
Abstract
Background
All bacterial genomes contain repetitive sequences which are members of specific DNA families. Such repeats may occur as single units, or found clustered in multiple copies in a head-to-tail configuration at specific loci. The number of clustered units per locus is a strain-defining parameter. Assessing the length variability of clusters of repeats is a versatile typing methodology known as multilocus variable number of tandem repeat analysis (MLVA).
Results
Stenotrophomonas maltophilia is an environmental bacterium increasingly involved in nosocomial infections and resistant to most antibiotics. The availability of the whole DNA sequence of the S. maltophilia strain K279a allowed us to set up fast and accurate PCR-based diagnostic protocols based on the measurement of length variations of loci carrying a variable number of short palindromic repeats marking the S. maltophilia genome. On the basis of the amplimers size, it was possible to deduce the number of repeats present at 12 different loci in a collection of S. maltophilia isolates, and therefore label each of them with a digit. PCR-negative regions were labelled 0. Co-amplification of two pairs of loci provided a 4-digit code sufficient for immediate subtyping. By increasing the number of loci analyzed, it should be possible to assign a more specific digit profile to isolates. In general, MLVA data match genotyping data obtained by PFGE (pulsed-field gel electrophoresis). However, some isolates exhibiting the same PCR profiles at all loci display distinct PFGE patterns.
Conclusion
The utilization of the present protocol allows to type several S. maltophilia isolates in hours. The results are immediately interpretable without the need for sophisticated softwares. The data can be easily reproducible, and compared among different laboratories.
Similar content being viewed by others

Background
After years of debate regarding its appropriate taxonomic position, the nonfermentative, gram-negative bacillus previously known as Pseudomonas maltophilia or Xanthomonas maltophilia, has been definitively classified as Stenotrophomonas maltophilia [1]. This species is found in a wide variety of environments, and has been isolated from different sources, including water, sewage, soil and plant rhizosphere environments [2]. S. maltophilia is increasingly prevalent in hospitals, and is often isolated in hospitalized patients, as well as in cystic fibrosis (CF), burn, and immunosuppressed patients. The presence of S. maltophilia in CF patients is not associated with a worse clinical outcome. However, the organism contributes to chronic airway inflammation [3]. Moreover, in mixed infection formed in the CF lungs, S. maltophilia has been shown to influence the architecture of Pseudomonas aeruginosa biofilms by producing a diffusable signal factor [4].
S. maltophilia isolates exhibit high genetic diversity. Genotypic profiles have been determined by a variety of methods, including AFLP (amplified fragment length polymorphism) fingerprinting [5], RFLP (restriction fragment length polymorphism) analysis of the gyrase B gene [6] or the intergenic region between smeD and smeT genes [7], ERIC-PCR [8], and PFGE (pulsed-field gel electrophoresis) analysis of Xba I genomic digests [8–11].
Genome-wide analyses showed that in many bacterial genomes short DNA segments are amplified in tandem at specific chromosomal loci http://minisatellites.u-psud.fr. Changes in the number of repeats among isolates can be monitored by PCR, and MLVA (Multi locus variable number of tandem repeat analysis) surveys are widely used for subtyping purposes [12–19].
The sequence of the genome of the S. maltophilia strain K279a has been completed [20]. Genome inspection allowed us to set up a simple, fast and accurate PCR-based diagnostic protocol which relies on the measurement of length heterogeneity of specific intergenic regions of the S. maltophilia genome.
The present protocol allows typing several S. maltophilia isolates in hours. The assignment of a digit code to each isolate could be used to easily compare data among different laboratories.
Results
Specific sequence repeats punctuate the genome of S. maltophilia
The whole DNA sequence of the S. maltophilia strain K279a has been determined [20]. The genome is 4,851,126 bp in length, and has an average G+C content of 66.3%. We found that the K279a chromosome hosts an abundant family of small, palindromic repeats fitting the consensus GTAGTGCCGGCCGCTGGCCGGCA (complementary residues are underlined) that we called SMAG (for Stenotrophomonas MAltophilia GTAG) because they carry the tetranucleotide GTAG at one terminus, similarly to small repetitive extragenic palindromic sequences (REPs) identified in the genomes of Escherichia coli and other microrganisms [21]. SMAGs make up approximately 0.5% of the K279a genome, and are spread throughout the chromosome either as single units, or in pairs, separated by 5–80 bp long spacers. The size of the SMAG family allows to hypothesize that some of these repeats may function as regulatory signals either at the DNA or the RNA level, as shown for REPs [21].
SMAGs and the PCR-based genotyping of S. maltophiliaisolates
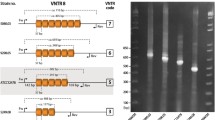
In the K279a chromosome, monomeric and dimeric SMAGs are reiterated in tandem at multiple chromosomal loci, along with tracts of variable length of flanking DNA. We exploited the occurrence of SMAG arrays to set up PCR-based typing protocols, and focused our attention on 12 such loci, labelled I to XII in accord to their location on the K279a chromosome (Table 2). No rule in the pattern of amplification of SMAG sequences at the different loci could be discerned (Fig. 1A). Thus, region I features 50 bp long repeats, resulting from the duplication of a monomeric 24 bp long SMAG along with 26 bp of flanking DNA. In contrast, region XII features repeats which are 103 bp in length, and results from the duplication of a dimeric 72 bp SMAG and 31 bp of flanking DNA. In pilot experiments, regions IX and X were amplified by PCR from the DNA of the control strain K279a and five different S. maltophilia isolates. As shown in Fig. 1B, the size of the SMAG-positive regions varies, and this correlates with changes in the number of repeating units as confirmed by sequence analysis. Isolates could thus be marked by a digit corresponding to the number of SMAG repetitions present at a given locus (Fig. 1B). Prompted by these results, we monitored the twelve SMAG-positive loci by PCR. Analyses were carried out on DNAs derived from 38 S. maltophilia strains, including the K279a strain, isolated from different sources (Table 1). On the basis of the amplimers size, it had been possible to deduce the number of repeats present at the loci in the various isolates, and therefore label each of them with a digit (Table 3). In some instances, we could not detect an amplification product for one or more loci in different isolates. PCR-negative regions were labelled 0. To confirm our findings, alternative primers were used for some of these regions, but no reliable amplification product could be detected. The lack of amplification may reflect either an extensive polymorphism or deletions occurred in the regions analyzed.

A) Organization of the repetitive SMAG-positive loci in the genome of the S. maltophilia K279a strain. The structure of loci I to XII is diagramatically shown. SMAG sequences are represented as boxes. Repeat units at each locus are highlighted and their size in bp is indicated. B) Allelic variations at SMAG+ loci. Amplimers spanning the loci IX and X derived from the DNA of the indicated S. maltophilia strains were electrophoresed on a 2% agarose gel. Numbers below bands mark the number of repeat units within each amplimer. The 100 bp ladder was used as molecular weight (MW) DNA marker.
A few PCR products, derived from the amplification of regions VII, XI and XII, were slightly different in length, and could not be assigned to a size class. In these instances, amplimers were assigned to the nearest size class, and marked with the letters a, b and c to denote size differences among them (Table 3). To clarify this issue, we determined the sequence of the PCR products derived by amplification of region VI in strains 915, 1029 and LMG959 (classified in Table 3 as 1a, 1b and 1c, respectively). In the control strain K279a, region VI contains four SMAG dimers. In the 915 strain, the amplified DNA (1a amplimer) was similar to the K279a interval, but only one SMAG dimer was present. In the LMG959 strain, the region amplified (1c amplimer) was 45 bp shorter, because the SMAG dimer was replaced by a SMAG monomer. In strain 1029, the size change of region VI (1b amplimer) was due to replacement of the SMAG dimer and 10 bp flanking sequence by a 66 bp palindromic element, that is a member of a distinct, less abundant family of GTAG+ repeats in S. maltophilia. Thus, size variations of loci analyzed may correlate with recombinational events which replace SMAGs with members of the same family, or related DNA families.
In order to assess the stability of the SMAG-positive regions, the strains 528, 916 and 1039 were sub-cultured for 5 days, and the DNA extracted from single colonies of each strain was analyzed by PCR (data not shown). No changes in the pattern of amplification at loci III, IV, IX and XII were observed.
As shown in Table 3, the information derived from the survey of loci II, V, I and VII was sufficient to obtain a 4-digit code, that assigned the 38 DNAs analyzed to 23 different PTs (PCR Types). Some strains, such as 528 and 571, belong to the same PT type, and exhibited the same PCR profile at all the other loci. The same holds true for the three PT type 14 strains 916, 1019 and 1053, and for 3 out of 7 of the PT-19 strains. In contrast, strains 714 an 262 belong to different PTs (17 and 19, respectively), but, aside from differences in region VII, were identical at all loci.
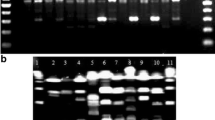
The finding that a comparatively relatively robust typing can be achieved by analysing only the four loci II, V, I and VII, highlighted in Table 3, is relevant, mostly in view of the fact that they can be co-amplified in pairs. In both instances, the amplimers corresponding to either locus can be easily distinguished because of their size range, allowing an immediate typing (Fig. 2).

Co-amplification of SMAG-positive loci. Amplimers deriving from the dual amplification of II and V, and I and VII loci from the DNA of the indicated strains were analyzed by elctrophoresis as in Fig. 1.
PFGE-typing of S. maltophiliaisolates
PFGE is the gold standard for strain genotyping also for S. maltophilia. PCR data shown in Table 3 partially complemented genotyping data obtained by PFGE. OBGTC9 and OBGTC10 strains exhibited the same PCR profile at all the SMAG-positive regions analyzed, and their PFGE profiles are undistinguishable (Fig. 3). Strains 916, 1019 and 1053, which belong to PFGE A-type, and strains 528 and 571, which belong to PFGE B-type (data not shown; for the PFGE relatedness of these strains, see Crispino et al., 2002) similarly exhibited the same PCR profile at all the loci (Table 3). Other strains exhibited similar PCR profiles, but different PFGE patterns. XbaI digests of OBGTC13, OBGTC23 and OBGTC30 DNAs produced undistinguishable PFGE patterns. Their PCR profiles were similar on the whole, but OBGTC23 featured a PCR type different from OBGTC13 and OBGTC30 (PT-10 vs PT-13), and the three isolates differed from each other at multiple additional loci. Moreover, K279a DNA was identical to OBGTC9 and OBGTC10 DNAs at all SMAG loci, but clearly differed from both DNAs when analyzed by PFGE (Fig. 3).

PFGE profiles of S. maltophilia strains. The DNA of the indicated isolates was restricted with XbaI. The digestion products were resolved on 1.2% agarose gels (see Methods). The percentage genetic similarity is shown above the dendrogram. Strain numbers and PT types are shown on the right of each PFGE profile. Numbers at the bottom refer to the size in kb of the MW DNA.
Discussion
The MLVA technique involves amplification and size analysis of polymorphic DNA regions containing variable numbers of tandemly repeated sequences, and is an established method to classify isolates of microbial species for which complete genome information is available [22]. The determination of the complete genome sequence of the S. maltophilia K279a strain allowed us to set up a MLVA assay also for this organism.
All the approaches so far utilized for the genotyping of S. maltophilia clinical isolates have led to the conclusion that S. maltophilia strains are highly heterogeneous [5–11]. According to AFLP fingerprinting [5] and gyrB RFLP [6] analysis, S. maltophilia can be assigned to 8–10 genomic groups. The majority of CF isolates grouped in two clusters [6], suggesting that isolates of specific groups have an increased potential for the colonization of the respiratory tract of CF patients. Considering the interest paid to S. maltophilia as an emerging opportunistic pathogen associated with nosocomial infections, we wanted to develop a fast, accurate and unexpensive method of genotyping which could be adopted for strain classification and comparisons. The chromosomal regions carrying clusters of SMAGs, an abundant sequence repeat spread in the S. maltophilia genome, vary in size among isolates, allowing MLVA-based typing surveys. The repeat units found at the various loci analyzed are sufficiently large to discriminate length variation among isolates by low molecular weight agarose gel electrophoresis. In most instances, PCR data enabled to set a direct correlation between the length of the amplimers and the number of SMAG repeats present at the analyzed loci. The latter could be directly marked by the number of repeats, setting the basis for a simple, numerical classification of the strains analysed. Broad sorting of a large number of isolates may suggest to restrict MLVA analyses to a few SMAG-positive loci. Allelic variants of the four loci II, V, I and VII (Table 3) are detectable by two PCR co-amplifications reaction, providing 4-digit typing profiles which could turn out to be effective for simple typing purposes. By increasing the number of loci analyzed, it could be possible to obtain a more discriminating digit profile, as in MLST analyses [23].
According to our typing scheme, regions not responding to the PCR approach have been assigned the digit 0. While uninformative on the integrity of the region under scrutiny, the lack of amplification of a certain locus is fully exploitable in a multi-typing system. A "caveat" may be represented by cases in which it is not possible to immediately correlate the size of the amplicon with the number of repeats as observed for amplimers slightly differing in length occasionally found at loci VII, XI and XII. However, this could represent a problem in the analyses of large populations of isolates, calling for a highly discriminating profiling. Minor size differences among amplicons allow discrimination for typing purposes as the major ones, and can be indicated by marking amplimers also with letters. However, accurate measuring of small size differences among amplimers which have been analyzed in different electrophoretic runs may be cumbersome. Thus, it would be advisable to assign to amplicons which may slightly differ in size the same digit, which should correspond to the most likely number of repeats present. This may ensure to rapidly proceed in classifying the different isolates, eventually further distinguishing them by means of additional analyses.
MLVA assays provide results that parallel PFGE data, although some differences have been noticed, since MLVA and PFGE measure different types of chromosomal modifications, and, for example, recombination events within a genome could be detected by PFGE, but overlooked by MLVA analyses [18, 24, 25]. The same holds true in our study, as strains such as K279a, OBGTC9 and OBGTC10, while identical according to MLVA data, differ when analyzed by PFGE. The method we have devised is simpler, less time-consuming and economically more advantageous than PFGE. As suggested by Tenover et al. [25], MLVA approaches could be particularly helpful to identify strains responsible for outbreaks in hospital settings, and to determine the relatedness of isolates collected over short periods of time. In contrast, PFGE could be priviliged for long time period analyses of bacterial populations.
Conclusion
The utilization of the present protocol will be useful for fast and efficient typing purposes. Several S. maltophilia isolates could be typed in hours, and the results interpreted de visu without the need for sophisticated software. Data would be easily reproducible, and immediately comparable among different laboratories.
Methods
S. maltophiliastrains
S. maltophilia strains analyzed in this study are listed in Table 1. Clinical isolates were identified as S. maltophilia by using the VITEK II system (bioMerieux, Morey-l'Etoile, France). The identification was confirmed by PCR amplification and sequence analysis of the 16S rDNA. Strains were routinely grown in brain heart infusion at 37°C, except for the environmental strains LMG959, LMG10871, LMG10879 and OBGN1 which were grown at 30°C. In order to analyze the stability of the genomic regions under scrutiny, a few strains were sub-cultured in brain heart infusion at 37°C 5 times for 18–24 hrs.
PCR amplification
The DNA of single colonies derived from the final subcultures was analyzed by PCR amplification of DNA regions of interest. Genomic DNA was extracted as described by De Gregorio et al. [26]. PCR reactions were carried out by incubating 20 ng of DNA with 160 ng of each primer in the presence of dXTPs (200 nanomoles), 1.5mM magnesium chloride and the Taq DNA polymerase Recombinant (Invitrogen). Because of the high GC content of the S. maltophilia genome (> 66%), all PCR reactions were carried out in GC-rich buffer (Roche). The oligomers used as primers, and the annealing temperatures, are listed in Table 2. Samples were incubated at 95°C for 5', and subsequently for 1' at 95°C, 1' at the annealing temperature and 1' at 72°C, for a total of 30 cycles. At the end of the cycle, samples were kept at 72°C for 7' before harvesting.
PCR products were electrophoresed on 1.5–2% agarose gels in 0.5×TBE buffer (45 mM Tris pH 8, 45 mM Borate, 0.5 mM EDTA) at 120 V (constant voltage). The 100 bp ladder (Fermentas) was used as molecular weight marker.
PFGE analysis
Preparation of agarose plugs containing chromosomal DNA for PFGE analysis was performed using the PulseNet standardized procedure http://www.cdc.gov/pulsenet The DNA plugs were digested with 60 U of XbaI (Roche Diagnostics) at 37°C fo 16 h. Genomic DNA fragments were separated by PFGE at 14°C on agarose 1.2% w/v gels in a clamped homogeneous field electrophoresis apparatus (CHEF-DRII system; Bio-Rad, Hemel Hempstead, UK), with pulse times ramped from 1 to 20 s over 21 h at 6.0 V/cm in 0.5× TBE. DNA fragments obtained from XbaI digestion of plugs containing chromosomal DNA of Salmonella braenderup strain H9B12 were used as molecular weight markers [27].
Electrophoretic patterns were analyzed by UPGMA (Unweighted Pair Group Method with Arithmetic mean) using the Gel Compar II version 4.5 software (Applied Maths).
References
Palleroni NJ, Bradbury JF: Stenotrophomonas, a new bacterial genus for Xanthomonas maltophilia (Hugh 1980) Swings et al. 1983. Int J Syst Bacteriol. 1993, 43: 606-609.
Denton M, Kerr KG: Microbiological and clinical aspects of infection associated with Stenotrophomonas maltophilia. Clin Microbiol Rev. 1998, 11: 57-80.
Waters VJ, Gómez MI, Soong G, Amin S, Ernst RK, Prince A: Immunostimulatory properties of the emerging pathogen Stenotrophomonas maltophilia. Infect Immun. 2007, 75: 1698-1703. 10.1128/IAI.01469-06.
Ryan RP, Fouhy Y, Garcia BF, Watt SA, Niehaus K, Yang L, Tolker-Nielsen T, Dow JM: Interspecies signalling via the Stenotrophomonas maltophilia diffusible signal factor influences biofilm formation and polymyxin tolerance in Pseudomonas aeruginosa. Mol Microbiol. 2008, 68: 75-86. 10.1111/j.1365-2958.2008.06132.x.
Hauben L, Vauterin L, Moore ERB, Hoste B, Swings J: Genomic diversity of the genus Stenotrophomonas. Int J Syst Bacteriol. 1999, 49: 1749-1760.
Coenye T, Vanlaere E, LiPuma JJ, Vandamme P: Identification of genomic groups in the genus Stenotrophomonas using gyrB RFLP analysis. FEMS Immunol Med Microbiol. 2004, 40: 181-185. 10.1016/S0928-8244(03)00307-9.
Gould VC, Okazaki A, Howe RA, Avison MB: Analysis of sequence variation among sme DEF multi drug efflux pump genes and flanking DNA from defined 16S rRNA subgroups of clinical Stenotrophomonas maltophilia isolates. J Antimicrob Chemother. 2004, 54: 348-353. 10.1093/jac/dkh367.
Gylmez D, Hascelik G: Stenotrophomonas maltophilia : antimicrobial resistance and molecular typing of an emerging pathogen in a Turkish university hospital. Clin Microbiol Infect. 2005, 11: 880-886. 10.1111/j.1469-0691.2005.01257.x.
Yao JDC, Conly JM, Krajden M: Molecular typing of Stenotrophomonas (Xanthomonas) maltophilia by DNA macrorestriction analysis and random amplified polymorphic DNA analysis. J Clin Microbiol. 1995, 33: 2195-2198.
Crispino M, Boccia MC, Bagattini M, Villari P, Triassi M, Zarrilli R: Molecular epidemiology of Stenotrophomonas maltophilia in a university hospital. J Hosp Infect. 2002, 52: 88-92. 10.1053/jhin.2002.1280.
Valdezate S, Vindel A, Martin-Davila P, Del Saz BS, Baquero F, Canton R: High genetic diversity among Stenotrophomonas maltophilia strains despite their originating at a single hospital. J Clin Microbiol. 2004, 42: 693-699. 10.1128/JCM.42.2.693-699.2003.
Onteniente L, Brisse S, Tassios PT, Vergnaud G: Evaluation of the polymorphisms associated with tandem repeats for Pseudomonas aeruginosa strain typing. J Clin Microbiol. 2003, 41: 4991-4997. 10.1128/JCM.41.11.4991-4997.2003.
Allix C, Supply P, Fauville-Dufaux M: Utility of fast mycobacterial interspersed repetitive unit-variable number tandem repeat genotyping in clinical mycobacteriological analysis. Clin Infect Dis. 2004, 39: 783-789. 10.1086/423383.
Lindstedt BA, Vardund T, Aas L, Kapperud G: Multiple-locus variable-number tandem-repeats analysis of Salmonella enterica subsp. Enterica serovar Typhimurium using PCR multiplexing and multicolor capillarelectrophoresis. J Microbiol Methods. 2004, 59: 163-172. 10.1016/j.mimet.2004.06.014.
Schouls LM, Heide van der HG, Vauterin L, Vauterin P, Mooi FR: Multiple-locus variable-number tandem repeat analysis of Dutch Bordetella pertussis strains reveals rapid genetic changes with clonal expansion during the late 1990s. J Bacteriol. 2004, 186: 5496-5505. 10.1128/JB.186.16.5496-5505.2004.
Koeck JL, Njanpop-Lafourcade BM, Cade S, Varon E, Sangare L, Valjevac S, Vergnaud G, Pourcel C: Evaluation and selection of tandem repeat loci for Streptococcus pneumoniae MLVA strain typing. BMC Microbiol. 2005, 5: 66.-10.1186/1471-2180-5-66.
Yazdankhah SP, Lindstedt BA, Caugant DA: Use of variable-number tandem repeats to examine genetic diversity of Neisseria meningitidis. J Clin Microbiol. 2005, 43: 1699-1705. 10.1128/JCM.43.4.1699-1705.2005.
Johansson A, Koskiniemi S, Gottfridsson P, Wistrom J, Monsen T: Multiple-locus variable-number tandem repeat analysis for typing of Staphylococcus epidermidis. J Clin Microbiol. 2006, 44: 260-265. 10.1128/JCM.44.1.260-265.2006.
Cho S, Boxrud DJ, Bartkus JM, Whittam TS, Saeed M: Multiple-locus variable-number tandem repeat analysis of Salmonella Enteritidis isolates from human and non-human sources using a single multiplex PCR. FEMS Microbiol Lett. 2007, 275: 16-23. 10.1111/j.1574-6968.2007.00875.x.
Crossman LC, Gould VC, Dow JM, Vernikos GS, Okazaki A, Sebaihia M, Saunders D, Arrowsmith C, Carver T, Peters N, Adlem E, Kerhornou A, Lord A, Murphy L, Seeger K, Squares R, Rutter S, Quail MA, Rajandream MA, Harris D, Churcher C, Bentley SD, Parkhill J, Thomson NR, Avison MB: The complete genome, comparative and functional analysis of Stenotrophomonas maltophilia reveals an organism heavily shielded by drug resistance determinants. Genome Biol. 2008, 9: R74-10.1186/gb-2008-9-4-r74.
Tobes R, Pareja E: Repetitive extragenic palindromic sequences in the Pseudomonas syringae pv. tomato DC3000 genome: extragenic signals for genome reannotation. Res Microbiol. 2005, 156: 424-433. 10.1016/j.resmic.2004.10.014.
van Belkum A: Tracing isolates of bacterial species by multi locus variable number of tandem repeat analysis (MLVA). FEMS Immunol Med Microbiol. 2007, 49: 22-27. 10.1111/j.1574-695X.2006.00173.x.
Maiden MC: Multi locus sequence typing of bacteria. Annu Rev Microbiol. 2006, 60: 561-588. 10.1146/annurev.micro.59.030804.121325.
Malachowa N, Sabat A, Gniadkowski M, Krzyszton-Russjan J, Empel J, Miedzobrodzki J, Kosowska-Shick K, Appelbaum PC, Hryniewicz W: Comparison of multiple-locus variable-number tandem-repeat analysis with pulsed-field gel electrophoresis, spa typing, and multi locus sequence typing for clonal characterization of Staphylococcus aureus isolates. J Clin Microbiol. 2005, 43: 3095-3100. 10.1128/JCM.43.7.3095-3100.2005.
Tenover FC, Vaughn RR, McDougal LK, Fosheim GE, McGowan JE: Multiple-locus variable-number tandem-repeat assay analysis of methicillin-resistant Staphylococcus aureus strains. J Clin Microbiol. 2007, 45: 2215-2219. 10.1128/JCM.02451-06.
De Gregorio E, Silvestro G, Venditti R, Carlomagno MS, Di Nocera PP: Structural organization and functional properties of miniature DNA insertion sequences in Yersiniae. J Bacteriol. 2006, 188: 7876-7884. 10.1128/JB.00942-06.
Hunter SB, Vauterin P, Lambert-Fair MA, van Duyne MS, Kubota K, Graves L, Wrigley D, Barrett T, Ribot E: Establishment of a universal size standard strain for use with the PulseNet standardized pulsed-field gel electrophoresis protocols: converting the national databases to the new size standard. J Clin Microbiol. 2005, 43: 1045-1050. 10.1128/JCM.43.3.1045-1050.2005.
Di Bonaventura G, Prosseda G, Del Chierico F, Cannavacciuolo S, Cipriani P, Petrucca A, Ammendolia MG, Concato C, Fiscarelli E, Casalino M, Piccolomini R, Nicoletti M, Colonna B: Molecular characterization of virulence determinants of Stenotrophomonas maltophilia strains isolated from patients affected by cystic fibrosis. Int J Immunopathol Pharmacol. 2007, 20: 529-537.
Acknowledgements
We are indebted to M.B. Avison for K279a strain and E. Fiscarelli for strains of the OBG collection. Research was supported in part by grant FFC 7/2007 of the Italian Cystic Fibrosis Research Foundation to BC and MC.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
ER and FR designed the PCR oligomers and carried out the DNA analyses, BC, MC and RF provided the S. maltophilia strains and performed the PFGE experiments, PPDN conceived the study and participated in its design and coordination, MSC and PPDN drafted the manuscript. All authors read and approved the final manuscript.
Emanuela Roscetto, Francesco Rocco contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Roscetto, E., Rocco, F., Carlomagno, M.S. et al. PCR-based rapid genotyping of Stenotrophomonas maltophiliaisolates. BMC Microbiol 8, 202 (2008). https://doi.org/10.1186/1471-2180-8-202
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-8-202