form mature biofilms.

Biofilm formation in the msa mutant in flow cells. The wild type strain COL, the msa mutant and the complemented msa mutant were used to inoculate flow cells. TSB supplemented with NaCl and glucose was provided at a flow rate of 0.5 ml/minute. Biofilm formation was monitored for 36 hours. Arrow indicates the direction of flow of medium.
Since expression of sarA is essential to biofilm formation, we wanted to examine the expression of sarA in cells forming a biofilm. We harvested biofilm from flow cells inoculated with msa mutant and wild-type COL at 24 h post-inoculation and measured the expression of sarA by RT-qPCR. We found that sarA expression levels were significantly reduced in the msa mutant, suggesting that msa is required for full expression of sarA in biofilm (Table 1). It is possible that the weak biofilm formation phenotype of the msa mutant could be due to a reduction in sarA expression.
Biofilm formation is a complex process that generally involves three stages: (1) primary adhesion to surfaces, (2) accumulation of multilayered clusters of cells, and (3) detachment. We carried out experiments to determine which stage of biofilm formation is disrupted in the msa mutant. Using two different adherence assays, we measured the ability of cells to attach to surfaces in the presence of host proteins by coating with human plasma and in their absence by not coating with plasma (Fig. 3). We found that the msa mutant had no defect in initial adherence to surfaces. In fact, the msa mutant adhered to surfaces significantly better than the wild-type COL strain especially in the catheter assay where no plasma was used (Fig. 3A). The complemented msa mutant showed a level of adhesion to surfaces that was similar to wild-type (Fig. 3). These results indicated that primary binding to surfaces was not responsible for the biofilm formation defect in the msa mutant. It was important to test initial adherence to surfaces with or without plasma coating because binding of host proteins is a major contributor to primary adhesion. In this case, however, we found that it does not play a role in the biofilm phenotype of the msa mutant.

Initial adherence assays. A. Catheter adherence assay. Standardized overnight cultures of the wild type strain COL, the msa mutant and the complemented msa mutant were incubated with catheters at 37°C for 30 minutes. Results represent the mean ± SEM of three independent experiments. Student's paired t test was used to compare the msa mutant and the complemented msa mutant to the wild type strain (*denotes statistical significance of P < 0.05). B. Standardized overnight cultures of the wild type strain COL, the msa mutant and the complemented msa mutant were incubated for 1 hour at 37°C in plasma-coated microtitre wells. Adherent cells were then fixed with ethanol and then stained with Crystal Violet. Ethanol was then used to elute the wells and absorbance was measured. Results represent the mean ± SEM from three independent experiments.
A recent study by O'Neill et al. [11] showed that in addition to primary adherence, fibronectin binding contributes to intercellular accumulation in biofilm. We examined the ability of the msa mutant to bind the immobilized ligands by coating microtitre wells with fibronectin or fibrinogen and compared the capacity of the wild type COL strain, the msa mutant and the complemented mutant to bind these host proteins. We found that the wild type strain and the complemented mutant bind both fibronectin and fibrinogen, however, the msa mutant binds fibronectin but not fibrinogen (data not shown). In an effort to explain these results, we examined the expression of fibronectin-binding protein A (fnbA) and clumping factor (clfA) in biofilm (Table 2). Consistent with the binding assays, there was no significant difference in expression levels of fnbA between the three strains, while the expression of clfA in the msa mutant was significantly reduced in biofilm. This indicated that the lack of fibrinogen binding by the msa mutant is primarily due to lack of expression of ClfA.
There are many other genes known to be involved in biofilm formation. The major autolysin, atl, has been shown to promote adherence of bacterial cells to solid surfaces [12, 13]. The Atl homolog of S. epidermidis, AtlE, has also been shown to play an important role in primary attachment to polystyrene surfaces [14]. Gene expression studies using RT-qPCR in the msa mutant revealed that atl levels were significantly reduced in biofilm (Table 2). Therefore, our studies with S. aureus strain COL msa mutant have demonstrated that primary adhesion to surfaces with or without a plasma coat does not require full expression of the major autolysin Atl. This is consistent with previous findings in which enhanced biofilm formation occurred in the absence of the major autolysin Atl [15]. Collectively, these results suggest that the strain COL msa mutant is not defective in primary adhesion to surfaces, but that the defect manifests in the accumulation stage of biofilm formation.
msa mutant is defective in the accumulation stage of biofilm formation
We investigated the possibility that mutation of the msa gene causes a growth defect that could explain a weak biofilm. We measured growth rates of the wild-type strain, the msa mutant, and the complemented msa mutant in planktonic cultures in TSB and found no significant difference between the msa mutant and the wild-type strain (data not shown). This was important to verify in order to eliminate the possibility of a growth defect caused by mutation of msa. We then measured the rate of accumulation of cells within the flow cell system for wild-type and msa mutant strains. In order to monitor cell deposition, we introduced into our strains plasmid pSB2019 (a kind gift from Dr. Phillip J. Hill; [16], which carries the constitutively expressed Gfp3), and used confocal microscopy to monitor biofilm formation at regular intervals. Consistent with our previous observations, there was no significant difference in initial adherence to the surface or formation of microcolonies between the msa mutant and the wild-type. However, the wild-type strain formed a thick biofilm as early as four hours after inoculation, while the msa mutant failed to develop a multilayered biofilm in the first six hours (Fig. 4). We observed a specific absence of biofilm "towers" in the msa mutant biofilms, as compared to the wild-type biofilms, further suggesting that the defect in biofilm formation in the msa mutant occurs in the accumulation stage.

Confocal microscopy images of biofilm. The msa mutant and the wild type strain COL were imaged 6 hours post inoculation of flow cells. The panels on the left are an overlay of multiple slices, and the side frames of the panels on the right show the z-stack showing the thickness and the architecture of the biofilm. The line in the z-stack indicates the level at which the photograph of the x-y plane was taken. Photographs were taken at a magnification of ×600.
To further characterize the biofilm formation defect in cells lacking msa, we used RT-qPCR to analyze the expression of icaA (ica operon), arcA (arginine deiminase), tcaR (transcription regulator), atlA (autolysin), alsS (alpha-acetolactate synthase), spxA (transcriptional regulator), genes that are reported to be involved in biofilm development (Table 2). Some of these selected loci are regulated by sarA (alsS, atlA, and icaA); the others are not directly associated with the sarA regulon (arcA, spx, and tcaR). Expression levels of these genes were analyzed in biofilm using the wild-type strain COL, the msa mutant, and the complemented msa mutant (Table 2). A change in expression level of twofold or higher was considered significant (Table 2).
Mutation of msa resulted in a significant increase in alsS expression in biofilm. The alsSD operon encodes acetolactate synthase and an acetolactate decarboxylase. Previous studies have reported that mutation of the alsSD operon in S. aureus resulted in a biofilm defect [17, 18]. The biofilm defect of the alsSD mutant was attributed to the role of this operon in the production of acetoin from pyruvate [18, 19]. Acetoin production is necessary for acid tolerance within biofilms [5, 20]. The effect of over-expression of the alsSD operon as observed in the msa S. aureus mutant on biofilm formation is not clear. One might speculate that premature build up acetoin in the medium could signal exhaustion of glucose and lead to detachment of cells from biofilm. Further studies are needed to explore this possibility.
Mutation of msa resulted in a significant decrease in expression of arcA in biofilm (Table 2). The arcA gene encodes arginine deiminase which is a member of the arginine deaminase (ADI) pathway. This pathway is used to generate energy using arginine under anaerobic conditions, [21–23]. The results of several studies point to the importance of the ADI pathway in biofilm formation and pathogenesis. Some oral bacteria have been shown generate ammonia via the ADI pathway to maintain pH homeostasis in biofilms [24]. Other studies have shown that the ADI pathway was induced during biofilm formation [5]. Additionally, bacteria in biofilm selectively utilize six amino acids, including arginine [25], further demonstrating the importance of the ADI pathway in biofilm formation and pathogenesis and may explain the msa mutant biofilm phenotype. However, when arcD was disrupted in the S. aureus strain UAMS-1, the mutant formed effective biofilms and were as virulent as wild-type in a catheter infection mouse model despite the fact that PIA was significantly reduced [25]. These discrepant results may be due to differences in strains, growth conditions, or infection model but they clearly indicated the need for more studies in the role of arginine metabolism in biofilm development and pathogenesis.
Biofilm accumulation relies on cell-cell adhesion mediated by the polysaccharide intercellular adhesin (PIA), which is produced by the icaADBC operon, and was shown to play a major role in biofilm accumulation [26]. Recent studies, however, have indicated that the icaADBC operon is not essential for biofilm formation in some strains [5, 15, 27, 28]. We studied the expression levels of genes encoded by this operon and found that the msa mutation reduced the expression of icaA in the post-exponential growth phase of planktonic cultures only (data not shown). In biofilm, however, the expression of icaA in the mutant was not significantly different from wild-type (Table 2). The ica-dependent pathway is primarily regulated by the icaR repressor [29]. When IcaR becomes activated by Spx, PIA levels are reduced [30, 31]. We found that there was no significant difference in expression of icaR or spx in the msa mutant compared to the wild-type (data not shown). This is consistent with findings by Tu Quoc et al. [32] that some biofilm-defective mutants did not show altered PIA levels. Additionally, O'Neill et al. [7], recently showed that glucose-induced biofilm formation in MRSA strains is ica-independent. This is relevant to our data, since the COL strain is a MRSA strain and glucose was added to the culture media to induce biofilm in this study suggesting that msa is involved in biofilm formation using an ica-independent mechanism as was previously described in some strains [5, 33, 34]. Another important locus, sasG, which is similar to accumulation- associated protein in S. epidermidis has been shown to play a role in cell-cell adhesion and accumulation of biofilm [35, 36], however, there was no change in expression of sasG between the msa mutant and wild type grown in planktonic cultures as determined by DNA microarray experiments in our lab (data not shown).
Conclusion
In summary, mutation of the msa gene in strain COL of S. aureus results in a weak biofilm at the accumulation stage resulting in an immature biofilm. This defect is likely mediated by the reduced expression of sarA in the msa mutant. Our results suggested that the weak/unstable biofilm defect in the msa mutant is an intermediate phenotype between the sarA mutant and wild-type. However, we cannot rule out the contribution of other loci that fall under the influence of msa in a sarA-independent manner (e.g. arcA). Our findings emphasize the complex nature of biofilms and indicated that several independent regulators and environmental stimuli contribute to the establishment of sessile communities of S. aureus. The intermediate phenotype of the biofilm formed by the msa mutant is a helpful clue in deciphering the sarA-mediated mechanism of biofilm formation, which is still unclear. Sequence analysis shows that Msa is a putative membrane protein [8, 37] suggesting that it may play a role in environmental sensing that feeds into the sarA regulon to contribute to biofilm formation.
Methods
Bacteria and growth conditions
The S. aureus biofilm-forming strain COL was used in this study. Strains were grown on tryptic soy agar (TSA) or in tryptic soy broth (TSB) at 37°C under constant aeration, supplemented with antibiotics where appropriate. Generalized transduction with phage Φ11 was used to generate an msa mutant and a complemented msa mutant in the COL strain, as described previously [8]. The media used in flow cells and microtitre plate assays was TSB supplemented with 3% sodium chloride and 0.5% glucose.
Biofilm assays
Biofilm assays were performed in microtitre plates and flow cells, as described previously [4, 5]. Briefly, flow cells (Stovall Life Science, Greensboro, NC) were pre-coated with human plasma. A suspension of bacteria from an overnight culture were then introduced into the flow cells by injection and allowed to incubate at 37°C for one hour. Media was then pumped through the flow cells at a flow rate of 0.5 ml/min. Flow cells were observed and photographed periodically. No antibiotic selection was used when growing the complemented msa mutant in biofilm.
Binding assays
Host proteins binding assays were performed as we described previously [5]. These assays were used to compare the capacity of the msa mutant to bind fibrinogen and fibronectin.
Initial adherence assay
We used two assays to measure the capacity of the msa mutant to bind surfaces with or without pre-coating with plasma. The first one was performed by modifying the microtitre biofim assay described previously [4]. Briefly, overnight cultures of S. aureus test strains were diluted to an OD560 of 0.1 in fresh TSB, and 200 μl was added to each well (polystyrene pre-coated with human plasma) in triplicate. Following a 1 hour incubation at 37°C, the microtitre wells were washed three times with 1× PBS. Adherent cells were then fixed with 200 μl of 100% ethanol for 10 min. Ethanol was removed and the wells were air dried for 2 min. Adherent cells were stained for 2 minutes with 200 μl of 0.41% Crystal Violet (w/v in 12% ethanol), then washed three times with 1× PBS. Wells were allowed to dry, and then ethanol was used to elute the wells. Absorbance readings were taken at 595 nm using a Synergy2™ Multi-Mode Microplate Reader (BioTek Instruments, Inc. Winooski, Vermont). Data shown is the average of three independent experiments. The second assay measures adherence to a catheter without pre-coating with plasma. This assay was performed as previously described [38]. Briefly, staphylococcal strains were grown overnight in TSB and cultures were standardized to an OD650 of 0.1. Catheters (PE10; Becton Dickinson and Co., Sparks, MD.) were cut to a length of 0.5 cm and placed into appropriate cultures. After incubating the cultures at 37°C for 30 min, the catheters were removed using sterile forceps and washed five times in sterile PBS. After washing, the catheters were placed in 1% proteose peptone (Difco, Ann Arbor, MI.) and continuously vortexed for two minutes to release bacterial cells from the catheter. For bacterial enumeration, serial dilutions were plated onto TSA plates. The mean and standard errors were calculated for the adherence of each strain.
Confocal microscopy
Confocal scanning laser microscopy was performed using a Carl-Zeiss LSM 510 META with excitation at 488 nm and emission collected at 500 to 530 nm (green channel). Image stacks of biofilms were taken from at least three distinct regions on the flow cell and were analyzed using the provided software (Carl-Zeiss, Inc., Peabody, Mass.). Thickness of the biofilm was measured starting from the z-section at the flow-cell/biofilm interface to the z-section at the top of the biofilm surface containing < 5% of total biomass.
RNA isolation
Total RNA was isolated from S. aureus planktonic cultures using Qiagen RNeasy Maxi column (Qiagen) as previously described [8], based on a method developed by Lindsay and Foster [39]. S. aureus cultures were grown without antibiotic selection and under low-aeration conditions (150 r.p.m. at a media volume:flask volume ratio of 0.5). Cells were harvested at optical densities (OD560) of 0.3, 1.5 and 4.0, which correspond to the mid-exponential, late-exponential, and post-exponential growth phases, respectively. RNA from biofilms was accomplished by harvesting cells from flow cells as previously described [5] and using using Qiagen RNeasy Maxi column. The data shown is from three independent experiments.
Real-time quantitative PCR
Gene expression analysis by real-time quantitative PCR (RT-qPCR) was performed as described previously [8], with all reactions done in triplicate. The constitutively expressed gyrase gene (gyr) was used as an endogenous control, as described previously [40]. Primer specificity and efficiency was measured as described previously [8]. Expression analysis for each gene was based on at least two independent experiments. Two-fold or higher changes in gene expression were considered significant.
References
Donlan RM, Murga R, Bell M, Toscano CM, Carr JH, Novicki TJ, Zuckerman C, Corey LC, Miller JM: Protocol for detection of biofilms on needleless connectors attached to central venous catheters. J Clin Microbiol. 2001, 39 (2): 750-753. 10.1128/JCM.39.2.750-753.2001.
Stewart PS: Mechanisms of antibiotic resistance in bacterial biofilms. Int J Med Microbiol. 2002, 292 (2): 107-113. 10.1078/1438-4221-00196.
Cramton S, Gotz F: Biofilm development in Staphylococcus. Microbial Biofilms. 2004, Washington, DC: ASM Press, 64-84.
Beenken KE, Blevins JS, Smeltzer MS: Mutation of sarA in Staphylococcus aureus limits biofilm formation. Infect Immun. 2003, 71 (7): 4206-4211. 10.1128/IAI.71.7.4206-4211.2003.
Beenken KE, Dunman PM, McAleese F, Macapagal D, Murphy E, Projan SJ, Blevins JS, Smeltzer MS: Global gene expression in Staphylococcus aureus biofilms. J Bacteriol. 2004, 186 (14): 4665-4684. 10.1128/JB.186.14.4665-4684.2004.
Valle J, Toledo-Arana A, Berasain C, Ghigo JM, Amorena B, Penades JR, Lasa I: SarA and not sigmaB is essential for biofilm development by Staphylococcus aureus. Mol Microbiol. 2003, 48 (4): 1075-1087. 10.1046/j.1365-2958.2003.03493.x.
O'Neill E, Pozzi C, Houston P, Smyth D, Humphreys H, Robinson DA, O'Gara JP: Association between methicillin susceptibility and biofilm regulation in Staphylococcus aureus isolates from device-related infections. J Clin Microbiol. 2007, 45 (5): 1379-1388. 10.1128/JCM.02280-06.
Sambanthamoorthy K, Smeltzer MS, Elasri MO: Identification and characterization of msa (SA1233), a gene involved in expression of SarA and several virulence factors in Staphylococcus aureus. Microbiology. 2006, 152 (Pt 9): 2559-2572. 10.1099/mic.0.29071-0.
Jacqueline C, Navas D, Batard E, Miegeville AF, Le Mabecque V, Kergueris MF, Bugnon D, Potel G, Caillon J: In vitro and in vivo synergistic activities of linezolid combined with subinhibitory concentrations of imipenem against methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2005, 49 (1): 45-51. 10.1128/AAC.49.1.45-51.2005.
Rigoulay C, Entenza JM, Halpern D, Widmer E, Moreillon P, Poquet I, Gruss A: Comparative analysis of the roles of HtrA-like surface proteases in two virulent Staphylococcus aureus strains. Infect Immun. 2005, 73 (1): 563-572. 10.1128/IAI.73.1.563-572.2005.
O'Neill E, Pozzi C, Houston P, Humphreys H, Robinson DA, Loughman A, Foster TJ, O'Gara JP: A novel Staphylococcus aureus biofilm phenotype mediated by the fibronectin-binding proteins, FnBPA and FnBPB. J Bacteriol. 2008
Biswas R, Voggu L, Simon UK, Hentschel P, Thumm G, Gotz F: Activity of the major staphylococcal autolysin Atl. FEMS Microbiol Lett. 2006, 259 (2): 260-268. 10.1111/j.1574-6968.2006.00281.x.
Yamada S, Sugai M, Komatsuzawa H, Nakashima S, Oshida T, Matsumoto A, Suginaka H: An autolysin ring associated with cell separation of Staphylococcus aureus. J Bacteriol. 1996, 178 (6): 1565-1571.
Heilmann C, Hussain M, Peters G, Gotz F: Evidence for autolysin-mediated primary attachment of Staphylococcus epidermidis to a polystyrene surface. Mol Microbiol. 1997, 24 (5): 1013-1024. 10.1046/j.1365-2958.1997.4101774.x.
Toledo-Arana A, Merino N, Vergara-Irigaray M, Debarbouille M, Penades JR, Lasa I: Staphylococcus aureus develops an alternative, ica-independent biofilm in the absence of the arlRS two-component system. J Bacteriol. 2005, 187 (15): 5318-5329. 10.1128/JB.187.15.5318-5329.2005.
Qazi SN, Rees CE, Mellits KH, Hill PJ: Development of gfp Vectors for Expression in Listeria monocytogenes and Other Low G+C Gram Positive Bacteria. Microb Ecol. 2001, 41 (4): 301-309.
Cassat J, Dunman PM, Murphy E, Projan SJ, Beenken KE, Palm KJ, Yang SJ, Rice KC, Bayles KW, Smeltzer MS: Transcriptional profiling of a Staphylococcus aureus clinical isolate and its isogenic agr and sarA mutants reveals global differences in comparison to the laboratory strain RN6390. Microbiology. 2006, 152 (Pt 10): 3075-3090. 10.1099/mic.0.29033-0.
Yang SJ, Dunman PM, Projan SJ, Bayles KW: Characterization of the Staphylococcus aureus CidR regulon: elucidation of a novel role for acetoin metabolism in cell death and lysis. Mol Microbiol. 2006, 60 (2): 458-468. 10.1111/j.1365-2958.2006.05105.x.
Kovacikova G, Lin W, Skorupski K: Dual regulation of genes involved in acetoin biosynthesis and motility/biofilm formation by the virulence activator AphA and the acetate-responsive LysR-type regulator AlsR in Vibrio cholerae. Mol Microbiol. 2005, 57 (2): 420-433. 10.1111/j.1365-2958.2005.04700.x.
Weinrick B, Dunman PM, McAleese F, Murphy E, Projan SJ, Fang Y, Novick RP: Effect of mild acid on gene expression in Staphylococcus aureus. J Bacteriol. 2004, 186 (24): 8407-8423. 10.1128/JB.186.24.8407-8423.2004.
Cunin R, Glansdorff N, Pierard A, Stalon V: Biosynthesis and metabolism of arginine in bacteria. Microbiol Rev. 1986, 50 (3): 314-352.
Ruepp A, Soppa J: Fermentative arginine degradation in Halobacterium salinarium (formerly Halobacterium halobium): genes, gene products, and transcripts of the arcRACB gene cluster. J Bacteriol. 1996, 178 (16): 4942-4947.
Schofield PJ, Edwards MR, Matthews J, Wilson JR: The pathway of arginine catabolism in Giardia intestinalis. Mol Biochem Parasitol. 1992, 51 (1): 29-36. 10.1016/0166-6851(92)90197-R.
Casiano-Colon A, Marquis RE: Role of the arginine deiminase system in protecting oral bacteria and an enzymatic basis for acid tolerance. Appl Environ Microbiol. 1988, 54 (6): 1318-1324.
Zhu Y, Weiss EC, Otto M, Fey PD, Smeltzer MS, Somerville GA: Staphylococcus aureus biofilm metabolism and the influence of arginine on polysaccharide intercellular adhesin synthesis, biofilm formation, and pathogenesis. Infect Immun. 2007, 75 (9): 4219-4226. 10.1128/IAI.00509-07.
Gotz F: Staphylococcus and biofilms. Mol Microbiol. 2002, 43 (6): 1367-1378. 10.1046/j.1365-2958.2002.02827.x.
Cucarella C, Tormo MA, Ubeda C, Trotonda MP, Monzon M, Peris C, Amorena B, Lasa I, Penades JR: Role of biofilm-associated protein bap in the pathogenesis of bovine Staphylococcus aureus. Infect Immun. 2004, 72 (4): 2177-2185. 10.1128/IAI.72.4.2177-2185.2004.
Fitzpatrick F, Humphreys H, O'Gara JP: The genetics of staphylococcal biofilm formation–will a greater understanding of pathogenesis lead to better management of device-related infection?. Clin Microbiol Infect. 2005, 11 (12): 967-973. 10.1111/j.1469-0691.2005.01274.x.
Conlon KM, Humphreys H, O'Gara JP: icaR encodes a transcriptional repressor involved in environmental regulation of ica operon expression and biofilm formation in Staphylococcus epidermidis. J Bacteriol. 2002, 184 (16): 4400-4408. 10.1128/JB.184.16.4400-4408.2002.
Frees D, Chastanet A, Qazi S, Sorensen K, Hill P, Msadek T, Ingmer H: Clp ATPases are required for stress tolerance, intracellular replication and biofilm formation in Staphylococcus aureus. Mol Microbiol. 2004, 54 (5): 1445-1462. 10.1111/j.1365-2958.2004.04368.x.
Pamp SJ, Frees D, Engelmann S, Hecker M, Ingmer H: Spx is a global effector impacting stress tolerance and biofilm formation in Staphylococcus aureus. J Bacteriol. 2006, 188 (13): 4861-4870. 10.1128/JB.00194-06.
Tu Quoc PH, Genevaux P, Pajunen M, Savilahti H, Georgopoulos C, Schrenzel J, Kelley WL: Isolation and characterization of biofilm formation-defective mutants of Staphylococcus aureus. Infect Immun. 2007, 75 (3): 1079-1088. 10.1128/IAI.01143-06.
Hennig S, Nyunt Wai S, Ziebuhr W: Spontaneous switch to PIA-independent biofilm formation in an ica -positive Staphylococcus epidermidis isolate. Int J Med Microbiol. 2007, 297 (2): 117-122. 10.1016/j.ijmm.2006.12.001.
Qin Z, Yang X, Yang L, Jiang J, Ou Y, Molin S, Qu D: Formation and properties of in vitro biofilms of ica -negative Staphylococcus epidermidis clinical isolates. J Med Microbiol. 2007, 56 (Pt 1): 83-93. 10.1099/jmm.0.46799-0.
Corrigan RM, Rigby D, Handley P, Foster TJ: The role of Staphylococcus aureus surface protein SasG in adherence and biofilm formation. Microbiology. 2007, 153 (Pt 8): 2435-2446. 10.1099/mic.0.2007/006676-0.
Conrady DG, Brescia CC, Horii K, Weiss AA, Hassett DJ, Herr AB: A zinc-dependent adhesion module is responsible for intercellular adhesion in staphylococcal biofilms. Proc Natl Acad Sci USA. 2008
Nagarajan V, Elasri MO: Structure and function predictions of the Msa protein in Staphylococcus aureus. BMC Bioinformatics. 2007, 8 (Suppl 7): S5-10.1186/1471-2105-8-S7-S5.
McKenney D, Hubner J, Muller E, Wang Y, Goldmann DA, Pier GB: The ica locus of Staphylococcus epidermidis encodes production of the capsular polysaccharide/adhesin. Infect Immun. 1998, 66 (10): 4711-4720.
Lindsay JA, Foster SJ: Interactive regulatory pathways control virulence determinant production and stability in response to environmental conditions in Staphylococcus aureus. Mol Gen Genet. 1999, 262 (2): 323-331. 10.1007/s004380051090.
Goerke C, Campana S, Bayer MG, Doring G, Botzenhart K, Wolz C: Direct quantitative transcript analysis of the agr regulon of Staphylococcus aureus during human infection in comparison to the expression profile in vitro. Infect Immun. 2000, 68 (3): 1304-1311. 10.1128/IAI.68.3.1304-1311.2000.
Acknowledgements
This work was supported by grant no.1R15AI062727-01A1 from the National Institute of Allergy and Infectious Diseases (NIAID) to MOE and by The Mississippi Functional Genomics Network (NIH/NCRR P20 RR016476).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
KS created the msa mutation, characterized the biofilm phenotype, analyzed gene expression and drafted the manuscript. AS carried out biofilm assays and analyzed gene expression. KS and AS contributed equally to this manuscript. VN analyzed sequence data of the msa mutant. MOE conceived of the study, participated in its design and coordination. All authors read and approved the final manuscript.
Karthik Sambanthamoorthy, Antony Schwartz contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Sambanthamoorthy, K., Schwartz, A., Nagarajan, V. et al. The Role of msa in Staphylococcus aureus Biofilm Formation. BMC Microbiol 8, 221 (2008). https://doi.org/10.1186/1471-2180-8-221
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-8-221