
Abstract
Background
Visceral leishmaniasis (VL) caused by Leishmania siamensis is an emerging disease continuously reported in six southern provinces of Thailand. To date, the phylogenetic relationships among Leishmania isolates from Thai patients and other Leishmania species are still unclear and the taxonomic diversity needs to be established. In this study, the phylogenetic inference trees were constructed based on four genetic loci (i.e., SSU-rRNA, ITS1, hsp70, and cyt b), using DNA sequences obtained from autochthonous VL patients from southern Thailand and reference sequences of reported Leishmania isolates from other studies deposited in GenBank.
Results
Phylogenetic analyses of hsp70 and cyt b loci supported a clade comprised of L. siamensis isolates, which is independent to the other members in the genus Leishmania. In combination with genetic distance analysis, sequence polymorphisms were observed among L. siamensis isolates and two different lineages could be differentiated, lineages PG and TR. Phylogenetic analysis of the cyt b gene further showed that L. siamensis lineage TR is closely related to L. enrietti, a parasite of guinea pigs.
Conclusion
The finding of this study sheds further light on the relationships of L. siamensis, both in intra- and inter-species aspects. This information would be useful for further in-depth studies on the biological properties of this important parasite.
Similar content being viewed by others
Background
Leishmaniasis, one of the most important neglected infectious diseases, is endemic in 88 tropical and subtropical countries. In the past, Thailand was thought to be free of leishmaniasis. From 1960–1986, sporadic cases were reported among Thais who had visited the endemic areas [1–3]. Since then, a few autochthonous cases of leishmaniasis caused by L. infantum and L. donovani were reported in 1996, 2005 and 2007; however, the sources of infection were not identified [4–6]. In 2008, based on sequence comparison of two genetic loci, Leishmania siamensis, a novel species causing autochthonous leishmaniasis (VL), was described for the first time in a Thai patient from a southern province of Thailand [7]. The analysis of three protein-coding genes revealed that the taxonomic position of L. siamensis is closely related to L. enrietti, a Leishmania of guinea pigs [8]. To date, more than ten autochthonous VL cases caused by L. siamensis were sporadically reported in six southern, one eastern and three northern provinces of Thailand [8, 9]. Due to the continually increasing number of cases, it is speculated that subclinical and clinical leishmaniasis in Thailand might exist in high numbers which needs prompt diagnosis.
The sequences of various genetic markers have been used to study the parasite diversity and relationships within Leishmania including the sequences of DNA polymerase α [10], RNA polymerase II [10], 7SL RNA [11], ribosomal internal transcribed spacer [12–14], the N-acetylglucosamine-1-phosphate transferase gene [15], mitochondrial cytochrome b gene [16] and heat shock protein 70 gene [17]. Building a database of sequences of new local isolates of Leishmania in Thailand, together with the published Leishmania sequences from GenBank, could be useful for future comparison studies. Therefore, this study aimed to genetically characterize L. siamensis isolated from five Thai VL patients, based on four genetic loci, i.e., small subunit ribosomal RNA (SSU-rRNA), internal transcribed spacer 1 (ITS1) region, heat shock protein 70 (hsp70), and cytochrome b (cyt b). In addition, we studied the phylogenetic relationships of L. siamensis within the genus Leishmania by comparison with retrieved sequences of other Leishmania species from GenBank.
Methods
Leishmaniafrom VL Thai patients
Samples used in this study were collected from five autochthonous VL patients reported from Phang-nga, Trang, Songkla, and Stun provinces, southern Thailand. All patients presented with hepatosplenomegaly and pancytopenia. Amastigotes were identified under microscope from Giemsa-stained bone marrow smears in all cases. Two axenic cultures of promastigotes were obtained using bone marrow aspirates in Schneider’s medium supplemented with 20% FBS. Genotypic characterization was processed on three positive clinical samples (i.e., Giemsa-stained bone marrow smears and buffy coat) and two cultured promastigotes. The information of these samples is shown in Table 1.
Ethics statement
The study was approved by the Ethics Committee of the Royal Thai Army Medical Department, Thailand. No information on the patients was presented in this study.
DNA preparation
DNA was extracted from the Giemsa-stained smears of bone marrow using modified FTA extraction paper (Whatman, Bioscience, USA) following the protocol as previously described [18]. The Genomic DNA Mini Kit (Tissue) (Geneaid, USA) was used to extract the DNA from other three remaining samples.
PCR amplification
PCR assays were used to amplify a fragment of four genetic loci using the previously described conditions, i.e., SSU-rRNA [19], ITS1 region [20], hsp70 [21], and cyt b [22]. The PCR products were subjected to electrophoresis on 1.5% agarose gels and stained with SYBR safe (Invitrogen, USA). Gels were photographed and documented on high-density printing paper using Uvisave gel documentation system I (Uvitech, UK).
Cloning and sequencing
PCR products amplified from the four loci were purified using a Wizard® SV Gel and PCR Clean-Up System (Promega, Madison, USA) according to the manufacturer instructions and then directly sequenced. For the PCR products that had insufficient amounts of DNA for direct sequencing, they were cloned in E. coli competent cells to produce a higher quantity of identical DNA. For cloning, the purified PCR product was ligated into pGEM-T Easy vector (Promega, Madison, USA). The ligated product was introduced into the E. coli strain JM109 by chemical transformation. One colony from each cloning reaction was selected. The recombinant plasmids were purified using Wizard® Plus SV Minipreps DNA purification system (Promega, Madison, USA) and bidirectional sequenced using universal primer T7 and SP6. DNA sequencing was conducted by 1st Base Pte. Ltd., Singapore. The chromatograms were validated and assembled in BioEdit version 7.0.1.
Phylogenetic analysis
The sequences were multiple-aligned with a set of Leishmania strains retrieved from the GenBank using ClustalX, version 2.0.12 [23]. The pairwise genetic distances among isolates were estimated using program MEGA (Molecular Evolutionary Genetics Analysis), version 4.0 [24]. To investigate the relationships among L. siamensis isolates and other Leishmania species, Leishmania sequences of each locus examined in this study from GenBank were included in the dataset. The evolutionary history was inferred by phylogenetic tree construction using three methods, i.e., Neighbor Joining (NJ), Maximum Parsimony (MP) and Bayesian inference. The NJ and MP trees were constructed using program MEGA, version 4.0 [24]. Reliability of the inferred trees was tested by 1000 bootstrap replications. For the Bayesian method, starting trees were random: four simultaneous Markov chains were run for 500,000 generations, burn-in values were set at 30,000 generations and trees were sampled every 100 generations. Bayesian posterior probabilities were calculated using a Markov Chain Monte Carlo sampling approach implemented in MrBAYES, version 3.1.2 [25]. The Akaike information criterion in Modeltest, version 3.06, was used to select a DNA substitution model of all phylogenetic analyses [26]. The following models were selected for the dataset of each gene: K2P (SSU-rRNA), TrN+Γ (ITS1 and hsp70), and GTR+Γ (cyt b). The nucleotide sequences generated in this study have been deposited in GenBank under accession no. JX195633-JX195637, JX195639-JX195640, and KC202880-KC202883.
Results
Sequence analysis
PCR amplification of each target locus resulted in amplicons of the expected sizes as follows: SSU-rRNA (540 bp), ITS1 (340–348 bp), hsp70 (1422 bp), and cyt b (865 bp). Due to the limited amount of DNA samples, studied loci of some samples were not successfully amplified. Twelve amplicons were successfully amplified and bidirectionally sequenced. As a result, a total of 15L. siamensis sequences were analyzed in this study. These consisted of four isolates from SSU-rRNA (CU1, PCM1, PCM2, and PCM4; sequences of PCM1 and PCM2 were reported by Bualert et al. [8]), four isolates from ITS1 (CU1, PCM1, PCM2, and sequences of PCM4; PCM1 were reported by Sukmee et al. [7]), four isolates from hsp70 (CU1, PCM2, PCM4, and PCM5), and three isolates from cyt b (CU1, PCM1, and PCM2).
The comparison of mean genetic distance among the five isolates of each respective target was not calculated since different sets of isolates were used for each marker (Table 1). However, our preliminary analysis using available L. siamensis isolates indicates that the overall mean genetic distance varied depending on the markers analyzed. The most variable marker was the ITS1 region, followed by the cyt b gene, and the hsp70 gene whereas the SSU-rRNA sequences were identical for all isolates.
Sequence analysis could divide the L. siamensis isolates into two groups; the first one consisted of four isolates (isolates CU1, PCM1, PCM4, and PCM5), and the second group consisted of only one isolate (isolate PCM2).
According to these results, the isolates of groups 1 and 2 could be considered as different lineages and primarily designated as lineages PG (isolates CU1, PCM1, PCM4, and PCM5) and TR (isolate PCM2), respectively. In addition, the genetic divergence between TR and PG lineages was much higher than usually observed within other species (data not shown).
Phylogenetic analysis
Three phylogenetic analyses using the NJ, MP, and Bayesian methods were performed to observe the relationships between two L. siamensis lineages. Using three different constructing methods, the trees showed similar phylogenetic topology for all four loci supported by related bootstrapping/posterior probability values.
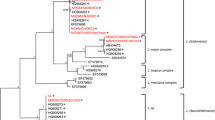
Regarding the phylogenetic tree inferred from each locus, the SSU-rRNA tree was constructed using four L. siamensis isolates and ten reference sequences of different Leishmania species (Figure 1a). The phylogenetic analyses grouped both L. siamensis lineages PG and TR together in a separated clade apart from other Leishmania species. Although lineages PG and TR were closely related according to the SSU-rRNA analysis, these two lineages formed separate clades in the phylogenetic tree inferred from other three markers. The ITS1 analysis of 13 Leishmania reference sequences and 14 L. siamensis sequences revealed a close relationship of L. siamensis to the members of L. braziliensis complex by forming a strongly supported cluster with both lineages PG and TR. Moreover, L. siamensis lineage TR formed a separate branch from the lineage PG but still shared a close relationship (Figure 1b). Interestingly, L. siamensis lineage PG clustered with the reference sequences previously isolated from Thai patients (GQ226034, GQ293226, JQ001751, and JQ001752), horse (JQ617283) in USA, and those isolated from a cow (CQ281282) and horses (CQ281278, CQ281279, CQ281280, and CQ281281) in Europe. Among these isolates, 100% sequence identity was revealed, except 99.6% identity of the isolate LECU1. For the hsp70 region, the phylogenetic tree was constructed using 15 reference sequences and four L. siamensis sequences. Both L. siamensis lineages apparently formed independent monophyletic clades outside the clusters of those other species while each L. siamensis lineage was still separated into different branches (Figure 1c). In addition, constructing the cyt b tree using 20 reference Leishmania sequences, two Endotrypanum sequences, and three L. siamensis sequences, similar results were observed. This tree showed high congruence to hsp70 tree since all taxa were concordantly clustered into the same species complex and placed L. siamensis at the basal branch of Leishmania in Euleishmania section.

The unrooted phylogenetic tree inferred from DNA sequences of four markers using Neighbor Joining method. The bootstrapping values less than 50 are omitted. The bootstrapping and posterior probability values estimated by Maximum parsimony and Bayesian inference methods are shown in parenthesis at each node, respectively. Asterisks indicate bootstrapping and posterior probability values that are below 50 or 0.5 or are not calculated by the analyses. Dense lines indicate Leishmania species complexes as described by Lainson and Shaw [30]. The species complex of L. adleri, L. turanica, L. gerbilli, and L. arabica are unclassified. Dot lines indicate the lineage sections suggested by Cupolillo et al. [35]. (a) SSU-rRNA, (b) ITS1, (c) hsp70, (d) cyt b.
In addition, the L. siamensis lineage TR was closely related to L. enrietti whereas lineage PG was furcated into a sister clade (Figure 1d). For sequence alignments of the ITS1, hsp70, and cyt b regions, see Additional files 1, 2, 3.
Discussion
This study characterized L. siamensis isolated from autochthonous VL Thai patients based on sequencing of four genetic loci. The construction of molecular evolutionary trees of Leishmania species has been extensively studied on various genetic markers both in conserved and variable regions [10–17]. The results of these studies allow us to view evolutionary processes, classify and discriminate species among Leishmania isolates. One of the widely used genetic markers for phylogenetic studies is the ribosomal RNA gene. This gene has proved to be useful for inferring the relationships of a wide range of organisms, including Leishmania[7, 27]. Even though the phylogenetic study based on the complete SSU-rRNA has shown that the variation of this gene limits the classification of this parasite at the subgenus level, studying the phylogenetic position using this gene is fundamentally required for a novel species, like L. siamensis[28, 29]. In this study, L. siamensis was grouped in the monophyletic branch of subgenus Leishmania (Leishmania) at a long distance in a unique subclade, primarily suggesting that this novel species is closely related to the members of L. (Leishmania) but evolved rapidly and nonrelative to the members in this subgenus. The incapability to discriminate between two lineages of L. siamensis proposed from the genetic distance analysis was not beyond our expectation since the studied region of this gene was remarkably conserved. Unfortunately, the sequences of the Paraleishmania section could not be included in this tree because the SSU-rRNA fragment amplified in this study is located in a different region than those deposited in the GenBank.
Recently, Asato et al. [16] and Fraga et al. [17] analyzed the phylogeny of genus Leishmania using the sequences obtained from the cyt b and the hsp70 regions and demonstrated the improvement of Leishmania classification from the traditional method proposed by Lainson and Shaw [30]. Their studies showed that these genes contained sufficient information for distinguishing species/subspecies and also human/nonhuman Leishmania. The high congruency between the cyt b and the hsp70 trees corresponding to the current classification were, thus, logically acceptable as the precise relationship of genus Leishmania. Employing the L. siamensis taxa into these trees provided more knowledge of this species in relation to other previously identified Leishmania species. Previous studies showed the early divergence of L. enrietti from other Leishmania groups, closely related to genus Endotrypanum, suggesting that this species may not belong to genus Leishmania [16]. In this study, grouping between both lineages of L. siamensis and L. enrietti rearranged the phylogenetic position of L. enrietti compared with a previous tree shown by Asato et al. [16]. The close relationship between lineage TR (previously described as Trang strain) and L. enrietti was supported by our previous work using concatenated sequences of three Leishmania protein-coding genes to construct the tree [8]. As shown in this study, L. enrietti and L. siamensis formed independent sister clades and shared the same branch of the members classified as Euleishmania, leaving the group of Paraleishmania completely separated. This finding distinctly indicated that they might be part of an unclassified subgenus of Leishmania. Unfortunately, the hsp70 sequences of L. enrietti and other species belonging to Paraleishmania were not available in the GenBank, and the alternative notion of this idea could not be obtained by the hsp70 tree in this study. However, the phylogenetic position of L. siamensis was in good agreement between the hsp70 and the cyt b trees in that these species were members of neither L. (Leishmania) nor L. (Viannia) and they should be regarded as an unclassified subgenus.
Since the identification of L. siamensis from a Thai VL case has been described using the comparison of mini-exon and ITS1 sequences in 2008 [7], more cases presumably caused by the same Leishmania species were reported on other continents. In 2009, autochthonous cutaneous leishmaniasis (CL) was reported in horses and a cow in Switzerland and Germany, followed by an additional case in a mare from the USA in 2012 [31–33]. These cases showed high ITS1 similarity compared with those previous reports of L. siamensis. To elucidate the relationship among the Leishmania detected from these cases and L. siamensis, these sequences were phylogenetically analyzed. The phylogenetic tree of ITS1 region, again, separated the L. siamensis lineage TR from lineage PG. The autochthonous Leishmania cases in livestock, previously reported in Europe and the USA, were closely related to the L. siamensis lineage PG, suggesting that lineage PG might not be indigenous. Although the relationship of these isolates was strongly supported by the posterior probability/bootstrapping values and nucleotide identity (99-100%), the studies on the isolates from Europe and the USA were limited only on the ITS1 region [31, 32]. Thus, the conclusion that the isolates from Thailand and other geographic areas share the same lineage is still premature. Further studies are needed to explore naturally infected reservoir animals like those found in Europe and the USA. More data of their biology, pathology and molecular biology as well as the transmission vectors are required before making conclusions about the relationship of Leishmania from these three different geographical areas.
Regarding the phylogenetic trees constructed in this study, the relationships between L. siamensis and other Leishmania species of SSU-rRNA and ITS1 apparently revealed conflicting phylogenetic signals to the other two markers examined in this study. These could be explained by the different evolutionary constraints displayed by each independent gene of each species [34]. Together, the immoderate sequence variations of the selected SSU-rRNA and ITS1 regions as well as the lack of data from the Paraleishmania group could impede the phylogenetic estimation to exhibit concordant relationships. Nevertheless, when cautiously considering the intra-species relationships within L. siamensis, the relatively high degree of genetic distance within species compared with other species complex in the genus Leishmania implied that lineages PG and TR of L. siamensis might not be a species complex. This analysis, on the other hand, strengthens the possibility that these two lineages might be of different species. Hence, further molecular studies on these two lineages using multilocus enzyme electrophoresis (MLEE) as the classical method/gold standard of Leishmania typing or MLST based on the protein genes used for MLEE would enhance the understanding of the phylogenetic basis of L. siamensis.
Conclusion
The genetic analysis conducted in this study brings more insight into the phylogenetic relationships of L. siamensis covering intra- and interspecies aspects. Two L. siamensis lineages were proposed based on the findings from this study, of which lineage PG was the predominant one responsible for VL in Thailand. The existence of this lineage seems to be not restricted only to Thailand but also prevalent on other continents, causing the disease to affect livestock. Little is known whether the two L. siamensis lineages designated in this study have different parasite characteristics such as geographical distribution, virulence in humans, host preference, transmission vector, as well as drug sensitivity. Knowing their taxonomy precisely will shed further light on parasite biological properties linked to closely related species.
At present, more VL cases caused by L. siamensis have been increasingly detected in southern Thailand and have also spread widely in other regions of the country. The disease burden is significantly underestimated and the true incidence is not well reflected, as only a few published case reports are available. Further study is required for a large scale molecular epidemiological study of emerging VL disease caused by L. siamensis in Thailand.
Consent
Written informed consent was obtained from the patient for publication of this report and any accompanying images.
References
Suttinont P, Thammanichanont C, Chantarakul N: Visceral leishmaniasis: a case report. Southeast Asian J Trop Med Public Health. 1987, 18 (1): 103-106.
Laohapaibul P, Siampakdi S: Kala-azar: report of one imported case. Siriraj Hosp Gaz. 1960, 12: 561-569. (In Thai)
Chutaputti A, Siripool P, Chitchang S, Radomyos P: Visceral leishmaniasis (Kala-azar): with hyper-splenism successfully treated with pentavalent antimony; report of 2 cases. Intern Med. 1986, 2: 262-265. (In Thai)
Kongkaew W, Siriarayaporn P, Leelayoova S, Supparatpinyo K, Areechokchai D, Duang-ngern P, Chanachai K, Sukmee T, Samung Y, Sridurongkathum P: Autochthonous visceral leishmaniasis: a report of a second case in Thailand. Southeast Asian J Trop Med Public Health. 2007, 38 (1): 8-12.
Maharom P, Siripattanapipong S, Mungthin M, Naaglor T, Sukkawee R, Pudkorn R, Wattana W, Wanachiwanawin D, Areechokchai D, Leelayoova S: Visceral leishmaniasis caused by Leishmania infantum in Thailand. Southeast Asian J Trop Med Public Health. 2008, 39 (6): 988-990.
Thisyakorn U, Jongwutiwes S, Vanichsetakul P, Lertsapcharoen P: Visceral leishmaniasis: the first indigenous case report in Thailand. Trans R Soc Trop Med Hyg. 1999, 93 (1): 23-24.
Sukmee T, Siripattanapipong S, Mungthin M, Worapong J, Rangsin R, Samung Y, Kongkaew W, Bumrungsana K, Chanachai K, Apiwathanasorn C: A suspected new species of Leishmania, the causative agent of visceral leishmaniasis in a Thai patient. Int J Parasitol. 2008, 38 (6): 617-622.
Bualert L, Charungkiattikul W, Thongsuksai P, Mungthin M, Siripattanapipong S, Khositnithikul R, Naaglor T, Ravel C, El Baidouri F, Leelayoova S: Autochthonous Disseminated Dermal and Visceral Leishmaniasis in an AIDS Patient, Southern Thailand, Caused by Leishmania siamensis. Am J Trop Med Hyg. 2012, 86 (5): 821-824.
Suankratay C, Suwanpimolkul G, Wilde H, Siriyasatien P: Autochthonous visceral leishmaniasis in a human immunodeficiency virus (HIV)-infected patient: the first in Thailand and review of the literature. Am J Trop Med Hyg. 2010, 82 (1): 4-8.
Croan DG, Morrison DA, Ellis JT: Evolution of the genus Leishmania revealed by comparison of DNA and RNA polymerase gene sequences. Mol Biochem Parasitol. 1997, 89 (2): 149-159.
Zelazny AM, Fedorko DP, Li L, Neva FA, Fischer SH: Evaluation of 7SL RNA gene sequences for the identification of Leishmania spp. Am J Trop Med Hyg. 2005, 72 (4): 415-420.
Davila AM, Momen H: Internal-transcribed-spacer (ITS) sequences used to explore phylogenetic relationships within Leishmania. Ann Trop Med Parasitol. 2000, 94 (6): 651-654.
Spanakos G, Piperaki ET, Menounos PG, Tegos N, Flemetakis A, Vakalis NC: Detection and species identification of Old World Leishmania in clinical samples using a PCR-based method. Trans R Soc Trop Med Hyg. 2008, 102 (1): 46-53.
Berzunza-Cruz M, Cabrera N, Crippa-Rossi M, Sosa Cabrera T, Perez-Montfort R, Becker I: Polymorphism analysis of the internal transcribed spacer and small subunit of ribosomal RNA genes of Leishmania mexicana. Parasitol Res. 2002, 88 (10): 918-925.
Waki K, Dutta S, Ray D, Kolli BK, Akman L, Kawazu S, Lin CP, Chang KP: Transmembrane molecules for phylogenetic analyses of pathogenic protists: Leishmania-specific informative sites in hydrophilic loops of trans- endoplasmic reticulum N-acetylglucosamine-1-phosphate transferase. Eukaryot Cell. 2007, 6 (2): 198-210.
Asato Y, Oshiro M, Myint CK, Yamamoto Y, Kato H, Marco JD, Mimori T, Gomez EA, Hashiguchi Y, Uezato H: Phylogenic analysis of the genus Leishmania by cytochrome b gene sequencing. Exp Parasitol. 2009, 121 (4): 352-361.
Fraga J, Montalvo AM, De Doncker S, Dujardin JC, Van der Auwera G: Phylogeny of Leishmania species based on the heat-shock protein 70 gene. Infect Genet Evol. 2010, 10 (2): 238-245.
Siripattanapipong S, Leelayoova S, Mungthin M, Thompson RC, Boontanom P, Saksirisampant W, Tan-Ariya P: Clonal diversity of the glutamate dehydrogenase gene in Giardia duodenalis from Thai isolates: evidence of genetic exchange or mixed infections?. BMC Microbiol. 2011, 11: 206-
Uliana SR, Nelson K, Beverley SM, Camargo EP, Floeter-Winter LM: Discrimination amongst Leishmania by polymerase chain reaction and hybridization with small subunit ribosomal DNA derived oligonucleotides. J Eukaryot Microbiol. 1994, 41 (4): 324-330.
El Tai NO, El Fari M, Mauricio I, Miles MA, Oskam L, El Safi SH, Presber WH, Schonian G: Leishmania donovani: intraspecific polymorphisms of Sudanese isolates revealed by PCR-based analyses and DNA sequencing. Exp Parasitol. 2001, 97 (1): 35-44.
Montalvo AM, Fraga J, Monzote L, Montano I, De Doncker S, Dujardin JC, Van der Auwera G: Heat-shock protein 70 PCR-RFLP: a universal simple tool for Leishmania species discrimination in the New and Old World. Parasitology. 2010, 137 (8): 1159-1168.
Kato H, Uezato H, Gomez EA, Terayama Y, Calvopina M, Iwata H, Hashiguchi Y: Establishment of a mass screening method of sand fly vectors for Leishmania infection by molecular biological methods. Am J Trop Med Hyg. 2007, 77 (2): 324-329.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R: Clustal W and Clustal X version 2.0. Bioinformatics. 2007, 23 (21): 2947-2948.
Kumar S, Nei M, Dudley J, Tamura K: MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform. 2008, 9 (4): 299-306.
Huelsenbeck JP, Ronquist F: MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001, 17 (8): 754-755.
Posada D, Crandall KA: Modeltest: testing the model of DNA substitution. Bioinformatics. 1998, 14 (9): 817-818.
Yang BB, Guo XG, Hu XS, Zhang JG, Liao L, Chen DL, Chen JP: Species discrimination and phylogenetic inference of 17 Chinese Leishmania isolates based on internal transcribed spacer 1 (ITS1) sequences. Parasitol Res. 2010, 107 (5): 1049-1065.
Fernandes AP, Nelson K, Beverley SM: Evolution of nuclear ribosomal RNAs in kinetoplastid protozoa: perspectives on the age and origins of parasitism. Proc Natl Acad Sci USA. 1993, 90 (24): 11608-11612.
van Eys GJ, Schoone GJ, Kroon NC, Ebeling SB: Sequence analysis of small subunit ribosomal RNA genes and its use for detection and identification of Leishmania parasites. Mol Biochem Parasitol. 1992, 51 (1): 133-142.
Lainson R, Shaw JJ: Evolution, classification and geographical distribution. The Leishmaniases in Biology and Medicine. Volume 1. Edited by: Peters W, Killick-Kendrick R. 1987, London: Academic Press, 1-120.
Muller N, Welle M, Lobsiger L, Stoffel MH, Boghenbor KK, Hilbe M, Gottstein B, Frey CF, Geyer C, von Bomhard W: Occurrence of Leishmania sp. in cutaneous lesions of horses in Central Europe. Vet Parasitol. 2009, 166 (3–4): 346-351.
Lobsiger L, Muller N, Schweizer T, Frey CF, Wiederkehr D, Zumkehr B, Gottstein B: An autochthonous case of cutaneous bovine leishmaniasis in Switzerland. Vet Parasitol. 2010, 169 (3–4): 408-414.
Reuss SM, Dunbar MD, Calderwood Mays MB, Owen JL, Mallicote MF, Archer LL, Wellehan JF: Autochthonous Leishmania siamensis in horse, Florida. USA Emerg Infect Dis. 2012, 18 (9): 1545-1547.
Phillipe H: Molecular phylogenetic in kinetoplats. Evolutionary Relationships among Protozoa. Edited by: Coomb GH, Vickerman K, Sleigh MA, Warren A. 1998, London: Systematics Association, 195-212.
Cupolillo E, Medina-Acosta E, Noyes H, Momen H, Grimaldi G: A revised classification for Leishmania and Endotrypanum. Parasitol Today. 2000, 16 (4): 142-144.
Acknowledgements
This work was financially supported by the Phramongkutklao College of Medicine. The authors would like to thank Dr. Mohamed Kasbari and Dr Francine Pratlong from the French Agency for Health and Safety and the French Reference Centre on Leishmaniasis, respectively, for the preliminary results of isoenzyme analysis.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SL participated in the study design, conceived the project, supervised the experiments, analyzed and interpreted the data, and co-wrote the manuscript. SS participated in the study design, performed the experiments, analyzed and interpreted the data, and co-wrote the manuscript. AH and HK performed the experiments. PT participated in the study design and conceived the project. PS and SO participated in specimen collection. MM participated in the study design, conceived the project, and co-wrote the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12866_2012_1924_MOESM1_ESM.jpeg
{kind=link}
Additional file 1: Sequence alignment of 348 bp of ITS1 region of L. donovani , L. infantum , Leishmania sp. (cow in Europe), Leishmania sp. (horse in Europe), L. siamensis (mare in the USA), L. siamensis lineage PG, and L. siamensis lineage TR. Bases that are identical to those of the L. siamensis lineage PG are indicated by dots, missing bases are indicated by hyphens, and bases that are different from those of the L. siamensis lineage PG are given. (JPEG 1 MB)
12866_2012_1924_MOESM2_ESM.jpeg
{kind=link}
Additional file 2: Sequence alignment of 1380 bp of hsp 70 region of L. donovani , L. infantum , L. siamensis lineage PG, and L. siamensis lineage TR. Bases that are identical to those of the L. siamensis lineage PG are indicated by dots, missing bases are indicated by hyphens, and bases that are different from those of the L. siamensis lineage PG are given. (JPEG 3 MB)
12866_2012_1924_MOESM3_ESM.jpeg
{kind=link}
Additional file 3: Sequence alignment of 816 bp of cyt b region of L. donovani , L. infantum , L. enrietti , L. siamensis lineage PG, and L. siamensis lineage TR. Bases that are identical to those of the L. siamensis lineage PG are indicated by dots, missing bases are indicated by hyphens, and bases that are different from those of the L. siamensis lineage PG are given. (JPEG 2 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Leelayoova, S., Siripattanapipong, S., Hitakarun, A. et al. Multilocus characterization and phylogenetic analysis of Leishmania siamensisisolated from autochthonous visceral leishmaniasis cases, southern Thailand. BMC Microbiol 13, 60 (2013). https://doi.org/10.1186/1471-2180-13-60
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-13-60