
Abstract
Background
Bacterial and cellular genotyping is becoming increasingly important in the diagnosis of infectious diseases. However, difficulties in obtaining sufficient amount of bacterial and cellular DNA extracted from the same human biopsy specimens is often a limiting factor. In this study, total DNA (host and bacterial DNA) was isolated from minute amounts of gastric biopsy specimens and amplified by means of whole genome amplification using the multiple displacement amplification (MDA) technique. Subsequently, MDA-DNA was used for concurrent Helicobacter pylori and human host cellular DNA genotyping analysis using PCR-based methods.
Results
Total DNA was isolated from gastric biopsy specimens of 12 subjects with gastritis and 16 control subjects having a normal mucosa. The DNA was amplified using a multiple displacement amplification (MDA) kit. Next, concurrent genotyping was performed using H. pylori-specific virulence gene PCR amplification assays, pyrosequencing of bacterial 16S rDNA and PCR characterisation of various host genes. This includes Interleukin 1-beta (IL1B) and Interferon-gamma receptor (IFNGR1) SNP analysis, and Interleukin-1 receptor antagonist (IL1RN) variable tandem repeats (VNTR) in intron 2. Finally, regions of the vacA-gene were PCR amplified using M13-sequence tagged primers which allowed for direct DNA sequencing, omitting cloning of PCR amplicons. H. pylori specific multiplex PCR assays revealed the presence of H. pylori cagA and vacA genotypic variations in 11 of 12 gastritis biopsy specimens. Using pyrosequencing, 16S rDNA variable V3 region signatures of H. pylori were found in 11 of 12 individuals with gastritis, but in none of the control subjects. Similarly, IL1B and IFNGR1-SNP and IL1RN-VNTR patterns could be established in all individuals. Furthermore, sequencing of M13-sequence tagged vacA-PCR amplicons revealed the presence of highly diverse H. pylori vacA-s/i/m regions.
Conclusion
The PCR-based molecular typing methods applied, using MDA-amplified DNA derived from small amounts of gastric biopsy specimens, enabled a rapid and concurrent molecular analysis of bacterial and host genes in the same biopsy specimen. The principles and technologies used in this study could also be applied to any situation in which human host and microbial genes of interest in microbial-host interactions would need to be sequenced.
Similar content being viewed by others

Background
The Gram-negative spiral shaped bacterium Helicobacter pylori [1] is associated with the development of a variety of gastroduodenal diseases such as chronic gastritis, peptic ulcer disease and gastric cancer [2, 3]. Virulence factors have been identified, including the production of urease (ureA) [4], a vacuolating cytotoxin (vacA) [5, 6], and a cytotoxin-associated antigen (cagA) [7, 8]. It has been implied that ureA [9], superoxide dismutase (sod) [10] and heat-shock protein 60 (hsp60) [11], found exclusively within the cytoplasm in other bacteria, are associated with the outer membrane in H. pylori. It is assumed that the variation in disease progression between patients is likely due to differences in bacterial virulence genes.
The H. pylori cytotoxin gene vacA is an important virulence marker. DNA sequence analysis has revealed that the vacA has a mosaic structure comprising allelic variations in the signal and midregion, each having two different alleles (s1/s2, m1/m2) with different biological activities [5, 12]. Furthermore, it has been shown that the repeated hydrophilic motif region (RHM) is a potential proteolytic cleavage-site that separates the VacA-94 kDa protein into a 58-kDa and a 37-kDa protein with different biological activities [6, 13]. Amino-acid substitutions, insertions and deletions within the RHM-region have been reported [14, 15]. It has been speculated that mutations such as truncations, insertions and deletions in the midregion of the vacA gene which lead to in-frame stop codons, are associated with non-toxic H. pylori strains [14]. Recently, it has been shown that the vacA intermediate region (vacA i-region) may provide an important and independent marker of VacA-associated pathogenicity [16].
It is well recognised that the development of gastric disease is strongly influenced by host genetic factors. Cytokines play an important role in H. pylori-induced disease. An association of increased risk of gastric cancer and IL1B polymorphisms [17–19] and IL1RN polymorphisms [17, 19–21] has been established. The IFNGR1 allele *2 has been associated with high H. pylori-reactive immunoglobulin G antibody concentrations [22] and an increased risk for atrophic gastritis [23]. Thus, for characterisation of bacterium-host interactions, it would appear that a concurrent typing of Helicobacter-specific virulence genes and cytokine polymorphisms in the same DNA, isolated only once from a biopsy specimen, is desirable.
Numerous PCR assays have been developed for the identification of H. pylori-specific virulence genes, reviewed in [15]. To reduce the number of H. pylori-specific virulence gene-based PCR amplification assays needed for such characterisation, we and others have developed multiplex PCR amplification assays which allow a highly sensitive detection and accurate genotyping of H. pylori-DNA [15, 24–26]. A recent study described a method along these lines which allowed for concurrent genotyping analysis of H. pylori 16S rDNA, 23S rDNA, cagA genes, and the cellular IL1B gene [27].
However, due to the low amount of bacterial DNA present in human biopsy specimens, the molecular identification of bacteria is often regarded impossible without culturing. To increase the specificity and sensitivity of the analysis of H. pylori infected biopsy specimens, nested PCR amplification in itself [28] or combined with Southern blot analysis of PCR amplicons has been used for molecular typing [29, 30]. Identification of PCR amplicons by DNA sequence analysis employing time-consuming and expensive cloning, and subsequent plasmid DNA isolation procedures has also been used. However, these methods are not well suited for large-scale epidemiological screening and in this context, alternative techniques such as direct sequencing of M13-sequence tagged PCR amplicons for molecular typing of i. e. Staphylococcus aureus protein A (SPA-typing) [31], and whole genome amplification (WGA) of DNA using multiple displacement amplification (MDA) have been developed [32, 33].
MDA is a PCR independent isothermal amplification technique that relies on the amplification of total genomic DNA, using a chemically modified random hexamer primer at 30°C and a bacteriophage Phi29 proofreading DNA polymerase, completing amplification within a few hours [32, 34]. This technique dramatically improves the high-fidelity production of large amounts of genomic DNA with uniform coverage of genes [35–37]. We have recently reported on the use of MDA for amplification of DNA isolated from human intestinal biopsies [38], human gallstones specimens [39], and archival plasma/serum samples [40] for subsequent use in molecular typing of H. pylori and host genes.
In this study, we report on the use of MDA-amplified DNA for the molecular analysis of bacterial and human host genes in minute amounts of gastric biopsy specimens. Moreover, we report on the successful PCR amplicon sequencing, using a single, universal M13 uni (-21) primer targeting all M13-sequence tagged PCR amplicons.
Methods
Study subjects, tissue collection and DNA isolation
Fresh frozen (-80°C) archival tissue samples from 28 subjects from a previous study which was approved by the local ethics committee and conducted in accordance with the Helsinki Declaration, including informed written consent, were used [41]. Based on histological findings and rapid urease test from a larger (n = 501) gastroscopic screening study in a randomly selected cohort of the population of Linköping, Sweden [41], a total of 28 individuals (14 men; median age 58, range 42–73 and 14 women; median age 65, range 43–76) were selected and included in the present study. Among the 12 individuals with gastritis, four had antrum gastritis, five had pangastritis and two subjects had corpus gastritis with H. pylori infection, respectively, and one individual had corpus gastritis without an obvious H. pylori infection. All sixteen control subjects were H. pylori-negative and had a normal gastric mucosa (for details see table 1). DNA from gastric biopsy specimens and bacterial strains Helicobacter pylori 26695 and J99, respectively, were extracted using a BioRobot M-48 Workstation and a MagAttract DNA Mini-48 kit as recommended by the manufacturer (Qiagen, Hilden, Germany).
Generation of MDA-amplified DNA
MDA of the isolated DNA was carried out using a GenomiPhi DNA amplification kit as recommended by the manufacturer (GE-Healthcare, Uppsala, Sweden). DNA concentrations were determined using a ND-1000 spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA). Three separate MDA reactions were performed from each sample. The integrity of the MDA-amplified DNA was analysed by Ethidium bromide stained agarose-gel electrophoresis. Appropriate amounts (usually 1 to 2 μl) were used in subsequent PCR amplification assays.
18S rRNA and 16S rRNA PCR amplification
To evaluate the integrity of the prepared MDA-amplified DNA derived from total biopsy DNA, subsequent 18S rDNA and 16S rDNA PCR amplification was performed. In brief, 18S rDNA PCR amplification was carried out using a Quantum 18S rDNA internal standard kit as recommended by the manufacturer (Ambion, Austin, TX, USA) yielding a 489 bp PCR amplicon. 16S rDNA PCR amplification yielding an approximately 450 bp amplicon was carried out using 16S rDNA broad-range primers [42] flanking the variable V3, V4 and V9 regions (Table 2) and PCR amplification condition No 3 (Table 3) in a final 25 μl reaction volume using thin walled tubes, a HotStarTaq Master-mix kit (Qiagen, Hilden, Germany), and a PTC-100 Thermocycler (MJ-Research; SDS-Biosciences, Falkenberg, Sweden) or Mastercycler gradient (Eppendorf, Hamburg, Germany) as previously described [38].
Pyrosequencing analysis
Pyrosequencing template generation was performed using 16S rDNA broad-range PCR amplicons as a template (see above). For that purpose, broad-range 16S rDNA PCR amplicons were purified using a GFX PCR DNA and Gel-band purification kit (GE Healthcare, Uppsala, Sweden) and pyrosequencing PCR amplicons were generated using an appropriate amount (usually 1 μl) of 16S rDNA template, a HotStarTaq Master mix kit (Qiagen AB, Solna, Sweden), 5 pmol each of Helicobacter-specific primer 5'-biotin HJ-HP-JBS.V3 and broad-range primer B-V3.AS (Table 2), both flanking the 16S rDNA V3 region only, and PCR condition No 1 (Table 3). Pyrosequencing analysis was carried out as described elsewhere [43] using a PSQ 96 MA System (Biotage AB, Uppsala, Sweden) and sequencing primer B-V3.AS (Table 2).
H. pylori-specific virulence-gene multiplex PCR amplification
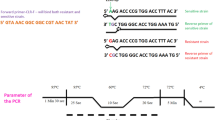
vacA, hsp60, ureI, sod, ureA and cagA (Table 2) multiplex PCR amplification was carried out using 5 pmol of each primer and PCR amplification condition No1 (Table 3) [15]. For vacA subtyping, a duplex PCR using primers VAI-F and VAI-R specific for vacA s1/s2 genotypes, primers VAG-F and VAG-R (Fig. 1; Table 2) specific for vacA m1/m2 genotypes [24, 26], and PCR condition No 1 were used (Table 3). PCR amplicons were analysed on the automated capillary electrophoresis QIAxcel system and a QIAxcel DNA high resolution kit (Qiagen, Hilden, Germany).

Cytokine polymorphism analysis
MDA-amplified DNA was analysed by pyrosequencing for the presence of IL1B- and IFNGR1-polymorphisms. Primers flanking the IL1B-511 [ref SNP ID:rs16944],IL1B-31 [rs1143627]and IFNGR1-56 [rs2234711] regions were designed based on the available genomic IL1B [GenBank:AY137079] and IFNGR1 sequences [GenBank:AY594694] using a PSQ Assay Design software (Biotage AB, Uppsala, Sweden). Templates for pyrosequencing SNP analysis were obtained by PCR amplification using 1 μl of MDA-amplified DNA, 5 pmol of each primer flanking the IL1B-511, IL1B-31, IL1B+3954 [rs1143634] and the IFNGR1-56 regions (Table 2), and PCR amplification condition No 2 for IL1B-511, IL1B-31 and IFNGR1 primers and No 3 for IL1B +3954 primers (Table 3). Pyrosequencing analysis was performed as described elsewhere [40, 43] with the modification of using a PSQ 96MA System, and the pyrosequencing primers specified in table 2.
The 86 bp variable number tandem repeat (VNTR) in intron 2 of the IL1RN gene was amplified using the primers IL1RNF1.SE and IL1RNR1.AS (Table 2) and PCR amplification No 5 (Table 3) followed by automatic capillary electrophoresis analysis using a QIAxcel system and a QIAxcel DNA high resolution kit (Qiagen, Hilden, Germany). So far, five VNTR alleles have been reported; allele *1–*5 having four, two, five, three, and six repeats, corresponding to amplicon sizes of 442 bp, 270 bp, 528 bp, 356 bp and 614 bp, respectively.
DNA sequence analysis of vacAPCR amplicons
Sequence data from Helicobacter pylori J99 [GenBank:AE001439], H. pylori 26695 [GenBank:AE000511], H. pylori NCTC 11638 [GenBank:U07145], and H. pylori unspecified [GenBank:U29401] for vacA primer design were retrieved from GenBank [44] and aligned using ClustalX [45]. Primers were designed and checked using Primer3 [46] and optimized by annealing temperature gradient PCR and Mg2+ concentration.
To identify and establish vacA s/i/m genotype, 2 μl MDA-amplified DNA derived from the 11 H. pylori-positive biopsies were PCR amplified with three new vacA-specific primer pairs, using 10 pmol of each primer and PCR condition No 4 (Table 3). The s-region was amplified with primer M13-SeqS.SE and SeqS.AS, the RHM and i-region with primers M13-seqVac.SE and seqVac.AS, and the m-region with primers M13-seqM.SE and VAG-R (Fig. 1; Table 2) yielding M13-sequence (M13 uni, -21) tagged PCR amplicons. PCR amplicons were treated with ExoSAP-IT to inactivate excess of oligonucleotide primers, following the manufacturer's instructions (USB Europe GmbH, Staufen, Germany), and lyophilised. Subsequent DNA sequence analysis was carried out using a custom DNA sequencing service (Eurofins MWG GmbH, Martinsried, Germany). H. pylori MDA-amplified DNA No 18 was further amplified using the primer combination M13-SeqVac.SE and VAG-R at the conditions described above. The obtained sequences corresponding to the 11 H. pylori-DNA vacA s-regions were aligned and compared with catalogued H. pylori vacA type s1a [GenBank:AY185128], s1b [GenBank:AB057223], s2 [GenBank:AY438687], and s1c [GenBank:AB057107] sequences using CLC DNA workbench 3 (CLC bio A/S, Aarhus, Denmark). Similarly, DNA sequences corresponding to the vacA i/m-regions were aligned and compared with catalogued H. pylori vacA type i1/m1 [GenBank:U05676] and type i2/m2 [GenBank:U29401] sequences.
Results
MDA-amplified DNA
Each biopsy yielded approximately 2–4 μg total DNA (A260/280 > 1.8) in a final volume of 150 μl water. On average, 1 μl genomic DNA generated approximately 12–22 μg MDA-amplified DNA. 18S rDNA and 16S rDNA PCR amplification using MDA-amplified DNA yielded PCR amplicons of the expected sizes, 490 bp and 450 bp, respectively (data not shown).
Presence of Helicobacterspp.-DNA in antrum biopsies
MDA-amplified DNA derived from 28 antrum biopsies was analysed for the presence of Helicobacter spp.-DNA. Pyrosequencing analysis revealed that 11 of 28 subjects were infected with H. pylori. H. pylori 16S rDNA sequences were detected only in biopsy specimens from patients with diagnosed gastritis (Table 1). 16S rDNA variable V3 region sequence motifs corresponding to H. pylori 26695 and J99 were detected in six and three samples, respectively. Two specimens had deviating V3 region sequence motifs (Fig. 2); H. pylori sequence No 6 corresponds to the V3 sequence motifs of H. pylori "strain A" [GenBank:DQ059082] and H. pylori sequence No 28 corresponds to a H. pylori 26695/J99-like combination of 26695 and J99. In the remaining 17 biopsies, Helicobacter spp.-DNA was not detected (Table 1).

H. pylori 16S V3 pyrosequencing results. DNA sequence alignment of the 16S rDNA variable V3 region of Helicobacter isolates derived from pyrosequencing analysis. Dashes indicate sequence identity with H. pylori 26695. Roman numerals indicate four Helicobacter-specific 16S rDNA variable V3 region sequence motif-groups [43]. The number of Helicobacter isolates in each group is indicated in parentheses. Group I, H. pylori strain 26695-like [GenBank:AE000620]: No 12, 14, 18, 23, 25, 27. Group II, H. pylori strain J99-like [GenBank:NC_000921]: No 9, 21, 22. Group III H. pylori strain A-like [GenBank:DQ059082]: No 6. Group IV, H. pylori 26695/J99-like: No 28 (see also table 1).
Multiplex PCR amplification
H. pylori-positive MDA-amplified DNA (based on 16S rDNA pyrosequencing analysis), and H. pylori 26695 and J99-DNA as references (Table 1) were further analysed by the H. pylori virulence gene-based multiplex PCR amplification assay. Capillary electrophoresis revealed hsp60, ureI, sodB, and ureA PCR amplicons of the expected sizes in all MDA-amplified DNA derived from the H. pylori positive biopsies and the control strains. However, multiplex PCR amplification generated only weak bands in No 12 and 22, indicating the presence of a low level of H. pylori DNA in these biopsies (Fig. 3a). The cagA+/vacA+ combination was present in six MDA-amplified DNA samples (No 9, 12, 22, 23, 25, 27) and was also found in the control strains. The cagA+/vacA- combination was present in three biopsies (No 6, 14, 18), and the vacA-/cagA- and vacA+/cagA- combinations were found in one biopsy each (No 21 and 28, respectively; Fig. 3a).

H. pylori multiplex PCR and vacA subtyping results. Results from the automatic capillary electrophoresis of multiplex PCR amplicons derived from 11 H. pylori-infected subjects and control strains H. pylori 26695 and J99. The positions of A)H. pylori-specific vacA, hspA, ureI, sodB, ureA, and cagA multiplex PCR amplicons and of B)vacA s1, vacA m1 and vacA m2 PCR amplicons are indicated. Neg represents a non-template control. A virtual internal reference marker is indicated in the left margin.
VacAsubtyping by duplex PCR amplification
Multiplex PCR amplification of the 11 H. pylori-positive MDA-amplified DNA samples yielded seven PCR amplicons with a vacA+ genotype (Fig. 3a). We also used another vacA-specific duplex PCR amplification assay which allows for the discrimination between signal region s1 and s2 alleles, and midregion m1 and m2 alleles, generating PCR amplicons of distinct sizes (Table 2). VacA s/m-profiles were generated from all H. pylori positive MDA-amplified DNA (Fig. 3b). The vacA type s1/m1 was observed in seven MDA-amplified DNA samples (No 9, 12, 22, 23, 25, 27, 28) and the two control strains, and the vacA type s1/m2 was observed in two biopsies (No 6 and 14). In one case each, only a single band corresponding to a vacA type s1 (No 18) or a vacA type m2 (No 21) was found (Fig. 3b; Table 4).
Cytokine SNP and IL1RN-VNTR analysis
MDA-amplified DNA derived from the 28 biopsies was used for pyrosequencing analysis of the human IL1B-511, IL1B-31, IL1B+3954 and IFNGR-56 SNPs (Table 5). In the 11 H. pylori-infected biopsy specimens, the SNP CC/CT/TT-genotype distribution was 4/5/2 for ILB1-511, 3/4/4 for IL1B-31, 8/3/0 for IL1B+3954, and 2/6/3 for IFNGR1-56. In the 17 H. pylori-negative biopsy specimens, the SNP CC/CT/TT-genotype distribution was 9/5/3 for IL1B-511, 3/5/9 for IL1B-31, 12/4/1 for IL1B +3954, and 2/6/9 for IFNGR1-56 (Table 5).
Capillary electrophoresis revealed an IL1RN-VNTR 1/1-genotype in six (No 9, 21, 22, 25, 27, 28), a 1/2-genotype in four (No 6, 12, 14, and 18), and a 2/2-genotype in one (No 23) of the H. pylori-infected gastritis biopsies. The biopsy specimen having gastritis without H. pylori infection (No 8) revealed an IL1RN-VNTR 2/2-genotype. An IL1RN-VNTR 1/1-genotype was found in eight (No 2, 3, 4, 5, and 7), a 1/2-genotype in four (No 11, 13, 20, 26), a 1/3-genotype in one (No 10), and a 2/2-genotype in three (No 1, 16, 17) of the histological normal biopsies (Fig. 4; Table 5).

IL1RN -VNTR genotyping results. Automatic capillary electrophoresis of IL1RN-VNTR PCR amplicons from all 28 biopsies. The number of each lane corresponds to the sample listed in table 1. The position of the virtual, internal marker and expected position of allele *1, *2 and *3 are indicated.
VacAsubtyping by DNA sequence analysis
DNA sequencing analysis revealed the presence of different vacA genotypes in all H. pylori strains, indicating that sequencing of M13-sequence tagged PCR amplicons is a more discriminating molecular typing approach than analysis of PCR amplicons by capillary gel electrophoresis as done in this study (Fig. 5, 6, 7, 8; Table 4). The apparent absence of vacA amplicons in the multiplex PCR analysis of sample no 6, 14, 18 and 21, is explained by a 70 bp deletion in the RHM region (Fig. 6). The PCR products for these samples are approximately 600 bp and coincide with the hsp60 amplicon of similar size (Fig. 3a; Table 2).

VacA s-region nucleotide sequence alignment. DNA sequence alignment of obtained vacA s-region. The vacA type s1a and s1b sequences are shaded in grey. Reference sequences are indicated with NCBI accession number [GenBank:AY185128] – s1a, [GenBank:AB057107] – s1c, [GenBank:AB057223] – s1b, and [GenBank:AY438687] – s2. Dots indicate nucleotide sequence identical to the one above and gaps indicate deletions. The gaps in the reference sequence are indicated by dashes.

VacA i- and RHM-region nucleotide sequence alignment. DNA sequence alignment of the obtained sequences containing intermediate A, B and C, and RHM regions, which are shaded in grey. Reference sequence [GenBank:U05675] (i1) and [GenBank:U29401] (i2) are obtained from Rhead et al. [16]. Dots indicate identical nucleotide sequences and gaps indicate nucleotide deletions. Primer binding sites are indicated with boxes.

VacA m-region protein sequence alignment. Alignment of deduced amino acid sequence, translated from the obtained vacA m-region DNA sequences. Samples with m2 genotype are shaded in grey. Reference sequence [GenBank:U05675] (m1) and [GenBank:U29401] (m2) are obtained from Rhead et al. [16]. Dots indicate identical nucleotide sequences and gaps indicate nucleotide deletions.

VacA i-region protein sequence alignment. Amino acid sequence alignment from the vacA variable i-regions. Shaded areas indicate the important B and C regions described by Rhead et al. [16].
DNA sequence alignment revealed ten vacA s1a genotypes (No 6, 9, 12, 14, 18, 23, 25, 27 and reference strain 26695), three s1b genotypes (No 22, 28, and strain J99) and one s2 genotype (No 21) when compared to H. pylori s1a [GenBank:AY185128], H. pylori s1b [GenBank:AB057223], and H. pylori s2 [GenBank:AY438687] as reference sequences (Fig. 5).
Nine H. pylori strains had a vacA type m1 (No 9, 12, 22, 23, 25, 27, 28, and reference strains 26695 and J99) and four had a type m2 (No 6 14, 18, 21) (Fig. 7; Table 4). Both i1 and i2 types, but also several chimeric types were identified. All the m1-type strains revealed an i1 genotype, No 21 (s2m2) had an i2, and the s1m2-strains (No 6, 14, and 18) had a chimeric i-region structure (Fig. 8).
DNA sequence analysis of a 900 bp PCR amplicon, produced using MDA-amplified DNA from biopsy No 18 and primers M13-seqVac.AS and VAG-R (Fig. 1), revealed a large deletion in the midregion of the vacA gene resulting in the absence of several primer target sites (Fig. 6). This explains the absence of a vacA m1/m2 genotype in the vacA-subtyping PCR amplification assay of biopsy No 18 (Fig. 3b).
Discussion
Rapidly and easily acquired information on bacterial and host genes is becoming increasingly important for diagnosis and decision-making when choosing suitable therapies for human infectious diseases. This makes new demands on clinical research and routine laboratories to develop molecularly based methods that can accurately identify and characterise bacterial virulence and host susceptibility and/or resistance genes. The amount of genomic DNA available for such genetic analyses is often limited. Quite often, only minute amounts of bacterial DNA can be found in human biopsies. Moreover, it is desirable to perform concurrent, multiple bacterial and host genotyping analyses from the same, limited amount of biopsy DNA. In such studies, the traditional approach is to purify DNA from cultured bacterial strains isolated from the biopsy specimens [27]. Sample preparation is commonly carried out using time-consuming cartridge or bead-based techniques. These methods do not allow an efficient high-throughput analysis of clinical samples and significant variations of DNA yield and purity can be observed. By contrast, an automated nucleic acid extractor combined with MDA-amplification yields DNA of high purity and integrity that can be used in downstream applications. Indeed, recent studies have demonstrated that PCR amplifications using MDA-amplified DNA can also be carried out under conditions where PCR amplifications normally are hampered due to the presence of PCR inhibitors [47].
In recent years, MDA has been tried out for the amplification of microbial DNA [36, 48, 49] and total DNA (bacterial and cellular) isolated from human biopsy specimens [38, 39]. This study shows the feasibility of using MDA-amplified total DNA, isolated from human biopsy specimens, for research and clinical diagnostic analysis of both host and infecting bacterium in the same DNA pool.
Pyrosequencing analysis of the16S rDNA variable V3 region revealed the presence of different H. pylori subspecies in the different biopsy specimens which is in agreement with previous reports showing that subtle DNA sequence variations occur in the 16S rDNA variable V3 region of H. pylori, providing a consistent system for subtyping [30, 43]. The taxonomy of these H. pylori strains may be a matter of debate. Subdivision of the species H. pylori into subspecies, based on biotypes, pathotypes or serotypes for taxonomic as well as clinical reasons has been suggested [50, 51].
Pro-inflammatory IL1B and IL1RN polymorphisms are associated with increased risk of gastric carcinoma in Caucasian populations [52]. Carriers of these pro-inflammatory polymorphisms revealed an increased IL1B gene expression pattern [19, 53] in the mucosa and increased prevalence of intestinal metaplasia and atrophic gastritis [19]. Similarly, a genome-wide linkage analysis identified SNPs in IFNGR1 affecting H. pylori infection [22]. Due to the limited number of biopsies analysed, we were not able to draw any statistically significant conclusion regarding allele frequencies in H. pylori-infected and histologically normal individuals. However, the primary goal of the present study was not to perform a clinical study at large but rather to establish new methodological approaches. In analogy with these findings we have recently shown that MDA-DNA derived from minute amounts of archival plasma/serum DNA allowed us to identify cytokine polymorphic SNP-sites by means of pyrosequencing analysis [40]. The use of IL1B-SNP analysis by means of PCR-restriction-fragment-length polymorphism where IL1B-511, IL1B-31 and IL1B+3954 PCR amplicons are digested with restriction enzyme AvaI, TaqI and AluI, respectively, is a widely accepted approach [19–21]. However, restriction enzyme digestion, followed by agarose gelelectrophoresis is a time consuming and, in a clinical routine laboratory context, a tedious process requiring up to 100 ng DNA in each assay [20]. Thus, we conclude that MDA-DNA derived from human biopsy specimens provides a reliable source for cytokine-SNP analysis and, therefore, the same approach may be applied for the characterisation of other host genetic factors in population studies at large
Incorporation of universal M13-sequencetags at the 5'-end of PCR primers facilitated straightforward sequencing of amplicons, which makes culturing of bacteria from human biopsy specimens and cloning of PCR amplicons prior to DNA sequencing unnecessary. This makes it possible for clinical routine laboratories using this technique to rapidly produce sequencing results. Sequencing of M13-tagged amplicons was first described for SPA-typing [31]. The technique has successfully been applied in our laboratory for direct sequencing of PCR amplicons in gene expression studies of vasopressin receptor mRNA splice-variants expressed in the human gastro-intestinal tract and surrounding tissues [54]. In the present study we have used M13 sequence-tags for successful partial sequencing of the H. pylori virulence vacA gene (Fig. 6). Multiple H. pylori strain infection was not seen in this study, although the number of samples included is small. Primer design is of utter importance since species-specific primers are essential to gain pure and reliable sequencing results directly from DNA isolated from a mixed flora.
According to a recent study, the intermediate region (i-region) of the H. pylori vacA gene is an important and independent marker of VacA-associated pathogenicity [16]. All i1-type but no i2-type strains induce vacuolation. Chimeric versions of the i-region (i1-i2) can induce low level of vacuolation in vitro. According to this criteria, our results show two tentatively non-toxic strains (s2/i2/m2- No 21 and s1/i2/m2- No 6) and one with reduced toxicity (s1/i1-i2/m2 No 14; No 18 is discussed below). The remaining biopsies contain toxic H. pylori s1/i1/m1 strains (Table 4).
False negative PCR results from the multiplex-PCR (Table 4) were revealed when using new primers, targeting conserved regions of the vacA gene. The negative results were caused by deletions in the RHM region, yielding a smaller amplicon than expected by previous analysis (Fig. 6). One sample (No 18) had large deletions in the RHM region of the gene, leaving out vacA-primer binding sites commonly used in vacA PCR amplification assays (Fig. 1 and 6). The sequence contained several premature stop codons downstream of the deletion (data not shown), indicating a possible inactive form of VacA. However, further analysis is needed in order to establish if this strain can produce an active protein.
The conclusions to be drawn from the present data is that the choice of primers in vacA PCR amplification assays influences the apparent prevalence vacA-positive H. pylori strains and, hence, precaution has to be taken in the interpretation of vacA-negative PCR amplification results. Thus, our results are in good agreement with previous reports that revealed a high level of vacA genotype variation such as single nucleotide mutations, deletion and in frame stop codons in the vacA alleles of non-toxic H. pylori strains [14, 15], but it seems questionable to what extent published vacA PCR amplification results can be compared between studies if DNA sequencing is not performed. From our and other studies it seems evident that it is necessary to establish partial DNA sequences from either vacA PCR amplicons (Fig. 3, 5, 6) or from full-length vacA open reading frames. Thus, high-throughput sequencing of M13 sequence-tagged PCR amplicons appears to be a more promising approach, both in research and clinical routine laboratories. This principle could be applied to any situation in which sequencing of PCR amplicons is desirable and thereby, one could abstain from tedious amplicon cloning and plasmid preparation procedures prior to DNA sequence analysis.
Histology is considered a sensitive method for detection of H. pylori in gastric biopsies. However, a recent study has demonstrated that PCR amplification using H. pylori-specific primers detected H. pylori-DNA in histological-negative gastric biopsies, indicating the clinical relevance of H. pylori detection by PCR amplification in biopsies with characteristic inflammatory changes [55]. Similarly, Kisa and co-workers [56] evaluated different diagnostic methods for the detection of H. pylori in gastric biopsy specimens. In their study the authors concluded that nested PCR amplification assays are necessary to detect H. pylori-DNA in gastric biopsy specimens. From this study we conclude that MDA-amplified DNA derived from minute amounts of biopsy specimen DNA is well suited for PCR-amplification and subsequent sequencing of M13 sequence-tagged amplicons. Thus, it is possible to avoid nested PCR amplification assays which often have to be combined with Southern blot analysis to increase the sensitivity and specificity [28, 30].
Conclusion
The highly sensitive PCR-based molecular typing methods described here enables reliable and concurrent genotyping analyses of bacterial and host cellular DNA from the same biopsy specimen at a reasonable cost and time. Although the power of this approach was demonstrated with concurrent H. pylori and cytokine SNP analysis, the principles and technologies could be applied to any situation in which microbial populations are analysed, and in particular for addressing questions concerning microbial-host interactions associated with human health in general.
References
Marshall BJ, Warren JR: Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984, 1: 1311-1315. 10.1016/S0140-6736(84)91816-6.
Cover TL, Blaser MJ: Helicobacter pylori and gastroduodenal disease. Annu Rev Med. 1992, 43: 135-145. 10.1146/annurev.me.43.020192.001031.
Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, Sibley RK: Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med. 1991, 325: 1127-1131.
Labigne A, Cussac V, Courcoux P: Shuttle cloning and nucleotide sequences of Helicobacter pylori genes responsible for urease activity. J Bacteriol. 1991, 173: 1920-1931.
Cover TL, Tummuru MKR, Cao P, Thompson SA, Blaser MJ: Divergence of genetic sequences for the vacuolating cytotoxin among Helicobacter pylori strains. J Biol Chem. 1994, 269 (14): 10556-10573.
Telford JL, Ghiara P, Dell'Orco M, Comanducci M, Burroni D, Bugnoli M, Tecce MF, Censini S, Covacci A, Xiang Z, Papini E, Montecucco C, Parente L, Rappuoli R: Gene structure of the Helicobacter pylori cytotoxin and evidence of its key role in gastric disease. J Exp Med. 1994, 179: 1653-1658. 10.1084/jem.179.5.1653.
Covacci A, Censini S, Bugnoli M, Petracca R, Burroni D, Macchia G, Massone A, Papini E, Xiang Z, Figura N, Rappuoli R: Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc Natl Acad Sci USA. 1993, 90: 5791-5795. 10.1073/pnas.90.12.5791.
Tummuru MK, Cover TL, Blaser MJ: Cloning and expression of a high-molecular-mass major antigen of Helicobacter pylori: evidence of linkage to cytotoxin production. Infect Immun. 1993, 61 (5): 1799-1809.
Hawtin PR, Stacey AR, Newell DG: Investigation of the structure and localization of the urease of Helicobacter pylori using monoclonal antibodies. J Gen Microbiol. 1990, 136: 1995-2000.
Spiegelhalder C, Gerstenecker B, Kersten A, Schlitz E, Kist M: Purification of Helicobacter pylori superoxide dismutase and cloning and sequencing of the gene. Infect Immun. 1993, 61 (12): 5315-5325.
Macchia G, Massone A, Burroni D, Covacci A, Censini S, Rappuoli R: The Hsp60 protein of Helicobacter pylori : structure and immune response in patients with gastroduodenal diseases. Mol Microbiol. 1993, 9: 645-652. 10.1111/j.1365-2958.1993.tb01724.x.
Atherton JC, Cao P, Peek RM, Tummuru MK, Blaser MJ, Cover TL: Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem. 1995, 270: 17771-17777. 10.1074/jbc.270.30.17771.
De Bernard M, Burroni D, Papini E, Rappuoli R, Telford J, Montecucco C: Identification of the Helicobacter pylori VacA toxin domain active in the cell cytosol. Infect Immun. 1998, 66: 6014-6016.
Ito Y, Azuma T, Ito S: Full-length sequence analysis of the vacA gene from cytotoxic and noncytotoxic Helicobacter pylori. J Infect Dis. 1998, 178: 1391-1398. 10.1086/314435.
Monstein H-J, Ellnebo-Svedlund K: Molecular typing of Helicobacter pylori by virulence-gene based multiplex PCR and RT-PCR analysis. Helicobacter. 2002, 7: 287-296. 10.1046/j.1523-5378.2002.00099.x.
Rhead JL, Letley DP, Mohammadi M, Hussein N, Mohagheghi MA, Eshagh Hosseini M, Atherton JC: A new Helicobacter pylori vacuolating cytotoxin determinant, the intermediate region, is associated with gastric cancer. Gastroenterology. 2007, 133: 926-936. 10.1053/j.gastro.2007.06.056.
El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, Lanyon G, Martin M, Fraumeni JF, Rabkin CS: Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000, 404: 398-402. 10.1038/35006081.
Furuta T, El-Omar EM, Xiao F, Shirai N, Takashima M, Sugimura H: Interleukin 1β polymorphisms increase risk of hypochlorhydria and atrophic gastritis and reduce risk of duodenal ulcer recurrence in Japan. Gastroenterol. 2002, 123: 92-105. 10.1053/gast.2002.34156.
Rad R, Dossumbekova A, Neu B, Lang R, Bauer S, Saur D, Gerhard M, Prinz C: Cytokine gene polymorphisms influence mucosal cytokine expression, gastric inflammation, and host specific colonization during Helicobacter pylori infection. GUT. 2004, 53: 1082-1089. 10.1136/gut.2003.029736.
Alpízar-Alpízar W, Perez-Perez GI, Une C, Cuenca P, Sierra R: Association of interleukin-1B and interleukin-1RN polymorphisms with gastric cancer in a high-risk population of Costa Rica. Clin Exp Med. 2005, 5: 169-176. 10.1007/s10238-005-0082-3.
Perez-Perez GI, Garza-Gonzalez E, Portal C, Olivares AZ: Role of cytokine polymorphisms in the risk of distal gastric cancer development. Cancer Epidemiol Biomarkers Prev. 2005, 14: 1869-1873. 10.1158/1055-9965.EPI-04-0889.
Thye T, Burchard GD, Nilius M, Müller-Myhsok B, Horstmann RD: Genome wide linkage analysis identifies polymorphism in the human interferon-γ receptor affecting Helicobacter pylori infection. Am J Hum Genet. 2003, 72: 448-453. 10.1086/367714.
Canzian F, Franceschi S, Plummer M, van Doorn LJ, Lu Y, Gioia-Patricola L, Vivas J, Lopez G, Severson RK, Schwartz AG, Munoz N, Kato I: Genetic polymorphism in mediators of inflammation and gastric precancerous lesions. Eur J Cancer Prev. 2008, 17: 178-183.
Chisholm SA, Teare EL, Patel B, Owen RJ: Determination of Helicobacter pylori vacA allelic types by single-step multiplex PCR. Lett Appl Microbiol. 2002, 35: 42-46. 10.1046/j.1472-765X.2002.01129.x.
Koehler CI, Mues MB, Dienes HP, Kriegsmann J, Schirmacher P, Odenthal M: Helicobacter pylori genotyping in gastric adenocarcinoma and MALT lymphoma by multiplex PCR analyses of paraffin wax embedded tissues. Mol Pathol. 2003, 56: 36-42. 10.1136/mp.56.1.36.
Chattopadhyay S, Patra R, Ramamurthy T, Chowdhury A, Santra A, Dhali GK, Bhattacharya SK, Berg DE, Nair GB, Mukhopadhyay AK: Multiplex PCR assay for rapid detection and genotyping of Helicobacter pylori directly from biopsy specimens. J Clin Microbiol. 2004, 42: 2821-2824. 10.1128/JCM.42.6.2821-2824.2004.
Hjalmarsson S, Alderborn A, Fock C, Muldin I, Kling H, Uhlén M, Engstrand L: Rapid combined characterization of microorganism and cellular genotypes using a single technology. Helicobacter. 2004, 9: 138-145. 10.1111/j.1083-4389.2004.00210.x.
Stärk KD, Nicolet J, Frey J: Detection of Mycoplasma hyopneumoniae by air sampling with a nested PCR assay. Appl Environ Microbiol. 1998, 64: 543-548.
Monstein H-J, Jonasson J: Differential virulence-gene mRNA expression in coccoid forms of Helicobacter pylori. Biochem Biophys Res Comm. 2001, 285: 530-536. 10.1006/bbrc.2001.5179.
Monstein H-J, Jonsson Y, Zdolsek J, Svanvik J: Identification of Helicobacter pylori DNA in human cholesterol gallstones. Scan J Gastroenterol. 2002, 37: 112-119. 10.1080/003655202753387455.
Frénay HM, Bunschoten AE, Schouls LM, van Leeuwen WJ, Vandenbroucke-Grauls CM, Verhoef J, Mooi FR: Molecular typing of Methicillin-resistant staphylococcus aureus on the basis of Protein A gene polymorphism. Eur J Microbiol Infect Dis. 1996, 15: 60-66. 10.1007/BF01586186.
Dean FB, Hosono S, Fang L, Wu X, Faruqi AF, Bray-Ward P, Sun Z, Zong Q, Du Y, Du J, Driscoll M, Song W, Kingsmore SF, Egholm M, Lasken RS: Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci USA. 2002, 99: 5261-5266. 10.1073/pnas.082089499.
Lasken RS, Egholm M: Whole genome amplification: abundant supplies of DNA from precious samples or clinical specimens. Trends Biotechnol. 2003, 21: 531-535. 10.1016/j.tibtech.2003.09.010.
Hosono S, Faruqi AF, Dean FB, Du y, Sun Z, Wu X, Du J, Kingsmore SF, Egholm M, Lasken RS: Unbiased whole-genome amplification directly from clinical samples. Genome Res. 2003, 13: 954-964. 10.1101/gr.816903.
Luthra R, Mederios LJ: Isothermal multiple displacement amplification. A highly reliable approach for generating unlimited high molecular weight genomic DNA from clinical specimens. J Mol Diagn. 2004, 6: 236-242.
Groathouse NA, Brown SE, Knudson DL, Brennan PJ, Slayden RS: Isothermal amplification and molecular typing of the obligate intracellular pathogen mycobacterium leprae isolated from tissues of unknown origin. J Clin Microbiol. 2006, 44: 1502-1508. 10.1128/JCM.44.4.1502-1508.2006.
Yokouchi H, Fukuoka Y, Mukoyama D, Calugay R, Takeyama H, Matsunaga T: Whole-metagenome amplification of a microbial community associated with scleractinian coral by multiple displacement amplification using Phi29 polymerase. Environm Microbiol. 2006, 8: 1155-1163. 10.1111/j.1462-2920.2006.01005.x.
Monstein H-J, Olsson C, Nilsson I, Grahn N, Benoni C, Ahrné S: Multiple displacement amplification of DNA from human colon and rectum biopsies: bacterial profiling and identification of Helicobacter pylori-DNA by means of 16S rDNA-based TTGE and pyrosequencing analysis. J Microbiol Methods. 2005, 63: 239-247. 10.1016/j.mimet.2005.03.012.
Nilsson I, Shabo I, Svanvik J, Monstein H-J: Multiple displacement amplification of isolated DNA from human gallstones: molecular identification of Helicobacter DNA by means of 16S rDNA-based pyrosequencing analysis. Helicobacter. 2005, 10: 592-600. 10.1111/j.1523-5378.2005.00361.x.
Sun Y-Q, Monstein H-J, Ryberg A, Borch K: Multiple strand displacement amplification of DNA isolated from human archival plasma/serum: identification of cytokine polymorphism by pyrosequencing analysis. Clin Chim Acta. 2007, 377: 108-113. 10.1016/j.cca.2006.09.003.
Borch K, Jönsson KA, Petersson F, Redéen S, Mårdh S, Franzén LE: Prevalence of gastroduodenitis and Helicobacter pylori infection in a general population sample: relations to symptomatology and life-style. Dig Dis Sci. 2000, 45: 1322-1329. 10.1023/A:1005547802121.
Grahn N, Olofsson M, Ellnebo-Svedlund K, Monstein H-J, Jonasson J: Identification of mixed bacterial DNA contamination in broad-range PCR amplification of 16S rDNA V1 and V3 variable regions by pyrosequencing of cloned amplicons. FEMS Microbiol Lett. 2003, 219: 87-91. 10.1016/S0378-1097(02)01190-4.
Monstein H-J, Nikpour-Badr S, Jonasson J: Rapid molecular identification and subtyping of Helicobacter pylori by pyrosequencing of the 16S rDNA variable V1 and V3 region. FEMS Microbiol Lett. 2001, 199: 103-107. 10.1111/j.1574-6968.2001.tb10658.x.
The GenBank database at NCBI. [http://www.ncbi.nlm.nih.gov/Genbank]
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG: The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research. 1997, 24: 4876-4882. 10.1093/nar/25.24.4876.
Rozen S, Skaletsky HJ: Primer3 on the WWW for general users and for biologist programmers. Bioinformatics Methods and Protocols: Methods in Molecular Biology. Edited by: Krawetz S, Misener S. 2000, Totowa, NJ: Humana Press, 365-386.
Gonzales JM, Portillio MC, Saiz-Jimenez C: Multiple strand displacement amplification as a pre-polymerase chain reaction (pre-PCR) to process difficult to amplify samples and low copy number sequences from natural environments. Environ Microbiol. 2005, 7: 1024-1028. 10.1111/j.1462-2920.2005.00779.x.
Abulencia CB, Wyborski DL, Garcia JA, Podar M, Chen W, Chang SH, Chang HW, Watson D, Brodie EL, Hazen TC, Keller M: Environmental whole-genome amplification to access microbial populations in contaminated sediments. Appl Environ Microbiol. 2006, 72: 3291-3301. 10.1128/AEM.72.5.3291-3301.2006.
Evans MF, Adamson CS, Cooper K: Evidence of HPV16 integration in low- and high-grade cervical lesions that regress demonstrated by multiple displacement amplification and Southern blot hybridisation. J Clin Pathol. 2008, 61: 541-543. 10.1136/jcp.2007.051797.
Blaser MJ: Heterogeneity of Helicobacter pylori. Eur J Gastroenterol Hepatol. 1997, 9: S3-7.
Blaser MJ: Not all Helicobacter pylori strains are created equal: should all be eliminated?. Lancet. 1997, 349: 1020-1022. 10.1016/S0140-6736(96)09133-7.
El-Omar EM: The importance of interleukin 1β in Helicobacter pylori associated disease. Gut. 2001, 48: 743-747. 10.1136/gut.48.6.743.
Hwang IR, Kodama T, Kikuchi S, Sakai K, Peterson LE, Graham DY, Yamaoka Y: Effect of interleukin-1 polymorphisms on gastric mucosal interleukin-1beta production in Helicobacter pylori infection. Gastroenterol. 123: 1793-1803. 10.1053/gast.2002.37043.
Monstein H-J, Truedsson M, Ryberg A, Ohlsson B: Vasopressin receptor mRNA expression in the human gastrointestinal tract. Euro Surg Res. 2008, 40: 34-40. 10.1159/000108655.
Zsikla V, Hailemariam S, Baumann M, Mund MT, Schaub N, Meier R, Cathomas G: Increased rate of Helicobacter pylori infection detected by PCR in biopsies with chronic gastritis. Am J Surg Pathol. 2006, 30: 242-248. 10.1097/01.pas.0000186397.18206.1b.
Kisa O, Albay A, Mas MR, Celasun B, Dognaci L: The evaluation of diagnostic methods for the detection of Helicobacter pylori in gastric biopsy specimens. Diagn Microbiol Infect Dis. 2002, 43: 251-255. 10.1016/S0732-8893(02)00409-1.
Dixon MF, Genta RM, Yardley JH, Correa P: Classification and grading of gastritis. The updated Sydney system. International Workshop on the Histopathology of Gastritis, Houston 1994. Am J Surg Pathol. 1996, 20: 1161-1181. 10.1097/00000478-199610000-00001.
Database of Single Nucleotide Polymorphisms. [http://www.ncbi.nlm.nih.gov/projects/SNP]
Acknowledgements
This study was supported by grants from the Research Council in the South East of Sweden (FORSS) and the Molecular Biology Program, Laboratory Medicine Centre-LMC, University Hospital Linköping, Sweden. The help of Dr Isabelle Nilsson and Niclas Grahn with 16S rDNA pyrosequencing analysis, and the critical reading and commenting on the manuscript by Dr Jon Jonasson is greatly appreciated. Part of this study has been presented at the 14th International Workshop on Campylobacter, Helicobacter and related Organisms in Rotterdam, The Netherlands, 2–5 September 2007; Poster P339.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
AR, KB Y-QS and HJM participated in the conception, design, drafting of the manuscript, and final approval of the version to be published. AR and HJM were responsible for the acquisition, analysis and interpretation of data. KB selected the biopsy specimens in the study. All authors have read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ryberg, A., Borch, K., Sun, YQ. et al. Concurrent genotyping of Helicobacter pylorivirulence genes and human cytokine SNP sites using whole genome amplified DNA derived from minute amounts of gastric biopsy specimen DNA. BMC Microbiol 8, 175 (2008). https://doi.org/10.1186/1471-2180-8-175
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-8-175