
Abstract
Background
SPI-18 is a pathogenicity island found in some Salmonella enterica serovars, including S. Typhi. SPI-18 harbors two ORFs organized into an operon, hlyE and taiA genes, both implicated in virulence. Regarding the hlyE regulation in S. Typhi, it has been reported that RpoS participates as transcriptional up-regulator under low pH and high osmolarity. In addition, CRP down-regulates hlyE expression during exponential growth. Previously, it has been suggested that there is another factor related to catabolite repression, different from CRP, involved in the down-regulation of hlyE. Moreover, PhoP-dependent hlyE up-regulation has been reported in bacteria cultured simultaneously under low pH and low concentration of Mg2+. Nevertheless, the relative contribution of each environmental signal is not completely clear. In this work we aimed to better understand the regulation of hlyE in S. Typhi and the integration of different environmental signals through global regulators.
Results
We found that Fis participates as a CRP-independent glucose-dependent down-regulator of hlyE. Also, Fis and CRP seem to exert the repression over hlyE through down-regulating rpoS. Moreover, PhoP up-regulates hlyE expression via rpoS under low pH and low Mg2+ conditions.
Conclusions
All these results together show that, at least under the tested conditions, RpoS is the central regulator in the hlyE regulatory network, integrating multiple environmental signals and global regulators.
Similar content being viewed by others


Background
The genus Salmonella includes two species, Salmonella bongori and Salmonella enterica. S. enterica is comprised of six subspecies where only a small fraction of the subspecies I serovars are involved in infections of humans and other warm-blooded animals [1, 2]. Most of these Salmonella serovars are “generalists”, infecting a wide range of hosts and causing different symptoms. This is the case for S. enterica serovar Typhimurium (S. Typhimurium) and S. enterica serovar Enteritidis (S. Enteritidis) [3]. In contrast, some serovars are able to infect a specific host, causing typhoid-like disease [4]. These include S. enterica serovar Typhi (S. Typhi), which infects only humans.
S. enterica infection begins with ingestion of contaminated water or food. Some environmental conditions found in the gut induce the expression of virulence factors that participate in the intestinal invasion and colonization, including genes found in the Salmonella Pathogenicity Island 1 (SPI-1) [5]. Among these conditions, high osmolarity, microaerobiosis and response to bile seem to be the most important signals at this stage of the infection [5, 6]. Some S. enterica serovars, such as S. Typhi in humans, can enter the host bloodstream, disseminate and survive inside the macrophages by expressing a different subset of genes, including SPI-2 genes [7]. The most important conditions found at this stage of the infection include nutrient depletion (especially Mg2+) and low pH [8]. It is thought that Salmonella virulence factors are specifically expressed at determined stages of the infection. Nevertheless, at present it is more obvious that several virulence factors are not restricted to a unique stage of the infection. For example, SipA, encoded by SPI-1 enhances entry efficiency to intestinal epithelial cells by promoting actin polymerization, but also contributes to proliferation of Salmonella inside macrophages [9].
The SPI-18 is a pathogenicity island found in a subset of S. enterica serovars able to produce a systemic disease in humans, including S. Typhi; but absent in generalist serovars such as S. Typhimurium [10]. The SPI-18 harbors only two ORFs organized into an operon, the taiA – hlyE genes [10, 11]. TaiA is a novel invasin involved in increased phagocytosis of the bacteria by macrophages [11]. On the other hand, HlyE (also called ClyA or SheA) is a well characterized hemolysin that is exported in outer membrane vesicles [12]. Human infections by S. Typhi cause a specific antibody response to HlyE, indicating effective toxin production during the normal infective cycle [13]. Moreover, heterologous expression of S. Typhi hlyE in S. Typhimurium improves the colonization of deep organs in mice [10].
Regarding hlyE regulation in S. Typhi, previously it has been reported that RpoS participates as a transcriptional positive regulator under low pH and high osmolarity [14]. In addition, the global regulator CRP, implicated in catabolite repression, down-regulates hlyE expression during exponential growth. Nevertheless, addition of glucose to the growth medium results in a decrease of hlyE mRNA in S. Typhi Δcrp mutant, suggesting that there is another factor related to catabolite repression, different from CRP, involved in down-regulation of hlyE in S. Typhi [14]. PhoP-dependent up-regulation of hlyE was reported when bacteria were cultured simultaneously under low pH and low concentration of Mg2+[11]. Nevertheless, the relative contribution of each environmental signal is not clear.
In this work we wanted to better understand the regulation of hlyE in S. Typhi and the integration of different environmental signals through global regulators. We found that Fis participates as a CRP-independent glucose-dependent down-regulator of hlyE in S. Typhi. Moreover, Fis and CRP seem to exert the repression over hlyE via down-regulating rpoS. On the other hand, PhoP up-regulates hlyE expression via rpoS under low pH and low Mg2+ conditions. All these results together show that, at least under the tested conditions, RpoS is the central regulator in the hlyE regulatory network, integrating multiple environmental signals.
Results
Fis participates in the repression of hlyE at transcriptional level
CRP is a regulator that acts as a cAMP receptor. The cAMP-CRP complex is activated when glucose is scarce in the culture medium, consequently, numerous genes are up-/down-regulated [15]. CRP is a transcriptional regulator previously reported as an activator of the hlyE transcription in E. coli[16]. Nevertheless, genetic experiments showed that CRP participates in the negative regulation of hlyE in S. Typhi since crp deletion led to increased β-galactosidase activity associated to the ΔhlyE::lacZ strain [14]. Moreover, hlyE presented a CRP-independent down-regulation when glucose is present, suggesting the presence of another down-regulator [14]. Fis (factor for inversion simulation) is a nucleoid-associated protein that participates in the structuring of the bacterial chromosome, also known to interact at specific promoters to regulate gene transcription in E. coli and S. enterica[17–19]. Since Fis has been implicated in catabolite repression in E. coli[20, 21], we assessed the role of Fis in hlyE transcription in S. Typhi by RT-PCR. As shown in Figure 1A, the Δfis mutant presented increased transcription of hlyE compared with the WT, similar to the levels detected in the Δcrp mutant (control). In presence of glucose, the increased transcriptional level of hlyE seen in the Δcrp mutant was reverted, consistently with the presence of additional unknown glucose-dependent down-regulation [14]. On the other hand, the presence of glucose was unable to restore the normal transcriptional levels of hlyE in the Δfis mutant (Figure 1A), showing that Fis is the CRP-independent down-regulator factor previously stated by Fuentes et al. [14], at least under laboratory conditions. In all cases, the complementation with the corresponding gene cloned into a plasmid reverted the mutant phenotype with respect to hlyE transcription (Figure 1A). Moreover, we found no epistasis between crp and fis with respect to hlyE transcription in LB with or without glucose added (Figure 1A). Further, the crp and fis mutations seemed to produce additive effects on hlyE transcription, strongly suggesting that these two repressors act through different pathways to repress hlyE expression.

Fis participates in down-regulation of hlyE expression in S. Typhi. A) RT-PCR assay for mRNAs examining transcription of the hlyE gene from S. Typhi wild-type (STH2370), Δcrp, Δfis, Δcrp Δfis double mutant, and the complemented strains Δcrp/pTCRP and Δfis/pTFIS. All the strains were cultured as described in Methods. *p < 0.05; **p < 0.01 (ANOVA). B) Relative hemolysis, obtained from the S. Typhi STH2370 wild type, Δfis and Δcrp mutants. All the strains were grown in LB and samples were taken to perform the hemolytic activity quantification (see Methods). The total hemolytic activity was calculated deducting relative hemolysis of S. Typhi ΔhlyE[10] from S. Typhi wild type, deducting relative hemolysis of S. Typhi ΔhlyE Δfis from S. Typhi Δfis or S. Typhi ΔhlyE Δcrp from S. Typhi Δcrp, respectively. C) Relative hemolysis, obtained from the S. Typhi STH2370 wild type, Δfis and Δcrp mutants grown in LB + 0.4% glucose. Hemolysis was calculated as described in B. D) Immunodetection of epitope-tagged HlyE from S. Typhi hlyE-3xFLAG (par.: parental strain), S. Typhi hlyE-3xFLAG Δcrp, and S. Typhi hlyE-3xFLAG Δfis. Bacteria were cultured as described in Methods. E) RT-PCR for fis transcript levels detection in S. Typhi wild type strain grown to logarithmic phase in LB or LB + glucose. In all the cases, the data correspond to mean values of three independent experiments performed in triplicate. Error bars correspond to the SD.
To corroborate whether an increase in the hlyE transcript due to the absence of fis in LB and in LB + glucose resulted in increased HlyE-associated hemolytic activity, erythrocyte damage produced by whole bacterial lysates of S. Typhi WT and Δfis mutants grown in LB or LB + glucose was compared by quantifying the release of hemoglobin. We used PBS and 5% sodium deoxycholate to obtain the value for 0% and 100% hemolysis respectively, as previously described [14]. As shown in Figure 1B and C, the Δcrp mutant appeared to be more hemolytic that the WT strain only when cultured in LB (control). As previously reported, this effect is abolished in presence of glucose [14]. On the other hand, the Δfis mutant appeared also to be more hemolytic than the WT strain. However, unlike the effect observed with the Δcrp mutant, the presence of glucose was unable to revert the hemolysis to the WT level in the Δfis strain, consistently with the results obtained with RT-PCR (Figure 1A). S. Typhi Δcrp ΔhlyE and S. Typhi Δfis ΔhlyE exhibited no hemolysis in any condition (data not shown). To corroborate the data obtained by RT-PCR and by the determination of the hemolytic activity, we constructed the S. Typhi hlyE-3xFLAG strain by placing a 3xFLAG at the C-terminus of hlyE. This procedure led to subsequent detection of the FLAG-tagged proteins via Western blotting as previously described [22]. Then, we constructed the S. Typhi hlyE-3xFLAG Δcrp and S. Typhi hlyE-3xFLAG Δfis derivatives by electrotransforming with gDNA from single mutants as described [23]. These strains were used to determine the HlyE accumulation. As shown in Figure 1D, all the results obtained by RT-PCR were confirmed by Western blot. These results together show that fis participates in the repression of hlyE at transcriptional level.
Glucose increases transcription of fis
As stated above, the effect of Δcrp mutant on hlyE transcriptional level is relieved when glucose is added into the culture medium, suggesting that a second repressor is activated in some way under a glucose-rich medium. Considering that Fis also participates as an hlyE repressor and considering that glucose is unable to relief the Δfis effect in the hlyE transcription (Figure 1A), we inferred that glucose might increase the transcription of fis. In order to test this hypothesis, we performed a RT-PCR to detect the fis transcript of S. Typhi cultured to OD600 = 0.4 in LB or LB + glucose. As shown in Figure 1E, the presence of glucose increased fis transcription compared with bacteria grown in standard LB. This result supports the fact that glucose increases the fis expression, thereby contributing to the repression of hlyE.
CRP and Fis participate in the repression of hlyE transcription by repressing rpoS transcription
To find out whether CRP is repressing hlyE expression through a direct binding to the respective promoter region, we performed a shift mobility assay using purified CRP activated with cAMP and the DNA corresponding to the taiA – hlyE promoter region. Nevertheless, we were unable to find direct binding (data not shown). This result suggested that CRP is participating in hlyE repression through an indirect pathway. Previously, it has been reported that rpoS expression is repressed by the cAMP-CRP complex in Vibrio vulnificus[24]. In Salmonella, previously reported contradictory data indicate that CRP is an rpoS activator or an rpoS repressor [19, 25]. RpoS seems to be one of the most important factors participating in hlyE expression, since the detection of these transcripts is not possible in ΔrpoS mutants when grown in pH 5.0, high osmolarity or even in standard LB [14]. Thus, CRP might repress hlyE expression by repressing rpoS in S. Typhi. To determine the role of CRP in rpoS expression, we assessed rpoS expression by RT-PCR. As shown in Figure 2A, the rpoS transcription increased in the Δcrp mutant compared with the WT strain. Nevertheless, the rpoS transcriptional levels obtained from the Δcrp mutant were comparable to those of the WT strain in presence of glucose, following the same pattern for hlyE transcript in Figure 1A. This result suggests that crp participates as a transcriptional repressor of rpoS in S. Typhi. Since rpoS is regulated at multiple levels [26], we determined whether the changes in the rpoS transcription also lead to changes in RpoS accumulation. For that, we performed a Western blot comparing RpoS accumulation in the S. Typhi rpoS-3xFLAG and S. Typhi rpoS-3xFLAG Δcrp strains. As shown in Figure 2B, RpoS from S. Typhi rpoS-3xFLAG was practically undetectable under the tested conditions, consistently with the fact that the RpoS accumulation is prevented at logarithmic phase [27]. On the other hand, the lack of crp led to an evident RpoS accumulation. Similarly to the data observed for hlyE expression (Figure 1), these results suggest that crp is an rpoS repressor and, in presence of glucose, there is another crp-independent repressor. The obvious candidate to be such a repressor is the transcriptional regulator Fis. Then, we assessed the role of fis in the rpoS transcription and RpoS accumulation as previously described for crp. As shown in Figure 2A, the Δfis mutant presented a slight increase in the rpoS transcriptional level compared with the WT strain. Again, the addition of glucose was unable to abolish the increase of rpoS transcript in the Δfis mutant (Figure 2A). Moreover, the lack of fis clearly contributed to the RpoS accumulation even in presence of glucose, as inferred from Figure 2B. All these results are consistent with the presence of CRP and Fis DNA-binding boxes at the rpoS promoter region in S. Typhimurium [19, 25], which are also conserved in S. Typhi rpoS promoter (Figure 2C). Altogether, these results suggest that CRP and Fis are able to repress the rpoS expression by direct binding, thereby regulating hlyE expression in S. Typhi.

CRP and Fis participate in the repression of rpoS . A) RT-PCR assay for mRNAs examining transcription of the rpoS gene from S. Typhi wild-type (STH2370), Δcrp, Δfis, Δcrp Δfis double mutant, and the complemented strains Δcrp/pTCRP and Δfis/pTFIS. All the strains were grown in LB buffered at pH 7.0 to an OD600 = 0.35, washed three times with PBS prior to be resuspended with LB (control) or LB + 0.4% glucose, and incubated 1 h at 37°C. After the incubation, total RNA were isolated for each strain. The same amount of RNA was applied for RT-PCR. The transcription levels were normalized against the 16 s transcript in all cases. The data correspond to mean values of three independent experiments performed in triplicate. Error bars correspond to the SD. *p < 0.05; **p < 0.001 (ANOVA). B) Immunodetection of epitope-tagged RpoS from S. Typhi hlyE-3xFLAG (par.: parental strain), S. Typhi rpoS-3xFLAG Δcrp, and S. Typhi hlyE-3xFLAG Δfis. Bacteria were grown in LB buffered at pH 7.0 to an OD600 = 0.35, washed three times with PBS prior to be resuspended with LB or LB + 0.4% glucose, and incubated 1 h at 37°C. After the incubation, total proteins were obtained, and 20 μg were used to detect RpoS accumulation as described in Methods. C) In silico sequence analysis of CRP and Fis binding boxes in S. Typhi CT18 rpoS promoter. The S. Typhi rpoS promoter contains conserved binding boxes of S. Typhimurium rpoS promoter according to the previous evidence [19, 25]. Segmented underlined sequences indicate putative CRP boxes. Underlined sequences indicate putative Fis boxes. Asterisk shows a shared region of each box at the marked position. In bold, transcription start site, and short underlined sequences indicate predicted −35 and −10 promoter regions.
PhoPQ two-component regulatory system participates in hlyE induction via rpoS
As shown above, we identified two genes, crp and fis, implicated in the down-regulation of hlyE. In addition, it has been described two global regulators implicated in the up-regulation of this gene: RpoS and PhoP. RpoS-dependent transcriptional up-regulation in low pH and high osmolarity was reported [14]. PhoP-dependent up-regulation was observed when bacteria were cultured simultaneously under low pH and low concentration of Mg2+ (10 μM MgCl2) [11]. Nevertheless, considering that the low pH is an environmental signal that activates both the RpoS- and PhoP-dependent up-regulation of hlyE, it is not clear the relative contribution of each global regulator to the up-regulation of this gene. Moreover, the relative contribution of low pH and low Mg2+ concentrations to the PhoP-dependent up-regulation is also unknown, since, as stated, these two conditions were tested at the same time [11]. In order to better understand the up-regulation of hlyE, we compared the transcription induction of this gene in LB pH 5.0 versus LB pH 7.0, and in minimal medium 9 (M9) 10 μM Mg2+ versus M9 10 mM Mg2+ by RT-PCR in different genetic backgrounds. As shown in Figure 3A, pH 5.0 is an inducer condition of the hlyE expression, as previously reported [14]. Nevertheless, the relative expression of hlyE in the ΔrpoS mutant is nearly zero, even under pH 5.0. This result suggests that rpoS is necessary for the induction of hlyE expression under low pH, but also to maintain a basal expression of hlyE. This result strongly suggests that phoPQ is unable to induce the expression of hlyE under low pH when rpoS is absent. The induction levels obtained from the ΔrpoS ΔphoPQ double mutant reinforces this hypothesis, highlighting the fact that rpoS is epistatic over phoPQ. On the other hand, the ΔphoPQ mutant shows the half of induction under low pH compared with the WT strain, suggesting that rpoS, by itself, is sufficient to exert a partial induction under low pH. Thus, both rpoS and phoPQ are necessary to achieve a complete induction under low pH of hlyE in S. Typhi. However, the phoPQ-dependent up-regulation occurs via RpoS. The Figure 3B shows that 10 μM Mg2+ is also an inducer condition, exhibiting approximately 25 fold compared with 10 mM Mg2+. Again, the relative expression of hlyE in the ΔrpoS mutant is nearly zero under high or low Mg2+ concentration, supporting the fact that rpoS is essential for the hlyE expression under different culture conditions. In addition, the induction under 10 μM Mg2+ is completely abolished in the ΔphoPQ mutant, strongly suggesting that phoPQ is essential to increase the hlyE transcript under low Mg2+. Moreover, rpoS alone is not sufficient for increase the hlyE transcript under the same tested conditions. Finally, rpoS is epistatic over phoPQ, reinforcing the fact that the induction of the hlyE expression under low concentration of Mg2+ depends on PhoPQ and occurs via RpoS. In all the cases, the induction was restored when the mutants were complemented with the corresponding genes cloned into a plasmid vector (Figure 3).

PhoPQ two-component regulatory system participates in the hlyE induction via rpoS. A) RT-PCR assay for mRNAs examining transcription of the hlyE gene from S. Typhi wild-type (STH2370), ΔrpoS, ΔphoPQ, and the ΔrpoS ΔphoPQ double mutant, and the complemented strains ΔrpoS/pBRPOS and ΔphoPQ/pBPHOPQ. All the strains were grown in LB buffered at pH 7.0 to an OD600 = 0.35, washed three times with PBS prior to be resuspended with LB buffered at pH 7.0 (control) or LB buffered at pH 5.0, and incubated 1 h at 37°C. After the incubation, total RNA were isolated for each strain. The same amount of RNA was applied for RT-PCR. The transcription levels were normalized against the 16 s transcript in all cases. The data correspond to mean values of three independent experiments performed in triplicate. Error bars correspond to the SD. B) RT-PCR assay for mRNAs examining transcription of the hlyE gene from S. Typhi wild-type (STH2370), ΔrpoS, ΔphoPQ, and the ΔrpoS ΔphoPQ double mutant. All the strains were grown in LB buffered at pH 7.0 to an OD600 = 0.35, washed three times with PBS prior to be resuspended with M9 10 mM Mg2+(control) or M9 10 μM Mg2+, and incubated 1 h at 37°C. After the incubation, total RNA were isolated for each strain. The same amount of RNA was applied for RT-PCR. The transcription levels were normalized against the 16 s transcript in all cases. The data correspond to mean values of three independent experiments performed in triplicate. Error bars correspond to the SD. In all the cases, the numbers above the bars represent the fold of induction. *p < 0.05 (ANOVA).
All these results together strongly suggest that RpoS is essential for hlyE transcription under all the conditions tested. On the other hand, phoPQ participates in hlyE induction under pH 5.0 and low Mg2+ in an RpoS-dependent manner.
Discussion
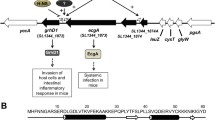
In this work we studied the regulation of hlyE expression and the integration of different environmental signals through global regulators (Figure 4).

Model showing that CRP, Fis and PhoPQ regulate taiA-hlyE operon expression via RpoS. RpoS integrates multiple environmental signals through different global regulators in order to regulate the taiA-hlyE operon expression. Like hlyE, taiA expression is up-regulated by RpoS and PhoPQ under low pH and low Mg2+ concentrations [11, 14].
As previously reported, CRP participates in the hlyE repression. Nevertheless, the mechanism involved in such repression has not been elucidated [14]. The absence of obvious CRP DNA-binding boxes at the promoter region of taiA – hlyE operon [14], and the fact that we were unable to find direct binding between CRP-cAMP and the taiA – hlyE promoter region, suggests that CRP might be acting indirectly, more likely regulating other transcriptional regulators. Here, we present genetic evidence indicating that CRP exerts down-regulation of hlyE by repressing rpoS (Figures 1 and 2), one of the most important hlyE activators in S. Typhi [14]. CRP has been also described as an RpoS repressor in other bacterial species. For instance, E. coli Δcrp is remarkably resistant to hydrogen peroxide and acid stress, due to an elevated catalase activity attributable to enhanced accumulation of the alternate sigma factor RpoS [28]. On the other hand, it has been reported that rpoS expression is repressed by direct binding of CRP-cAMP to its promoter region in Vibrio vulnificus[24]. In that same work, the authors suggest that CRP could repress RpoS expression in S. enterica by direct binding arguing that S. enterica rpoS promoter presents similar CRP binding boxes than V. vulnificus[24]. Nevertheless, the available studies of S. enterica include experiments performed only with S. Typhimurium, and the data are controversial. Cheng and Sun demonstrated direct binding of CRP to the rpoS promoter region in S. Typhimurium, where it is able to bind to two sites in this sequence. Both sites are conserved in S. Typhi rpoS promoter, suggesting a direct binding by CRP. Moreover, the authors showed that base-substitutions performed in the CRP-binding sites at the rpoS promoter region, led to a decreased rpoS transcription, concluding that CRP is an rpoS up-regulator [25]. Nevertheless, this result can be also explained by the fact that the base-substitution may be affecting unknown overlapped regulators’ binding sites. On the other hand, Hirsch and Elliott showed that S. Typhimurium rpoS-lacZ Δcrp exhibited twice more β-galactosidase activity compared with the otherwise isogenic strain S. Typhimurium rpoS-lacZ, suggesting that CRP is an RpoS repressor [19], supporting our findings in S. Typhi. In addition, CRP has been described as a pleiotropic factor in S. enterica implicated in carbon source utilization, synthesis of flagella, reduced growth rate, and attenuation of virulence, among other functions [29–32]. This versatility can be explained, at least in part, by the fact that CRP is regulating rpoS.
As previously stated, hlyE presents a CRP-independent down-regulation when glucose is present in the culture medium, suggesting the presence of another glucose-dependent down-regulator [14]. In this work, we found that fis participates in hlyE repression especially in presence of glucose (Figure 1). It has been stated that Fis regulates gene expression by increasing/decreasing its level in E. coli[20]. In this work, we found that glucose induces the fis expression in S. Typhi, suggesting that Fis is also regulated by its level in S. enterica (Figure 1E). On the other hand, Fis precludes the RpoS accumulation by repressing the rpoS transcription (Figure 2). Hirsch and Elliott showed that fis represses rpoS expression by direct binding in S. Typhimurium [19], supporting our observations. As with CRP, Fis might participate in down-regulation of hlyE via RpoS, and this regulation could be exerted directly due to the presence of several putative Fis binding boxes in S. Typhi rpoS promoter (Figure 2C). Considering that crp and fis deletions seem to produce additive effects in hlyE transcription (Figure 1A), these two repressors probably act through different pathways to repress the hlyE expression (Figure 4).
We also explored the relative participation of rpoS and phoPQ in the up-regulation of hlyE under pH 5.0 and low Mg2+ concentrations. We found that RpoS is essential for hlyE expression under all the tested conditions, strongly suggesting that RpoS accumulation is the most relevant event in hlyE regulation. Moreover, both rpoS and phoPQ are necessary to achieve a complete induction of hlyE under low pH in S. Typhi, where the phoPQ-dependent induction occurs via RpoS. The induction pathway of RpoS by low pH is not fully understood. One component of induction is RpoS stabilization, where PhoPQ participates by serving as a transcriptional activator of the iraP (yaiB) gene in S. Typhimurium. IraP enhances RpoS stability by interacting with RssB, the protein that normally delivers RpoS to the ClpXP protease for degradation [33], supporting the fact that phoPQ requires rpoS to induce hlyE expression (Figure 4). On the other hand, it has been reported that mutant strains unable to degrade RpoS still exhibited induction under acid shock in S. enterica. Moreover, rpoS translation is enhanced under low pH [34]. This phenomenon can explain the fact that rpoS is able to partially induce hlyE transcription under pH 5.0, independently of the presence of phoPQ (Figure 3A).
In the case of the hlyE induction under low Mg2+ concentrations, we found that phoPQ is essential to increase hlyE transcript in an rpoS-dependent manner. In S. enterica RpoS accumulates when bacteria are grown under low Mg2+ conditions [33]. This process requires PhoPQ, which is specifically activated under low Mg2+ by increasing the transcription of iraP, as previously reported [33, 35]. It is possible that PhoPQ is participating in the increase of hlyE transcription under low Mg2+ via IraP-dependent RpoS stabilization. Nevertheless, in the case of low Mg2+ we found that rpoS is unable to induce an increased transcription of hlyE in a ΔphoPQ background. Previous studies reported that low Mg2+ concentrations are unable to induce RpoS accumulation in a ΔphoPQ background [33], supporting our results.
In this paper we showed that RpoS is the most important activator of hlyE expression. According to our results, RpoS sits atop a hierarchical network that integrates diverse environmental and physiological cues. PhoPQ, CRP, and Fis fine-tune these global inputs to precisely control the dosage of HlyE, but also the time in the infection cycle in which this gene is expressed. For HlyE, the amount of protein produced can determine the function. For instance, sublytic concentrations of HlyE affect the Ca2+ homeostasis in epithelial cells by induction of slow, intracellular Ca2+ oscillations, while increased concentrations act cytolytically [36]. Thus, HlyE belongs to the novel group of pore-forming toxins shown to elicit such biphasic responses [36]. On the other hand, HlyE apparently is participating in different stages of the infective process. Previously, it has been reported that S. Typhi hlyE mutants are impaired in invasion of human epithelial cells HEp-2 in vitro[10]. Consistently, other pore-forming hemolysins play critical roles in invasion of eukaryotic epithelial cells [37–39]. These data show that hlyE could participate in the intestinal stage of the S. Typhi infection. On the other hand, HlyE is capable to reduce or control bacterial growth, as hlyE deletion enhanced S. Typhi survival within macrophages without affecting cytotoxicity [11], suggesting that this cytolysin could also be participating in the systemic dissemination of bacteria inside macrophages. In addition, hlyE increase their expression when bacteria were cultured under high osmolarity, a condition normally associated to the gut [14, 40], but also when bacteria were cultured under low Mg2+ concentrations or low pH (Figure 3), conditions found inside the macrophages [41, 42]. RpoS, PhoP, CRP and Fis may be important for the proper hlyE expression inside macrophages, since the RpoS and PhoP respective regulons are activated inside these cells; and both crp and fis expression is repressed in the same environment [41, 43]. Furthermore, hlyE induction by high osmolarity is RpoS dependent [14], reinforcing the fact that this sigma factor is an integrator of different signaling pathways. All this evidence together strongly suggests that HlyE is not circumscribed to a single stage of the infective process, as previously proposed [14]. This work show that hlyE presents a complex regulation network involving different environmental signals and global regulators, presumably because this gene is actually participating in different stages of the S. Typhi infections process.
Conclusions
We studied the hlyE regulation in S. Typhi using genetic approaches. All our results together show that, at least under the tested conditions, RpoS is the most important up-regulator in the hlyE regulatory network. RpoS integrates multiple environmental signals and global regulators, including CRP, Fis, and PhoP signaling pathways, to control S. Typhi hlyE expression.
Methods
Bacterial strains, media and culture conditions
A clinical isolate of S. Typhi, named STH2370, was obtained from the Infectious Diseases Hospital Lucio Córdova in Santiago, Chile. S. Typhi STH2370 strain was grown to an optical density at 600 nm (OD600) of 1.4, and frozen as a primary stock. S. Typhi STH2370 and derivative strains were grown routinely in liquid culture in Luria-Bertani (LB) broth (Bacto tryptone, 10 g/liter; bacto yeast extract, 5 g/liter; NaCl, 5 g/liter) at 37°C, with aeration provided by shaking. When required, medium was supplemented with chloramphenicol (20 μg/ml), ampicillin (100 μg/ml), gentamicin (20 μg/ml) or glucose (0.4% w/v). LB medium was solidified by the addition of agar (15 g/l). The culture medium was modified in order to simulate the environmental conditions during S. Typhi infection process. To determine the effect of glucose in gene expression, bacteria were grown in LB buffered at pH 7.0 to an OD600 = 0.35, washed three times with PBS prior to be resuspended with LB buffered at pH 7.0 (control) or LB buffered at pH 7.0 + 0.4% w/v glucose. To determine the effect of low pH in gene expression, bacteria were grown in LB buffered at pH 7.0 to an OD600 = 0.35, washed three times with PBS prior to be resuspended with LB buffered at pH 7.0 (control) or LB buffered at pH 5.0 with buffer citrate. To determine the effect of low Mg2+ in gene expression, bacteria were grown in LB buffered at pH 7.0 to an OD600 = 0.35, washed three times with PBS prior to be resuspended with M9 10 mM Mg2+(control) or M9 10 μM Mg2+. In all the cases, bacteria were incubated 1 h at 37°C with shaking prior to isolate total RNA or total proteins of each strain. It is important to underline that all the strain tested are in logarithmic phase at OD600 = 0.35 when cultured in LB buffered at pH 7.0.
Construction of S. Typhi mutant strains
Mutant strains with substitution of the hlyE, rpoS, crp, phoPQ or fis genes by resistance cassettes (cat: resistance to chloramphenicol) or FRT scar were constructed using the Red/Swap method [44]. PCR primers with 60 bases long overlapping the internal regions of the genes were synthesized with 40 bases at the 5’ ends corresponding to the regions flanking the desired substitutions (Table 1). S. Typhi hlyE-3xFLAG and S. Typhi rpoS-3xFLAG mutants were constructed using the primers listed in Table 1 as previously described [22]. All the double mutants were constructed by electrotransformation with gDNA from single mutants as described [23]. The presence of each substitution was confirmed by PCR using primers complementary to the DNA genome flanking the sites of substitution.
Plasmid construction
PCR amplifications for plasmid construction were performed using an Eppendorf thermal cycler and Taq (Fermentas) DNA polymerase. Reaction mixtures contained 1x PCR buffer, 1.5 mM MgCl2, each dNTP (200 mM), primers (1 mM), 100 ng of template DNA, and 2 U of Taq DNA polymerase. Standard conditions for amplification were 30 cycles at 96°C for 40 s, 60°C for 45 s, and 72°C for 2 min, followed by a final extension step at 72°C for 10 min. Template S. Typhi STH2370 chromosomal DNA was prepared as described [45]. For complementing S. Typhi ΔrpoS or S. Typhi ΔphoPQ, we constructed the pBRPOS and pBPHOPQ plasmids by cloning the PCR product generated with RPOS-C + RPOS-NP or PHOPQ-C + PHOPQ-NP, respectively, into pBBR5. For complementing S. Typhi Δcrp or S. Typhi Δfis, we constructed the pTCRP and pTFIS plasmids by cloning the PCR product generated with CRP-C + CRP-NP or FIS-C + FIS-NP, respectively, into pCR TOPO 2.1 TA (Invitrogen) according to the manufacturer’s instructions. In all the cases, the cloned amplicons contained the ORF and the corresponding promoter region.
RNA isolation, reverse transcription and real-time quantitative PCR (RT-PCR)
Total mRNA from the strains grown under the tested conditions was extracted using TRIzol reagents (Invitrogen) as described by the manufacturer. RNA was precipitated with isopropanol for 10 min at room temperature, washed with ice cold 70% v/v ethanol and resuspended in DEPC-treated water prior to treatment with DNase I to remove any trace of DNA. Purity of extracted RNA was determined by spectrometry. Reverse transcription was performed on 2 μg of DNase-treated RNA using Superscript II RT (Invitrogen) at 50°C for 50 min followed by 70°C for 10 min in 20 μl with 2.5 mM of the corresponding reverse primer (RT-HLYE-C, RT-RPOS-C and RT-FIS-C for hlyE, rpoS and fis mRNA detection, respectively). The 16sRV reverse primer for 16 s mRNA by quantitative detection was used under each condition to normalize against a reference transcript. Relative quantification of each mRNA was performed using Brilliant II SYBR Green QPCR Master Reagent Kit and the Mx3000P detection system (Stratagene). The reaction mixture was carried out in a final volume of 20 μl containing 1 μl of diluted cDNA (1:1000), 0.24 μl of each primer (120 nM) (RT-HLYE-C + RT-HLYE-N for hlyE mRNA detection, RT-RPOS-C + RT-RPOS-N for rpoS mRNA detection, RT-FIS-C + RT-FIS-N for fis mRNA detection and 16sFW + 16sRV for 16 s mRNA detection), 10 μl of 10X Master Mix, 0.14 μl of diluted ROX (1:200) and 8.38 μl of nuclease-free H2O. The reaction was performed under the following conditions: 10 min at 95°C followed by 40 cycles of 30 s at 95°C, 30 s at 53°C and 45 s at 72°C. Finally a melting cycle from 53 to 95°C was performed to check for amplification specificity. Amplification efficiency was calculated from a standard curve constructed by amplifying serial dilutions of RT-PCR products for each gene. These values were used to obtain the fold change in expression for the gene of interest normalized with 16 s levels as previously described [46]. Experiments were performed in three biological and technical replicates for each culture condition. When appropriate, the statistical significance of differences in the relative expression data was determined using the one way ANOVA test.
Immunodetection analysis
3xFLAG fusion proteins were immunodetected by the use of anti-FLAG M2 mAbs from Sigma. Strains carrying the epitope-tagged gene were grown under the conditions described above and washed twice with 100 mM Tris–HCl pH 8.0. Bacterial pellets were resuspended with 100 mM Tris–HCl and sonicated 60 s on ice. Then, the samples were mixed with 1 volume of Laemmli buffer [47]. Suspensions were incubated at 98°C for 5 min. The resulting lysates were quickly centrifuged to remove cell debris and used, straight or suitably diluted, for SDS-PAGE. Twenty μg of bacterial proteins, previously quantified following the Bradford method [48], were resolved by SDS-PAGE and then transferred to poly (vinylidenedifluoride) membranes and probed with mAbs (1:1.000) and horseradish peroxidase-conjugated goat antimouse IgG [1:5.000 (Sigma)]. Detection was performed by enhanced chemioluminescence (ECL, Amersham Pharmacia).
Hemolytic activity quantification
S. Typhi and derivative strains were grown under the tested conditions, the OD600 was recorded, and the cultures were chilled to 4°C. Samples of each strain were washed twice with PBS, resuspended in 1 ml of PBS and then sonicated for 60 s on ice. This suspension was gently mixed with 1 volume of sheep erythrocytes (109 cells/ml) previously washed twice with PBS, and incubated 30 min at 37°C without shaking. To measure the hemolytic activity, samples were cleared by centrifugation and the OD545 was determined. Instead of the bacterial suspension, PBS or 5% sodium deoxycholate (DC) were used as reference hemolysis values for 0 and 100%, respectively. The arbitrary hemolytic units were calculated using the formula: (OD545Sample – OD545PBS)/(OD545DC – OD545PBS).
References
Brenner FW, Villar RG, Angulo FJ, Tauxe R, Swaminathan B: Salmonella nomenclature. J Clin Microbiol. 2000, 38 (7): 2465-2467.
Porwollik S, Boyd EF, Choy C, Cheng P, Florea L, Proctor E, McClelland M: Characterization of Salmonella enterica subspecies I genovars by use of microarrays. J Bacteriol. 2004, 186 (17): 5883-5898.
Pascopella L, Raupach B, Ghori N, Monack D, Falkow S, Small PL: Host restriction phenotypes of Salmonella typhi and Salmonella gallinarum. Infect Immun. 1995, 63 (11): 4329-4335.
Barrow PA, Huggins MB, Lovell MA: Host specificity of Salmonella infection in chickens and mice is expressed in vivo primarily at the level of the reticuloendothelial system. Infect Immun. 1994, 62 (10): 4602-4610.
Rychlik I, Barrow PA: Salmonella stress management and its relevance to behaviour during intestinal colonisation and infection. FEMS Microbiol Rev. 2005, 29 (5): 1021-1040.
Altier C: Genetic and environmental control of Salmonella invasion. J Microbiol. 2005, 43 Spec No: 85-92.
Ochman H, Soncini FC, Solomon F, Groisman EA: Identification of a pathogenicity island required for Salmonella survival in host cells. Proc Natl Acad Sci U S A. 1996, 93 (15): 7800-7804.
Deiwick J, Hensel M: Regulation of virulence genes by environmental signals in Salmonella typhimurium. Electrophoresis. 1999, 20 (4–5): 813-817.
Brawn LC, Hayward RD, Koronakis V: Salmonella SPI1 effector SipA persists after entry and cooperates with a SPI2 effector to regulate phagosome maturation and intracellular replication. Cell Host Microbe. 2007, 1 (1): 63-75.
Fuentes JA, Villagra N, Castillo-Ruiz M, Mora GC: The Salmonella Typhi hlyE gene plays a role in invasion of cultured epithelial cells and its functional transfer to S. Typhimurium promotes deep organ infection in mice. Res Microbiol. 2008, 159 (4): 279-287.
Faucher SP, Forest C, Beland M, Daigle F: A novel PhoP-regulated locus encoding the cytolysin ClyA and the secreted invasin TaiA of Salmonella enterica serovar Typhi is involved in virulence. Microbiology. 2009, 155 (Pt 2): 477-488.
Wai SN, Lindmark B, Soderblom T, Takade A, Westermark M, Oscarsson J, Jass J, Richter-Dahlfors A, Mizunoe Y, Uhlin BE: Vesicle-mediated export and assembly of pore-forming oligomers of the enterobacterial ClyA cytotoxin. Cell. 2003, 115 (1): 25-35.
von Rhein C, Hunfeld KP, Ludwig A: Serologic evidence for effective production of cytolysin A in Salmonella enterica serovars typhi and paratyphi A during human infection. Infect Immun. 2006, 74 (11): 6505-6508.
Fuentes JA, Jofre MR, Villagra NA, Mora GC: RpoS- and Crp-dependent transcriptional control of Salmonella Typhi taiA and hlyE genes: role of environmental conditions. Res Microbiol. 2009, 160 (10): 800-808.
Kolb A, Busby S, Buc H, Garges S, Adhya S: Transcriptional regulation by cAMP and its receptor protein. Annu Rev Biochem. 1993, 62: 749-795.
Westermark M, Oscarsson J, Mizunoe Y, Urbonaviciene J, Uhlin BE: Silencing and activation of ClyA cytotoxin expression in Escherichia coli. J Bacteriol. 2000, 182 (22): 6347-6357.
Azam TA, Ishihama A: Twelve species of the nucleoid-associated protein from Escherichia coli. Sequence recognition specificity and DNA binding affinity. J Biol Chem. 1999, 274 (46): 33105-33113.
McLeod SM, Johnson RC: Control of transcription by nucleoid proteins. Curr Opin Microbiol. 2001, 4 (2): 152-159.
Hirsch M, Elliott T: Fis regulates transcriptional induction of RpoS in Salmonella enterica. J Bacteriol. 2005, 187 (5): 1568-1580.
Browning DF, Grainger DC, Beatty CM, Wolfe AJ, Cole JA, Busby SJ: Integration of three signals at the Escherichia coli nrf promoter: a role for Fis protein in catabolite repression. Mol Microbiol. 2005, 57 (2): 496-510.
Galan B, Manso I, Kolb A, Garcia JL, Prieto MA: The role of FIS protein in the physiological control of the expression of the Escherichia coli meta-hpa operon. Microbiology. 2008, 154 (Pt 7): 2151-2160.
Uzzau S, Figueroa-Bossi N, Rubino S, Bossi L: Epitope tagging of chromosomal genes in Salmonella. Proc Natl Acad Sci U S A. 2001, 98 (26): 15264-15269.
Toro CS, Mora GC, Figueroa-Bossi N: Gene transfer between related bacteria by electrotransformation: mapping Salmonella typhi genes in Salmonella typhimurium. J Bacteriol. 1998, 180 (17): 4750-4752.
Lee HJ, Park SJ, Choi SH, Lee KH: Vibrio vulnificus rpoS expression is repressed by direct binding of cAMP-cAMP receptor protein complex to its two promoter regions. J Biol Chem. 2008, 283 (45): 30438-30450.
Cheng Y, Sun B: Polyphosphate kinase affects oxidative stress response by modulating cAMP receptor protein and rpoS expression in Salmonella typhimurium. J Microbiol Biotechnol. 2009, 19 (12): 1527-1535.
Navarro Llorens JM, Tormo A, Martinez-Garcia E: Stationary phase in gram-negative bacteria. FEMS Microbiol Rev. 2010, 34 (4): 476-495.
Hengge-Aronis R: Signal transduction and regulatory mechanisms involved in control of the sigma(S) (RpoS) subunit of RNA polymerase. Microbiol Mol Biol Rev. 2002, 66 (3): 373-395. table of contents
Donovan GT, Norton JP, Bower JM, Mulvey MA: Adenylate cyclase and the cyclic AMP receptor protein modulate stress resistance and virulence capacity of uropathogenic Escherichia coli. Infect Immun. 2013, 81 (1): 249-258.
Chen ZW, Hsuan SL, Liao JW, Chen TH, Wu CM, Lee WC, Lin CC, Liao CM, Yeh KS, Winton JR, Huang C, Chien MS: Mutations in the Salmonella enterica serovar Choleraesuis cAMP-receptor protein gene lead to functional defects in the SPI-1 Type III secretion system. Vet Res. 2010, 41 (1): 5-
Garmory HS, Brown KA, Titball RW: Salmonella vaccines for use in humans: present and future perspectives. FEMS Microbiol Rev. 2002, 26 (4): 339-353.
Zhang X, Kelly SM, Bollen WS, Curtiss R: Characterization and immunogenicity of Salmonella typhimurium SL1344 and UK-1 delta crp and delta cdt deletion mutants. Infect Immun. 1997, 65 (12): 5381-5387.
Rosu V, Chadfield MS, Santona A, Christensen JP, Thomsen LE, Rubino S, Olsen JE: Effects of crp deletion in Salmonella enterica serotype Gallinarum. Acta Vet Scand. 2007, 49: 14-
Tu X, Latifi T, Bougdour A, Gottesman S, Groisman EA: The PhoP/PhoQ two-component system stabilizes the alternative sigma factor RpoS in Salmonella enterica. Proc Natl Acad Sci U S A. 2006, 103 (36): 13503-13508.
Audia JP, Foster JW: Acid shock accumulation of sigma S in Salmonella enterica involves increased translation, not regulated degradation. J Mol Microbiol Biotechnol. 2003, 5 (1): 17-28.
Garcia Vescovi E, Soncini FC, Groisman EA: Mg2+ as an extracellular signal: environmental regulation of Salmonella virulence. Cell. 1996, 84 (1): 165-174.
Söderblom T, Oxhamre C, Wai SN, Uhlen P, Aperia A, Uhlin BE, Richter-Dahlfors A: Effects of the Escherichia coli toxin cytolysin A on mucosal immunostimulation via epithelial Ca2+ signalling and Toll-like receptor 4. Cell Microbiol. 2005, 7 (6): 779-788.
Hertle R, Schwarz H: Serratia marcescens internalization and replication in human bladder epithelial cells. BMC Infect Dis. 2004, 4: 16-
Strauss EJ, Ghori N, Falkow S: An Edwardsiella tarda strain containing a mutation in a gene with homology to shlB and hpmB is defective for entry into epithelial cells in culture. Infect Immun. 1997, 65 (9): 3924-3932.
Wood GE, Dutro SM, Totten PA: Target cell range of Haemophilus ducreyi hemolysin and its involvement in invasion of human epithelial cells. Infect Immun. 1999, 67 (8): 3740-3749.
Bajaj V, Lucas RL, Hwang C, Lee CA: Co-ordinate regulation of Salmonella typhimurium invasion genes by environmental and regulatory factors is mediated by control of hilA expression. Mol Microbiol. 1996, 22 (4): 703-714.
Eriksson S, Lucchini S, Thompson A, Rhen M, Hinton JC: Unravelling the biology of macrophage infection by gene expression profiling of intracellular Salmonella enterica. Mol Microbiol. 2003, 47 (1): 103-118.
Rhen M: Hierarchical gene regulators adapt to its host milieus. Int J Med Microbiol. 2005, 294 (8): 487-502.
Faucher SP, Porwollik S, Dozois CM, McClelland M, Daigle F: Transcriptome of Salmonella enterica serovar Typhi within macrophages revealed through the selective capture of transcribed sequences. Proc Natl Acad Sci U S A. 2006, 103 (6): 1906-1911.
Datsenko KA, Wanner BL: One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000, 97 (12): 6640-6645.
Santiviago CA, Toro CS, Bucarey SA, Mora GC: A chromosomal region surrounding the ompD porin gene marks a genetic difference between Salmonella typhi and the majority of Salmonella serovars. Microbiology. 2001, 147 (Pt 7): 1897-1907.
Pfaffl MW: A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29 (9): e45-
Laemmli UK: Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970, 227 (5259): 680-685.
Bradford MM: A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976, 72: 248-254.
Acknowledgements
This work was supported by FONDECYT (Fondo Nacional de Desarrollo Científico y Tecnológico, Government of Chile) Grants 1110120 (GCM) and 11121506 (JAF) and UNAB-Grant DI-13-11/I (MRJ) and DI-222-12/N (JAF); MRJ is a MECESUP fellow program “Fortalecimiento del programa de Doctorado en Biociencias Moleculares” Grant UAB-0802. AAH is a FONDECYT Postdoctoral, fellow Grant 3130523.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MRJ designed the studies, performed the experiments and contributed to write the manuscript. LMR performed the experiments of Figure 1. NAV performed the hemolysis assays. AAH participated in discussing the results and revising the manuscript. GCM participated in the revision of the manuscript. JAF designed the studies and wrote the paper. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Jofré, M.R., Rodríguez, L.M., Villagra, N.A. et al. RpoS integrates CRP, Fis, and PhoP signaling pathways to control Salmonella Typhi hlyE expression. BMC Microbiol 14, 139 (2014). https://doi.org/10.1186/1471-2180-14-139
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-14-139