
Abstract
Background
Mycobacterium avium subsp. paratuberculosis (MAP), the causative agent of Johne's disease (JD) persistently infects and survives within the host macrophages. While it is established that substantial genotypic variation exists among MAP, evidence for the correlates that associate specific MAP genotypes with clinical or sub-clinical disease phenotypes is presently unknown. Thus we studied strain differences in intracellular MAP survival and host responses in a bovine monocyte derived macrophage (MDM) system.
Results
Intracellular survival studies showed that a bovine MAP isolate (B1018) and a human MAP isolate (Hu6) persisted in relatively higher numbers when compared with a sheep MAP isolate (S7565) at 24-hr, 48-hr and 96-hr post infection (PI). MDMs stimulated with B1018 up-regulated IL-10 at the transcript level and down-regulated TNFα at the protein and transcript levels compared with stimulations by the S7565 and Hu6. MDMs infected with Hu6 showed a down regulatory pattern of IL-10 and TNFα compared to stimulations by S7565. Cells stimulated with B1018 and Hu6 had low levels of matrix metalloprotease-3 (MMP3) and high levels of tissue inhibitor of metalloprotease-1 (TIMP1) at 96-hr PI relative to MDMs stimulated by S7565.
Conclusion
Taken together, results suggest that the bovine (B1018) and the human (Hu6) MAP isolates lead to anti-inflammatory and anti-invasive pathways in the macrophage environment whereas the sheep (S7565) MAP isolate induces a pro-inflammatory pathway. Thus the infecting strain genotype may play a role in polarizing the host immune responses and dictate the clinicopathological outcomes in this economically important disease.
Similar content being viewed by others

Background
Johne's disease (JD) is caused by the intracellular pathogen, Mycobacterium avium subsp. paratuberculosis (MAP). Several molecular techniques have been applied to differentiate and characterize MAP isolates from diverse hosts and geographic locations [1–3]. A recent study applied highly discriminatory molecular markers termed short sequence repeats (SSR) to analyze the diversity among MAP isolates from a variety of hosts [4]. The results provided evidence for interspecies transmission of several MAP genotypes with some showing host specificity. Intriguingly, all genotyping studies addressing diversity using a variety of methods show that MAP isolated from human Crohn's disease cases are a subset of MAP genotypes widespread in distribution among animal populations. These findings raise several questions regarding the association of specific genotypes with human disease and/or chronic sub-clinical versus overt clinical disease in animals. Since no information on disease phenotypes was obtained when the isolates were acquired for genotyping in our laboratory, logical interpretation of genotype-phenotype associations was not possible. In the absence of clinical data associated with genotypes, and a suitable animal model to rapidly identify strain-associated differences, studying MAP interactions in a cellular (macrophage) interphase provides an indirect tool to help dissect the early molecular events that occur during host-pathogen interactions.
Despite the fact that hosts have only a limited number of pathways in which they respond to pathogens, macrophages show both pathogen-specific gene expression profiles and a shared gene expression pattern when infected with diverse bacterial pathogens [5]. An in vitro study of human macrophage responses to a repertoire of genotypically and epidemiologically well defined clinical isolates of Mycobacterium tuberculosis (MTB) showed a strain dependent host response [6]. A more recent study has shown a shared and a unique gene expression signature by human macrophages stimulated with four isolates of M. avium that varied in growth characteristics [7]. Significant differences in cytokine-chemokine profiles or global gene expression profiles in either well-established cell lines (THP-1 or U937) or peripheral blood mononuclear cells (PBMCs) in response to diverse pathogenic and non pathogenic mycobacteria, have also been documented by several recent studies [8–11]. Taken together, available data in the current literature strongly suggests that macrophages infected with mycobacteria have differential gene expression profiles and that the infecting genotypes may dictate down stream host responses. Surprisingly, there have been no reports so far about comparative analyses of diverse clinical isolates of MAP within a host/host macrophage, which is a well established fact in other mycobacteria. We believe that this crucial piece of evidence is important in order to understand complex mechanisms underlying the virulence of this economically important pathogen.
Towards these long term goals, in this study we asked if genotypically distinct strains of MAP derived from different host species elicit differential responses in bovine monocyte derived macrophages. To test our hypothesis that there would be no strain-specific variation in host responses, we studied the growth kinetics of genotypically distinct strains of MAP in both BOMAC cells [12] and bovine monocyte derived macrophages (MDM). We compared the modulation of cytokines such as IL-10, TNFα and matrix metalloproteinase (MMP) genes such as MMP3 and MMP9 as a function of infecting genotype. The importance of these cytokines in JD has been reported elsewhere [13, 14]. Cytokines IL-10 and TNFα were evaluated because the relative balance in expression of these cytokines indicates macrophage activation. PBMCs isolated from cattle infected with JD have been shown to up regulate MMP9 and TIMP after stimulation with MAP [15]. MMPs when secreted in lower quantities help in leukocyte migration but when secreted in larger quantities cause tissue destruction [16]. A balance between MMP and TIMP is important in the extent of tissue degradation [16]. In summary, in this study we report a differential response of bovine monocyte derived macrophages to a variety of MAP isolates.
Results
Intracellular survival kinetics of MAP strains
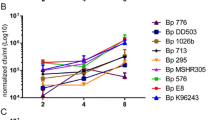
B1018 (bovine MAP isolate) was efficiently phagocytosed by bovine MDMs and persisted at fairly high numbers when compared to other isolates at all time points (Figures 1, 2, 3, 4). S7565 (sheep MAP isolate) decreased in bacterial numbers until 24-hr PI, started to multiply by 48-hr PI and dropped in total intracellular bacterial numbers by 96-hr PI. Hu6 (human MAP isolate) declined in numbers until 16-hr post infection and began replicating until 48-hr after infection and started to drop until 96-hr PI. M. avium showed a persistent trend until 24 hrs after infection of MDMs and replicated thereafter, until 48 hrs post infection.

IL-10 mRNA expressed by MDM cells exposed to M. paratuberculosis strains over time was measured by Real time RT PCR and fold change in gene expression relative to β actin was calculated by 2-ΔΔCT method. Mean fold change in gene expression is plotted on Y-axis (note that the Y1-axis scales have been optimized for magnitude of IL-10 expression for each strain). Intracellular bacterial numbers based on the amplification of hsp65 were calculated based on genome size of MAP and represented as genome equivalents (GE) on second Y-axis. MDMs stimulated with B1018 and Hu6 MAP isolates increased IL-10 mRNA by 96 hrs PI relative to cell stimulations by S7565 MAP isolate and M. avium.

IL-10 protein secreted by MDM cells exposed to MAP over time was measured by ELISA. Total amount of protein (pg/ml) is plotted on Y-axis (note that the Y1-axis scales have been optimized for magnitude of IL-10 expression for each strain). Intracellular bacterial numbers were calculated based on genome size of MAP and represented as genome equivalents (GE) on second Y-axis. MDMs stimulated with B1018 and Hu6 MAP isolates gradually up regulated IL-10 secretion from 2-hr until 96-hr PI.

TNFα mRNA expressed by MDM cells exposed to MAP over time was measured by Real time RT PCR and fold change in gene expression relative to β actin was calculated by 2-ΔΔCT method. Mean fold change in gene expression is plotted on Y-axis (note that the Y1-axis scales have been optimized for magnitude of IL-10 expression for each strain). Intracellular bacterial numbers based on the amplification of hsp65 were calculated based on the genome size of MAP and represented as genome equivalents (GE) on second Y-axis. MDMs stimulated with B1018 expressed lower levels of TNFα mRNA relative to other cell stimulations.

TNFα protein secreted by MDM cells exposed to MAP over time was measured by ELISA. Total amount of protein (pg/ml) is plotted on Y-axis (note that the Y1-axis scales have been optimized for magnitude of TNFα expression for each strain). Intracellular bacterial numbers were calculated based on the genome size of MAP and represented as genome equivalents (GE) on second Y-axis. MDMs stimulated with B1018 secreted low amounts of TNFα protein relative to other cell stimulations.
In BOMAC cells S7565 multiplied more rapidly and stayed in higher numbers at 96 hr PI relative to B1018. However, in bovine MDM cells B1018 was efficiently internalized and stayed in higher numbers relative to S7565 and Hu6 MAP isolates (data not shown).
IL-10 expression profile
As intracellular bacteria began replication (2–96 hr PI), MDMs stimulated with bovine (B1018) and human (Hu6) MAP isolates up-regulated IL-10 mRNA (fig 1) and protein (fig 2) levels over the entire infection period and this peaked from 48-hr to 96-hr PI. There was a positive correlation between IL-10 mRNA gene expression and IL-10 protein secretion (data not shown) observed in the cells stimulated with B1018 and Hu6. Cells stimulated with S7565 and M. avium down regulated IL-10 mRNA by 96-hr PI (fig 1). Cells stimulated with M. avium showed a gradual increase in IL-10 protein secretion until 48-hr PI and started to drop until 96-hr PI (fig 2). Interestingly, cells stimulated with S7565 showed a sustained increase in IL-10 protein secretion until 96-hr PI (fig 2).
TNFα expression profile
MDMs stimulated with B1018 and Hu6 isolates showed a down regulatory trend in TNFα mRNA expression from 2-hr to 48-hr PI that switched course to an up-regulatory trend from 48-hr until 96-hr PI (fig 3). Cells stimulated with B1018 gradually up-regulated TNFα protein secretion from 2-hr until 48-hr PI that dropped thereafter (fig 4). Cells stimulated with Hu6 increased TNFα protein secretion until 24-hr PI that showed a down-regulatory trend until 96-hr PI (fig 4). Cells stimulated with S7565 up-regulated TNFα mRNA and protein levels by 96-hr PI (fig 3 &4). At 96-hr PI, the magnitude of TNFα mRNA and protein level was significantly lower (P < 0.05) in cells stimulated with B1018 relative to cell stimulations by S7565, Hu6 and M. avium (fig 3). Although, cells stimulated with M. avium down regulated TNFα protein secretion from 16-hr until 96-hr PI, the amount of protein detected was significantly higher (P < 0.05) relative to other stimulations (fig 4).
MMP3 mRNA expression
MDMs stimulated with B1018 increased the production of MMP3 mRNA at 16-hr which was followed by a rapid decline until 96 hrs PI (table 1). Cells stimulated with S7565 produced a peak level of MMP3 at 24-hr that declined by 96-hr. While MMP3 mRNA production by cells stimulated with B1018 was high at 16-hr relative to cells stimulated by S7565 and Hu6, the data was not statistically significant (P = 1.0). At 24-hr there was a significant increase (P < 0.05) in the production of MMP3 mRNA by cells stimulated with S7565 relative to other stimulations. There was a gradual up-regulation of MMP3 mRNA production by MDMs stimulated with Hu6 until 48 hrs and declined by 96-hr.
MMP9 mRNA expression
Cells stimulated with B1018 showed lower levels of MMP9 mRNA production at 48-hr and 96-hr PI relative to cell stimulations with other MAP isolates (table 2). Cells stimulated with S7565 had a peak MMP9 mRNA production at 48-hr after infection, which was significantly higher (P < 0.05) when compared with stimulations by other MAP isolates (table 2). Cells stimulated with Hu6 also showed an up-regulatory trend in MMP9 mRNA production until 96-hr (table 2). Cells stimulated with Hu6 had significantly (P < 0.05) low MMP9 mRNA levels relative to cells stimulated with S7565 at 24-hr and 48-hr PI (table 2).
TIMP1 mRNA expression
LSMean values of TIMP1 mRNA levels suggested that MDMs stimulated with B1018 showed higher levels relative to cell stimulations by S7565 (data not statistically significant) (p = 1.0). There was a peak production of TIMP1 mRNA observed at 96-hr in MDMs stimulated with B1018 (table 3).
Discussion
Macrophages are the first line of host defense against any invading bacteria. Despite the fact that macrophages offer a hostile environment to several pathogenic bacteria, MAP is able to persistently survive and replicate within the phagosome environment of host macrophages. Studying the biochemical processes operating at the host-pathogen interface will help elucidate the mechanisms by which MAP has developed expertise to survive and replicate inside macrophages. Over the years many researchers have employed techniques such as microarray, semi-quantitative PCR, Q-RT-PCR to study the gene expression profiles in a cellular model after infection with pathogenic mycobacteria including type strains of MAP [9, 17, 18]. However, a possible MAP genotype-disease phenotype association has not been established despite the evidence that there is diversity in the genotypes of MAP strains isolated from several different hosts [19, 20].
In the absence of a well characterized experimental animal model to study host pathogen interactions of MAP in JD, cellular models have served as a helpful surrogate to researchers [21–24]. While BOMAC cells provide an easy to use immortalized cell line to study host-pathogen interactions, our studies with this cell line support earlier observations by Sager et al. [25] further highlighting that this bovine monocytoid cell line may differ in their behavior compared with MDMs. Thus, we chose to characterize MAP strain dependent host response only in bovine MDM cells.
Bovine monocyte derived macrophages infected with MAP have been previously employed to study changes in cytokine profiles [21, 26]. In the present study we have demonstrated a MAP genotype dependent phenotype characterized by differences in the cytokine profiles such as IL-10, TNFα and MMPs in MDMs. The importance of IL-10 and TNFα in JD has been reported elsewhere [13, 14]. IL-10 inhibits macrophage activation and is one of the major anti inflammatory cytokines [27]. TNFα is a major inflammatory cytokine produced by activated macrophages and is involved in controlling bacterial replication [28–30].
Our results based on the data generated using MDM cells obtained from two different animals consistently showed that B1018 (bovine MAP isolate) efficiently entered and remained in higher numbers within MDM cells relative to other MAP isolates. Cells stimulated with B1018 up-regulated expression of IL-10 mRNA (P < 0.005) while expression of TNFα mRNA was down-regulated relative to other MAP isolates. This was also evident in the relatively low proteins identified in the culture supernatant. Previous studies also reported a similar phenotype for the ATCC 19698 strain of MAP in bovine macrophages [21]. Khaleifh et. al [31] reported an up regulatory pattern of IL-10 secretion in bovine macrophages following infection with a type strain of MAP which is consistent with our findings, although the magnitude of up regulation at the protein level was much lower compared with their IL-10 levels. When compared to B1018, Hu6 and S7565 strains significantly down regulated IL-10 mRNA and up regulated TNFα mRNA. MDMs stimulated with S7565 and Hu6 MAP strains had significantly high amounts (P < 0.05) of TNFα and IL-10 secreted into culture supernatants relative to B1018 strain. A similar proinflammatory response by dendritic cells to whole cell MAP (strain 316F) infection has been recently demonstrated [32]. The study also documented that stimulation of dendritic cells by a recombinant immunodominant antigen of MAP included severe anti-inflammatory responses invoking the hypothesis that the differential in macrophage responses seen in our study may have occurred due to differences in expression of specific virulence genes by the strains studied. Our studies with SSR analysis [4, 19, 33, 34] and SNP analysis (Zhu and Sreevatsan, unpublished) suggest that specific genotypes may be associated with subclinical disease while others may lead to clinically overt disease. Additionally, in vitro analysis of MAP survival within primary macrophage cells in the present study show clear distinction in entry, survival and persistence as a function of genotypes. DNA microarray analysis of the genome content of MAP isolates employed in this study using MAA104 array revealed that several large sequence polymorphisms (LSPs) were missing in S7565 when compared to B1018 (Semret M, presented at 8th International Colloquium on Paratuberculosis). While this may explain the variations between sheep (S7565) and bovine (B1018) isolates, the variations in host response to bovine (B1018) or human (Hu6) genotypes of MAP may exist in SNPs and/or variations in bacterial gene regulation within the host. Confirmation of this finding will require analysis of a larger genotypically diverse collection for both host and pathogen gene expression. Comparisons of infections with a type strain of MAP and M. avium avium in bovine MDMs have revealed an increased expression of TNFα in cells infected with M. avium avium and a down regulation of IL-10 [21]. The M. avium intracellulare strain employed in our study showed similar trends in TNFα production when compared to other MAP strains studied. However, an increased IL-10 level was detected in culture supernatants infected with M. avium intracellulare compared to other strains at 96-hr PI. The differences observed could be because the M. avium intracellulare strain utilized in this study was unique in that this strain carried IS900, an insertion element that was once considered unique to MAP.
Matrix metalloproteinases (MMPs) are a family of calcium-dependent proteinases [35] involved in cell migration, tissue remodeling and destruction. Tissue inhibitors of MMP (TIMP) inhibit the activity of MMP. A balance between MMP and TIMP produced at the site of inflammation influences the ability of immune cells to migrate and the amount of tissue destruction caused [36]. PBMCs isolated from cattle infected with JD have been documented to up-regulate MMP9 and TIMP after stimulation with MAP [15, 37]. MMP and TIMP are reported to play a functional role in infections caused by pathogenic mycobacteria [38]. In our study, cells stimulated with B1018 down-regulated MMP9 while up-regulating TIMP1 production relative to MDMs stimulated with Hu6 and S7565 strains. This is consistent with the idea that these MAP genotypes (finger print: 7G4GGT) lead to an anti-inflammatory and anti-invasive milieu allowing their persistence and survival within macrophages. THP-1 cells infected with MTB have shown significant production of MMP9 and TIMP1 but not MMP1 [16]. MMP9 when secreted at low levels aid leukocyte migration to sites of inflammation. However, in large amounts, MMP9 causes tissue destruction [16]. It has also been shown that TNFα production at the site of inflammation correlates with MMP9 production [39]. Our observations are consistent with the idea that S7565 strain may elicit a relatively more invasive pathway during infection. Taken together, our studies demonstrate a MAP genotype-dependent response in a bovine monocyte derived macrophage model.
Conclusion
The present findings provide key insights into the MAP genotype-disease phenotype associations. Further analysis of this complex "ancient dialogue" between MAP and macrophages derived from its natural host (bovines) will help elucidate the pathogenesis associated with different genotypes isolated from diverse host species. These studies will aid in understanding the proximal events involved in the progression of JD and the virulence of MAP isolates thus enabling design of early intervention strategies. Future studies with a broader range of MAP isolates with common and unique genotypes associated with JD are warranted.
Methods
Preparation of monocyte derived macrophages (MDM)
Two colostrum-deprived Holstein bull calves obtained from a Johne's disease free herd served as a source for peripheral blood and MDMs. The calves were tested 4 and 8 weeks after birth by fecal culture and serum ELISA and were confirmed to be JD free. The protocol used for the preparation of MDMs is described elsewhere [40]. Briefly, peripheral blood was drawn from jugular vein into acid-citrate dextrose containing vacutainers (BD Vacutainer, Rutherford, NJ). Blood was centrifuged at 2000 rpm for 20 minutes to obtain a clean buffy coat. Buffy coats were re-suspended in sterile 1× PBS (1:10 dilution) and overlaid on Histopaque (Sigma Aldrich, St. Louis, Mo) following manufacturer's recommendations. The tubes were centrifuged at 400 × g for 30 minutes at room temperature to separate mononuclear cells from other polymorphonuclear leukocytes. Mononuclear cells from the interphase were harvested carefully using a sterile Pasteur pipette and transferred to a second sterile 50-ml conical tube. The cells were washed with 10-ml of 1× PBS at 250 × g for 10 minutes. Supernatant was discarded and the cell pellet was re-suspended in a small volume of 1× PBS. The mononuclear cells were transferred to TEFLON jars (Savillex Corporation, Minnetonka, MN) containing RPMI 1640 medium supplemented with L-glutamine, HEPES and 20% autologous serum. The jars were incubated at 37°C, 95% air and 5% CO2 for 4 days. After four days monocytes differentiated and became larger in size. Differentiated monocytes were counted using a hemacytometer and seeded onto the tissue culture plates at appropriate dilutions and incubated at 37°C, 95% air and 5% CO2 for 2 hours. Plates were washed twice with sterile 1× PBS to remove the non-adherent cells. The adherent cells were used for all in-vitro infections.
M. paratuberculosis isolates
The selected MAP strains included B1018 (SSR fingerprint: 7G4GGT9nG), S7565 (SSR fingerprint: 15G3GGT), Hu6 (SSR fingerprint: 7G5GGT11nG) and Ma6043 (IS900 positive isolate identified as M. avium intracellulare by multiple target analyses; no SSR data) (11). B1018 was isolated from a cow with clinical JD and carried a fingerprint which is common to about 12% of bovine M. paratuberculosis strains in a national collection (Harris and Sreevatsan, Unpublished) and in greater than 45% of isolates from Ohio (21), S7565 was isolated from sheep and Hu6 was isolated from a Crohn's disease patient.
Bacterial cultures
All the MAP cultures were incubated at 37°C on MB7H9 plates supplemented with OADC enrichment medium and Mycobactin J. After 3–4 weeks of growth on MB7H9 plates, cultures were confirmed to be free of other contaminating organisms as determined by nil growth on BHI or blood agar plates incubated overnight at 37°C. Few colonies from MB7H9 plate cultures were inoculated into MB7H9 broth culture supplemented with OADC enrichment medium and Mycobactin J and incubated at 37°C for 3 days to achieve actively growing MAP. Three day-old cultures were used to obtain an optical density at 600 nm (OD600) to determine the colony forming units (cfu) of bacteria using the formula: 0.3 at OD600 = 109cfu/ml, and applied in all in vitro infections of bovine MDM. Bacteria were used at a 5:1 multiplicity of infection.
Experimental design
MDM monolayers were grown on six well tissue culture plates. Cells were stimulated with three MAP isolates (B1018: bovine, S7565: sheep, and Hu6: human) and one IS900 positive non-MAP isolate (6043: identified as M. avium intracellulare). Lipopolysachharide (LPS; 100 μg/ml) (Sigma Aldrich, St. Louis, Mo) stimulated and LPS (100 μg/ml) in conjunction with recombinant bovine IFNγ (14 ng/ml) (Serotec, Raleigh, NC) stimulated cells served as positive controls where as, nil stimulated MDM served as negative controls in the experiment. All the stimulations were carried out in triplicates and were performed simultaneously. Cell stimulations were repeated twice on MDMs from each animal to evaluate consistency in the data generated. The culture plates were incubated at 37°C, 95% air and 5% CO2 until used. At each time point (2-hr, 16-hr, 24-hr, 48-hr and 96-hr) cells and culture supernatants from all the treatments were harvested. The culture supernatant was collected and stored at -70°C until used for ELISA. Monolayers were immediately washed twice with sterile, pyrogen-free 1× PBS and used in RNA extractions.
RNA extraction and real time Q-RT-PCR
RNA extractions were carried out using TRIzol reagent (Invitrogen, Carlsbad, CA) following manufacturers' recommendations. All the RNA samples were treated with RNAse free DNAse I (Ambion, Austin, TX) according to manufacturer's recommendations and stored at -70°C until utilized in QRT-PCR. Prior to their use, RNA was assessed for the quality and quantity using a spectrophotometer (GeneQuant pro by Amersham Bio sciences Corp, Piscataway, NJ). Subsequently, all the samples were diluted using nuclease free water at a concentration of 10 ng/μl. Later, Real time Q-RT-PCR was performed using Light Cycler system (Roche Diagnostics, Indianapolis, IN). Single step RT PCR was performed using Quantitect SYBR Green RT PCR kit (Qiagen Inc., Valencia, CA). Briefly, each reaction mixture contained 10 μl of master mix, 0.2 μl of RT mix, 2 μl of template RNA and gene specific (IL-10, TNFα, MMP3, MMP9, or TIMP1) primers. Reactions were performed in 20 μl light cycler capillaries (Roche, Indianapolis, IN). Primers used to analyze all the transcripts have been reported else where. The Q-RT-PCR data was analyzed by using 2-ΔΔCT method as previously described [41, 42].
Survival analysis of MAP strains
MDMs or BOMAC cells [12] were infected with three MAP isolates and an M. avium at an MOI of 1:5 as described above. All the stimulations were carried in triplicates and were performed simultaneously. Infected cells were incubated at 37C, 5%CO2 until desired. At each time point (0, 2, 16, 24, 48 and 96 hrs post infection) culture plates were removed and monolayers were vigorously washed for three times with 1× PBS to get rid of loosely adherent cells and external bacteria. DNA from the monolayers was extracted using TRIzol (Invitrogen, Carlsbad, CA) following manufacturer's recommendations.
Briefly, Monolayers were lysed on culture plates using 1 ml of Trizol reagent. Samples were then mixed with zirconium beads (0.1 mm) and homogenized for 3 minutes using mini bead beater (Biospec products, Bartlesville, OK). This effectively released mycobacterial DNA. Later, samples were mixed with 200 μl of chloroform and centrifuged at 1500 rpm for 5-min at 4C. Pellet was washed twice with 0.1 M sodium citrate in 10% ethanol. Finally pellet was washed with 75% ethanol, air dried and dissolved in sterile distilled water and stored at -20C until used. DNA was used in the real time PCR to quantitate hsp65 gene of MAP. Since hsp65 is a single copy gene, it provided a good estimate of the total number of organisms that survived macrophage infection over time. All the amplifications were carried out in Light cycler (Roche, Indianapolis, IN) using quantitect SYBR Green (Qiagen, Valencia, CA).
Briefly, each reaction mixture contained 10 μl of master mix, 2 μl of template DNA and gene specific primers (forward – 5' GCC GCT GCT GAT CAT CGC CGA 3') (reverse – 5' CCT TGG TGA CGA CCT T 3'). Reactions were performed in 20 μl light cycler capillaries (Roche, Indianapolis, IN). Obtained ct (crossing time points) values from the real time PCR were converted to genome equivalents. Based on the genome size of MAP one genome equivalent was calculated to be equal to about 9.9 fmg of MAP DNA.
Development of hsp65 standard for quantification purposes
DNA extracted from the broth cultures of MAP was pooled and the concentrations were determined. Five, ten-fold dilutions of the DNA was made and used as template in the real time PCR for amplification of hsp65 gene. Obtained ct values were imported onto an Excel spread sheet. Ct vales were plotted against genome equivalents (Y axis) and regression analysis was performed. This regression equation was used to estimate the bacterial numbers (as genome equivalents) in all treatment samples.
Quantitation of extracellular cytokine production by ELISA
Cytokine sandwich ELISA was used to detect IL-10 and TNFα from the culture supernatants. The protocol was adopted and modified to our conditions as described [43, 44]. Briefly, 96 well micro titer plates (NUNC, Rochester, NY) were coated with mouse anti bovine IL-10 (Serotec Inc, Raleigh, NC) or mouse anti bovine TNFα (generous gift from Dr. Paape, USDA, Ames, IA) for overnight at 4°C. Next day plates were washed with PBS Tween20 (0.05%) and blocked with PBS/BSA (0.5%) for one hour at room temperatures. Culture supernatants were added to the plates. The standard protein was serially diluted and added to the corner wells. Standard protein for TNFα was purchased from Endogen where as standard protein for IL-10 was a generous gift from Dr. Howard, Animal Research Center, UK. The plates were incubated at 4°C overnight. The plates were then washed briefly with PBS Tween20 (0.05%) and mouse anti bovine IL-10 labeled with biotin (Serotec Inc, Raleigh, NC) or rabbit anti 1 bovine TNFα (generous gift from Dr. Paape, USDA, Ames, IA) was added and incubated at 37°C for 3 hours. Plates were then washed and streptavidin HRP (Serotec Inc, Raleigh, NC) was added to plates that were used for IL-10 detection, incubated for an hour at room temperature. Anti rabbit IgG1 labeled with HRP (Biorad, CA) was added to plates for detecting TNFα and incubated at room temperature for 2 hours. All the plates were then added with color developing solution (Biorad, CA) and plates were read using ELISA plate reader at 413 nm.
Estimation of protein concentration of IL-10 standard protein
The standard protein used for quantitating IL-10 in culture supernatants was gifted by Dr. Howard, UK. The source of recombinant bovine IL-10 was culture supernatant obtained from Cos-7 cells transfected with a plasmid encoding bovine IL-10. The IL-10 supplied had 3000 biological units per ml. One biological unit of IL-10 supplied to us corresponded to an equivalent of 600 ng of protein.
References
Muskens J, Bakker D, de Boer J, van Keulen L: Paratuberculosis in sheep: its possible role in the epidemiology of paratuberculosis in cattle. Vet Microbiol. 2001, 78 (2): 101-109. 10.1016/S0378-1135(00)00281-9.
Pavlik I, Bejckova L, Pavlas M, Rozsypalova Z, Koskova S: Characterization by restriction endonuclease analysis and DNA hybridization using IS900 of bovine, ovine, caprine and human dependent strains of Mycobacterium paratuberculosis isolated in various localities. Vet Microbiol. 1995, 45 (4): 311-318. 10.1016/0378-1135(94)00130-O.
Whittington RJ, Marsh IB, Whitlock RH: Typing of IS 1311 polymorphisms confirms that bison (Bison bison) with paratuberculosis in Montana are infected with a strain of Mycobacterium avium subsp. paratuberculosis distinct from that occurring in cattle and other domesticated livestock. Mol Cell Probes. 2001, 15 (3): 139-145. 10.1006/mcpr.2001.0346.
Ghadiali AH, Strother M, Naser SA, Manning EJ, Sreevatsan S: Mycobacterium avium subsp. paratuberculosis strains isolated from Crohn's disease patients and animal species exhibit similar polymorphic locus patterns. J Clin Microbiol. 2004, 42 (11): 5345-5348. 10.1128/JCM.42.11.5345-5348.2004.
Nau GJ, Richmond JF, Schlesinger A, Jennings EG, Lander ES, Young RA: Human macrophage activation programs induced by bacterial pathogens. Proc Natl Acad Sci U S A. 2002, 99 (3): 1503-1508. 10.1073/pnas.022649799.
Hoal-van Helden EG, Stanton LA, Warren R, Richardson M, van Helden PD: Diversity of in vitro cytokine responses by human macrophages to infection by mycobacterium tuberculosis strains. Cell Biol Int. 2001, 25 (1): 83-90. 10.1006/cbir.2000.0680.
Blumenthal A, Lauber J, Hoffmann R, Ernst M, Keller C, Buer J, Ehlers S, Reiling N: Common and unique gene expression signatures of human macrophages in response to four strains of Mycobacterium avium that differ in their growth and persistence characteristics. Infect Immun. 2005, 73 (6): 3330-3341. 10.1128/IAI.73.6.3330-3341.2005.
Manca C, Reed MB, Freeman S, Mathema B, Kreiswirth B, Barry CE, Kaplan G: Differential monocyte activation underlies strain-specific Mycobacterium tuberculosis pathogenesis. Infect Immun. 2004, 72 (9): 5511-5514. 10.1128/IAI.72.9.5511-5514.2004.
McGarvey JA, Wagner D, Bermudez LE: Differential gene expression in mononuclear phagocytes infected with pathogenic and non-pathogenic mycobacteria. Clin Exp Immunol. 2004, 136 (3): 490-500. 10.1111/j.1365-2249.2004.02490.x.
Theus SA, Cave MD, Eisenach KD: Activated THP-1 cells: an attractive model for the assessment of intracellular growth rates of Mycobacterium tuberculosis isolates. Infect Immun. 2004, 72 (2): 1169-1173. 10.1128/IAI.72.2.1169-1173.2004.
Theus SA, Cave MD, Eisenach KD: Intracellular macrophage growth rates and cytokine profiles of Mycobacterium tuberculosis strains with different transmission dynamics. J Infect Dis. 2005, 191 (3): 453-460. 10.1086/425936.
Stabel JR, Stabel TJ: Immortalization and characterization of bovine peritoneal macrophages transfected with SV40 plasmid DNA. Vet Immunol Immunopathol. 1995, 45 (3-4): 211-220. 10.1016/0165-2427(94)05348-V.
Alzuherri HM, Woodall CJ, Clarke CJ: Increased intestinal TNF-alpha, IL-1 beta and IL-6 expression in ovine paratuberculosis. Vet Immunol Immunopathol. 1996, 49 (4): 331-345. 10.1016/0165-2427(95)05477-4.
Adams JL, Czuprynski CJ: Ex vivo induction of TNF-alpha and IL-6 mRNA in bovine whole blood by Mycobacterium paratuberculosis and mycobacterial cell wall components. Microb Pathog. 1995, 19 (1): 19-29. 10.1006/mpat.1995.0041.
Coussens PM, Jeffers A, Colvin C: Rapid and transient activation of gene expression in peripheral blood mononuclear cells from Johne's disease positive cows exposed to Mycobacterium paratuberculosis in vitro. Microb Pathog. 2004, 36 (2): 93-108. 10.1016/j.micpath.2003.09.007.
Friedland JS, Shaw TC, Price NM, Dayer JM: Differential regulation of MMP-1/9 and TIMP-1 secretion in human monocytic cells in response to Mycobacterium tuberculosis. Matrix Biol. 2002, 21 (1): 103-110. 10.1016/S0945-053X(01)00175-5.
Weiss DJ, Evanson OA, Deng M, Abrahamsen MS: Gene expression and antimicrobial activity of bovine macrophages in response to Mycobacterium avium subsp. paratuberculosis. Vet Pathol. 2004, 41 (4): 326-337. 10.1354/vp.41-4-326.
Coussens PM, Colvin CJ, Rosa GJ, Perez Laspiur J, Elftman MD: Evidence for a novel gene expression program in peripheral blood mononuclear cells from Mycobacterium avium subsp. paratuberculosis-infected cattle. Infect Immun. 2003, 71 (11): 6487-6498. 10.1128/IAI.71.11.6487-6498.2003.
Motiwala AS, Amonsin A, Strother M, Manning EJ, Kapur V, Sreevatsan S: Molecular epidemiology of Mycobacterium avium subsp. paratuberculosis isolates recovered from wild animal species. J Clin Microbiol. 2004, 42 (4): 1703-1712. 10.1128/JCM.42.4.1703-1712.2004.
Whittington RJ, Taragel CA, Ottaway S, Marsh I, Seaman J, Fridriksdottir V: Molecular epidemiological confirmation and circumstances of occurrence of sheep (S) strains of Mycobacterium avium subsp. paratuberculosis in cases of paratuberculosis in cattle in Australia and sheep and cattle in Iceland. Vet Microbiol. 2001, 79 (4): 311-322. 10.1016/S0378-1135(00)00364-3.
Weiss DJ, Evanson OA, Moritz A, Deng MQ, Abrahamsen MS: Differential responses of bovine macrophages to Mycobacterium avium subsp. paratuberculosis and Mycobacterium avium subsp. avium. Infect Immun. 2002, 70 (10): 5556-5561. 10.1128/IAI.70.10.5556-5561.2002.
Zurbrick BG, Czuprynski CJ: Ingestion and intracellular growth of Mycobacterium paratuberculosis within bovine blood monocytes and monocyte-derived macrophages. Infect Immun. 1987, 55 (7): 1588-1593.
Tessema MZ, Koets AP, Rutten VP, Gruys E: How does Mycobacterium avium subsp. paratuberculosis resist intracellular degradation?. Vet Q. 2001, 23 (4): 153-162.
Momotani E, Whipple DL, Thiermann AB, Cheville NF: Role of M cells and macrophages in the entrance of Mycobacterium paratuberculosis into domes of ileal Peyer's patches in calves. Vet Pathol. 1988, 25 (2): 131-137.
Sager H, Davis WC, Jungi TW: Bovine monocytoid cells transformed to proliferate cease to exhibit lineage-specific functions. Vet Immunol Immunopathol. 1999, 68 (2-4): 113-130. 10.1016/S0165-2427(99)00015-X.
Weiss DJ, Evanson OA, McClenahan DJ, Abrahamsen MS, Walcheck BK: Regulation of expression of major histocompatibility antigens by bovine macrophages infected with Mycobacterium avium subsp. paratuberculosis or Mycobacterium avium subsp. avium. Infect Immun. 2001, 69 (2): 1002-1008. 10.1128/IAI.69.2.1002-1008.2001.
Bogdan C, Vodovotz Y, Nathan C: Macrophage deactivation by interleukin 10. J Exp Med. 1991, 174 (6): 1549-1555. 10.1084/jem.174.6.1549.
Eriks IS, Emerson CL: Temporal effect of tumor necrosis factor alpha on murine macrophages infected with Mycobacterium avium. Infect Immun. 1997, 65 (6): 2100-2106.
Kindler V, Sappino AP, Grau GE, Piguet PF, Vassalli P: The inducing role of tumor necrosis factor in the development of bactericidal granulomas during BCG infection. Cell. 1989, 56 (5): 731-740. 10.1016/0092-8674(89)90676-4.
Sano C, Sato K, Shimizu T, Kajitani H, Kawauchi H, Tomioka H: The modulating effects of proinflammatory cytokines interferon-gamma (IFN-gamma) and tumour necrosis factor-alpha (TNF-alpha), and immunoregulating cytokines IL-10 and transforming growth factor-beta (TGF-beta), on anti-microbial activity of murine peritoneal macrophages against Mycobacterium avium-intracellulare complex. Clin Exp Immunol. 1999, 115 (3): 435-442. 10.1046/j.1365-2249.1999.00838.x.
Khalifeh MS, Stabel JR: Effects of gamma interferon, interleukin-10, and transforming growth factor beta on the survival of Mycobacterium avium subsp. paratuberculosis in monocyte-derived macrophages from naturally infected cattle. Infect Immun. 2004, 72 (4): 1974-1982. 10.1128/IAI.72.4.1974-1982.2004.
Langelaar MF, Hope JC, Rutten VP, Noordhuizen JP, van Eden W, Koets AP: Mycobacterium avium ssp. paratuberculosis recombinant heat shock protein 70 interaction with different bovine antigen-presenting cells. Scand J Immunol. 2005, 61 (3): 242-250. 10.1111/j.1365-3083.2005.01559.x.
Motiwala AS, Strother M, Amonsin A, Byrum B, Naser SA, Stabel JR, Shulaw WP, Bannantine JP, Kapur V, Sreevatsan S: Molecular epidemiology of Mycobacterium avium subsp. paratuberculosis: evidence for limited strain diversity, strain sharing, and identification of unique targets for diagnosis. J Clin Microbiol. 2003, 41 (5): 2015-2026. 10.1128/JCM.41.5.2015-2026.2003.
Motiwala AS, Strother M, Theus NE, Stich RW, Byrum B, Shulaw WP, Kapur V, Sreevatsan S: Rapid Detection and Typing of Strains of Mycobacterium avium subsp. paratuberculosis from Broth Cultures. J Clin Microbiol. 2005, 43 (5): 2111-2117. 10.1128/JCM.43.5.2111-2117.2005.
Snoek-van Beurden PA, Von den Hoff JW: Zymographic techniques for the analysis of matrix metalloproteinases and their inhibitors. Biotechniques. 2005, 38 (1): 73-83.
Brew K, Dinakarpandian D, Nagase H: Tissue inhibitors of metalloproteinases: evolution, structure and function. Biochim Biophys Acta. 2000, 1477 (1-2): 267-283.
Coussens PM, Pudrith CB, Skovgaard K, Ren X, Suchyta SP, Stabel JR, Heegaard PM: Johne's disease in cattle is associated with enhanced expression of genes encoding IL-5, GATA-3, tissue inhibitors of matrix metalloproteinases 1 and 2, and factors promoting apoptosis in peripheral blood mononuclear cells. Vet Immunol Immunopathol. 2005, 105 (3-4): 221-234. 10.1016/j.vetimm.2005.02.009.
Quiding-Jarbrink M, Smith DA, Bancroft GJ: Production of matrix metalloproteinases in response to mycobacterial infection. Infect Immun. 2001, 69 (9): 5661-5670. 10.1128/IAI.69.9.5661-5670.2001.
Price NM, Gilman RH, Uddin J, Recavarren S, Friedland JS: Unopposed matrix metalloproteinase-9 expression in human tuberculous granuloma and the role of TNF-alpha-dependent monocyte networks. J Immunol. 2003, 171 (10): 5579-5586.
Coussens PM, Colvin CJ, Wiersma K, Abouzied A, Sipkovsky S: Gene expression profiling of peripheral blood mononuclear cells from cattle infected with Mycobacterium paratuberculosis. Infect Immun. 2002, 70 (10): 5494-5502. 10.1128/IAI.70.10.5494-5502.2002.
Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001, 25 (4): 402-408. 10.1006/meth.2001.1262.
Coussens PM, Verman N, Coussens MA, Elftman MD, McNulty AM: Cytokine Gene Expression in Peripheral Blood Mononuclear Cells and Tissues of Cattle Infected with Mycobacterium avium subsp. paratuberculosis: Evidence for an Inherent Proinflammatory Gene Expression Pattern. Infect Immun. 2004, 72 (3): 1409-1422. 10.1128/IAI.72.3.1409-1422.2004.
Paape MJ, Rautiainen PM, Lilius EM, Malstrom CE, Elsasser TH: Development of anti-bovine TNF-alpha mAb and ELISA for quantitating TNF-alpha in milk after intramammary injection of endotoxin. J Dairy Sci. 2002, 85 (4): 765-773.
Kwong LS, Hope JC, Thom ML, Sopp P, Duggan S, Bembridge GP, Howard CJ: Development of an ELISA for bovine IL-10. Vet Immunol Immunopathol. 2002, 85 (3-4): 213-223. 10.1016/S0165-2427(02)00007-7.
Acknowledgements
This study was supported by state and federal funds appropriated to the Ohio Agricultural Research and Development Center (OARDC). We would like to thank the laboratory of Dr. Larry Schlesinger for helping optimize cell culture protocols used in this study. Mycobacterial research in SS laboratory is supported by funds from Johne's Disease Integrated Research Program and funds from USDA-NRI (Animal Protection).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
Dr. Janagama, performed cell infections, and analyzed the data. Dr. Jeong, helped in the design and analysis of ELISAs for cytokines studied. He also provided intellectual input during cell-infection study design. Dr. Kapur provided intellectual help during the study design and data analysis. Dr. Coussens was responsible for the original concept development with Dr. Sreevatsan. He also helped in the performance of MAP survival studies. Dr. Sreevatsan developed the concepts, designed MAP infection studies, and analyzed the data with Dr. Janagama. Dr. Sreevatsan also helped prepare the manuscript for consideration of publication.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Janagama, H.K., il Jeong, K., Kapur, V. et al. Cytokine responses of bovine macrophages to diverse clinical Mycobacterium avium subspecies paratuberculosis strains. BMC Microbiol 6, 10 (2006). https://doi.org/10.1186/1471-2180-6-10
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-6-10