
Abstract
Background
Bats (Chiroptera) are one of the most diverse groups of mammals which carry out important ecological and agricultural functions that are beneficial to humans. However, they are increasingly recognized as natural vectors for a number of zoonotic pathogens and favourable hosts for zoonotic infections. Large populations of the Straw-Coloured Fruit Bat (Eidolon helvum) colonize the main campus of the Obafemi Awolowo University (OAU), Ile-Ife, Nigeria, but the public health implications of faecal contamination and pollution by these flying mammals is unknown. This study characterized S. aureus obtained from faecal samples of these migratory mammals with a view to determining the clonal types of the isolates, and to investigate the possibility of these flying animals as potential reservoir for zoonotic S. aureus infections.
Results
One hundred and seven (107) S. aureus isolates were recovered from 560 faecal samples in eleven roosting sites from January 2008 to February 2010. A large proportion of the isolates were susceptible to antibiotics, and molecular characterization of 70 isolates showed that 65 (92.9%) were assigned in coagulase type VI, while accessory gene typing classified 69 isolates into the following: type I (12; 17.1%), type II (3; 4.3%), type III (1; 1.4%) and type IV (53; 75.7%). On the whole, the isolates were grouped in five (A-E) main genotypes. Of the ten representative isolates selected for multilocus sequence typing (MLST), nine isolates were assigned with new sequence types: ST1725, ST1726, ST1727, ST2463-ST2467 and ST2470. Phylogenetic analysis provided evidence that S. aureus isolates in group C were closely related with ST1822 and associated clones identified in African monkeys, and group D isolates with ST75, ST883 and ST1223. The two groups exhibited remarkable genetic diversity compared to the major S. aureus clade.
Conclusions
Antibiotic resistance in faecal S. aureus isolates of E. helvum is low and multiple unique S. aureus lineages co-existed with E. helvum. The Straw-Coloured Fruit Bat in Ile-Ife, Nigeria is colonized predominantly by ST1725, ST1726, ST2463 and ST2470 with distinct genotypic characteristics that are rarely found in humans. This study has demonstrated on the possible existence of a reservoir of indigenous and anciently-divergent S. aureus clones among mammals in Africa.
Similar content being viewed by others

Background
Bats (Order: Chiroptera) are the only mammals capable of true sustainable flight and one of the most diverse and species rich mammals on earth [1]. They assist in the regulation of insect populations in their habitats, pollination of flowers and dispersal of seeds of economically important tress, and these ecological roles support forest regeneration and maintenance [2]. However, they roost near human habitation and their association with emerging infections has increased attention on these flying mammals as vectors of zoonotic pathogens [3–5]. The bat species Eidolon helvum is grouped under the suborder Megachiroptera, and it is the most widely distributed Straw-Coloured Fruit Bat which is found in the forest and savannah zones of sub-Saharan Africa [6, 7]. The prime habitats for E. helvum are the tropical forest and typically roost in colonies on tall trees like Eucalyptus saligna and Cocos nucifera[8].
Staphylococcus aureus is part of the normal flora of the skin and mucous membrane of a wide variety of mammals and birds, and recent studies have indicated that animals could be a source of S. aureus infections in humans [9–11]. The main campus of the Obafemi Awolowo University, Ile-Ife (OAU) Nigeria, is colonized by a large population of E. helvum[12, 13], but faecal contamination and pollution of the environment by these migratory mammals is a problem, moreover, the public health implications of their activities are not known. This study characterized S. aureus obtained from faecal samples of bats that colonize the main campus of the institution, with a view to understanding the clonal nature and diversity of the isolates, and to determine the possible risk of dissemination of S. aureus from bats to humans in the community through faecal shedding.
Results and Discussion
A total of 107 S. aureus isolates were obtained from 560 faecal samples of E. helvum based on phenotypic identification. Moreover, they were all genotypically confirmed by hsp60 partial sequencing, and there was excellent agreement between the phenotypic and molecular methods in the identification of the isolates. The number of samples and S. aureus isolates in each sampling site are indicated in Figure 1. Antibiotic susceptibility testing is paramount for monitoring resistance in commensal bacteria and various pathogens of clinical importance. In this study, all the isolates were susceptible to oxacillin, cefoxitin, tetracycline, chloramphenicol, gentamicin and mupirocin. However, four (3.7%) isolates were resistant to penicillin, while six (5.6%) and eight (7.4%) isolates were resistant to ciprofloxacin and erythromycin, respectively. None of the isolates exhibited inducible resistance however, 3.7% were constitutively resistant to clindamycin (Table 1). Studies have reported faecal carriage of methicillin-resistant S. aureus (MRSA) in animals [14, 15]. However, MRSA was not detected in this study which is similar to recent reports on analysis of faecal samples from swine and feedlot cattle [16, 17]. The low rate of resistance to different classes of antibiotics observed among the isolates in this study suggests that these migratory mammals may not have been exposed to the selective pressure of antimicrobial agents.

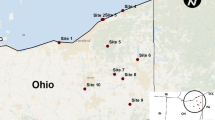
Map of Obafemi Awolowo University (OAU) campus showing the sampling site/roosting habitat of the Straw-Coloured Fruit Bat ( E. helvum ). The number of samples (in each site) and S. aureus isolates (in parenthesis) are indicated.
Molecular typing has been useful in understanding the epidemiology of S. aureus from animal and human hosts [18]. S. aureus is highly clonal in nature and though some are exclusively adapted to specific hosts [19], others are able to colonize multiple hosts [20–22]. Of the 107 S. aureus isolates, 70 (representing isolates obtained from faecal samples in the various sites) were randomly selected and further characterized. All the isolates were PVL-negative and 65 (92.9%) were grouped with coagulase (coa) type VI, but 5 (7.1%) were non-typeable. The accessory gene regulator (agr) typing classified 69 of the 70 isolates into the following: type I (12; 17.1%), type II (3; 4.3%), type III (1; 1.4%) and type IV (53; 75.7%). Based on their genotypic characteristics, ten representative isolates were selected for MLST and nine new sequence types: ST1725, ST1726, ST1727, ST2463-ST2467 and ST2470 were identified, and the sequences for the housekeeping genes have been deposited in the MLST database (http://www.mlst.net), while one representative isolate (Q22) was assigned with ST15. Overall, the 70 isolates were assigned into five main genotypes A to E (Table 2).
As shown in Figure 2, there was a clear phylogenetic out-group among the S. aureus taxon consisting of isolates in the hsp60-allele types C and D, which suggests that these genotypes diverged long before clones belonging to the major S. aureus clades exhibited the current size of genetic divergence. Moreover, based on concatenated sequences of seven genes used in MLST, isolates in hsp60-allele type C were closely related with S. aureus ST1822 and associated clones, and type D isolates with ST75, ST883 and ST1223 (Figure 3). We have tentatively designated these isolates as anciently-diverged S. aureus. Some studies had previously reported that divergent S. aureus ST75 (agr type I) and ST883 (agr type IV) originated in northern Australia, while ST1223-related clones were found in South East Asia [23–25]. Moreover, S. aureus isolates assigned with ST1822-related clones have been identified in African monkeys [26]. In this study, we identified divergent clones (ST2463-ST2467, ST2470) among Straw-Coloured Fruit Bats in Nigeria, which suggests that anciently-diverged S. aureus have not only been distributed in Australia and South East Asia, but also among mammals in Africa. These lineages evolved independently from major S. aureus populations over an extended period of time, and may be a new subspecies of S. aureus. A recent study had reported that chromosomal recombination had occurred at coa and agr loci at a uniform rate [27]. Therefore, it is difficult to identify the prototype of these genes. The agr type I or IV and the coa type VI, which were found most frequently in the anciently-diverged S. aureus isolates, may be the closest relation to the origin of agr and coa genes, respectively.

Phylogenetic tree based on hsp60 partial sequences of 70 S. aureus isolates from E. helvum. This tree was constructed by the neighbor-joining method, using MEGA ver. 5.05.

Phylogenetic tree based on concatenated arcC, aroE, glpF, gmk, pta, tpi and yqiL sequences of representative S. aureus isolates (F10, AC19, R5, AC10, F9, P1, Q15, R3, F16 and Q22). This tree was constructed by the neighbor-joining method, using MEGA ver. 5.05.
Conclusions
This study isolated S. aureus from faecal samples of E. helvum, a migratory mammal with an abundant population in OAU, Ile-Ife, Nigeria, and represents the first molecular study on S. aureus colonization of bats in Africa. The isolates were largely susceptible to a number of antibiotics. The combination of coagulase gene type VI and agr type IV are rare among S. aureus isolates associated with humans [28–31], and the evidence that isolates in group C were closely related with divergent ST1822-related clones identified in African monkeys, and group D isolates with ST75, ST883 and ST1223 indicate that there is the possible existence of a reservoir of indigenous and anciently-diverged clones among mammals in Africa.
Methods
Sample sites
A total of eleven roosting sites located in the academic area and the students’ hostel in OAU, Ile-Ife were identified for the study (Figure 1), and the duration for sample collection was from January 2008 to September 2008, February to May 2009, and February 2010. The faecal samples were obtained once a month in a designated sampling site between 6-7am by a non-invasive method in which three sterilized piece (36 × 45 inches) of cotton material were spread under the roosting trees. Fresh faecal samples were collected with sterile swab sticks and conveyed promptly to the Department of Microbiology Laboratory (OAU) for microbiological analysis.
Isolation and identification of S. aureusisolates
The swab stick was inserted into a test tube containing 3 ml of sterile nutrient broth (Biolab, supplied by Merck, Johannesburg, South Africa), swirled briefly to discharge the contents into the medium, and the culture was incubated at 37°C overnight. Thereafter, a loopful was streaked on mannitol salt agar (MSA) (Biolab, supplied by Merck, Johannesburg, South Africa) and incubated at 37°C for 48 hours. Preliminary identification of S. aureus was based on positive Gram stain, and positive results for catalase, coagulase (tube method) and DNase tests. The procedure described previously [32] was employed for DNA isolation. In summary, a single colony was suspended to a McFarland 1.0 standard in 100 μl of TE buffer (10 mM Tris, 1 mM EDTA, pH 8.0) with 10 U of achromopeptidase (Wako Chemical, Co. Ltd.), and the suspension was incubated at 55°C for 10 min. The supernatant was used as crude DNA for PCR. Molecular identification and confirmation of the isolates was based on sequencing analysis of the hsp60 gene as previously reported [33]. PCR products were sequenced by using a Big Dye Terminator (version 3.1) cycle sequencing kit (Applied Biosystems, Foster City, CA) with an ABI Prism 3100 genetic analyzer (Applied Biosystems).
Antibiotic susceptibility testing
The susceptibility testing of the isolates to 11 antibiotics was performed using the disk diffusion method and the following antibiotics were tested: penicillin (10 units), oxacillin (1 μg), cefoxitin (30 μg), erythromycin (15 μg), clindamycin (2 μg), tetracycline (30 μg), ciprofloxacin (5 μg), chloramphenicol (30 μg), fusidic acid (10 μg) gentamicin (10 μg) and mupirocin (5 μg and 200 μg). S. aureus ATCC 25923 was the control strain for the susceptibility testing. The result was interpreted as resistant or susceptible based on the interpretative standard according to the Clinical Laboratory Standards Institute (CLSI) manual for bacterial isolates from animals [34]. Interpretative zone diameter for resistance and susceptibility breakpoints to fusidic acid and mupirocin which are not stated in the CLSI guidelines were considered as described previously [35, 36]. The D-test for determining inducible resistance of clindamycin using erythromycin was performed. A truncated or blunted clindamycin zone of inhibition (D-Shape) indicated inducible resistance. Constitutive resistance was recognized by a clindamycin zone diameter of ≤14 mm [37].
Molecular characterization of the S. aureusisolates
Characterization of 70 isolates was determined by detection of the Panton Valentine Leukocidin (PVL) gene [38], agr[39] and coa gene typing [40]. The MAFFT program was used for multiple alignment of the hsp60 partial sequences, and a phylogenetic tree was constructed by the neighbor-joining and bootstrap methods, using MEGA ver. 5.05 [41]. Furthermore, MLST [42] was carried out on representative S. aureus isolates (based on hsp60 allelic type, coagulase and agr typing). The amplified PCR products were sequenced, and STs were determined for each isolate based on the alleles identified at each of the seven loci using the S. aureus MLST database (http://www.mlst.net). For six representative isolates (AC10, F9, P1, F16, Q15 and R13), we were unable to amplify the aroE and or glpF genes using the standard MLST primers. Therefore degenerate primers CC75dege-aroE-F (5’-WTGCAGTWATHGGWRRYCC-3’), CC75dege-aroE-R (5’-GGWWTATAAAYAATRT CACT-3’), CC75aroEseq-F (5’-CCAATTGAGCATTCYTTATC-3’), CC75dege-glpF-F (5’-GCWGAATTYHT DGGWACWGC-3’), CC75dege-glpF-R (5’-ATWGGYA AWATHGCATGWGC’), and CC75glpF-seq-R (5’-GCAT GTGCAATTCTTGGDC’), were designed by multiple alignment of amino acid sequences of each gene with complete genomes of S. aureus, S. epidermidis, S. haemolyticus and S. lugdunensis from the KEGG database (http://www.genome.jp/kegg/). Sequences of arcC, aroE, glpf, gmk, pta, tpi and yqiL in S. simiae, which was used as an outgroup, were obtained from the draft genome sequence of S. simiae CCM7213 [43]. A phylogenetic tree was constructed based on concatenated arcC, aroE, glpF, gmk, pta, tpi and yqiL sequences using the neighbor-joining method, using MEGA ver. 5.05.
Abbreviations
- OAU:
-
Obafemi Awolowo University
- PVL:
-
Panton Valentine Leukocidin
- Agr:
-
Accessory gene regulator
- Coag:
-
Coagulase
- MLST:
-
Multilocus sequence typing
- ST:
-
Sequence type
- E. helvum :
-
Eidolon helvum
- S. aureus :
-
Staphylococcus aureus
- MSA:
-
Mannitol salt agar
- DNase:
-
Deoxyribonuclease
- CLSI:
-
Clinical Laboratory Standards Institute
- MRSA:
-
Methicillin resistant Staphylococcus aureus.
References
Eick GN, Jacobs DS, Matthee CA: A Nuclear DNA Phylogenetic Perspective on the Evolution of Echolocation and Historical Biogeography of Extant Bats (Chiroptera). Mol Biol Evol. 2005, 22: 1869-1886. 10.1093/molbev/msi180.
Mildenstein T, de Jong C: Natural history, ecology and socio-economic value of bats. Investigating the Role of Bats in Emerging Zoonoses: Balancing Ecology, Conservation and Public Health Interest. Edited by: Newman SH, Field HE, Jong CE, Epstein JH. 2011, Rome: FAO Animal Production and Health Manual No. 12, 15-28.
Hayman DTS, Suu-Ire R, Breed AC, McEachern JA, Wang L, Wood JLN, Cunningham AA: Evidence of henipavirus infection in West Africa Fruit Bats. PLoS One. 2008, 23: e2739-
Mühldorfer K, Wibbelt G, Haensel J, Riehm J, Speck S: Yersinia species isolated from Bats, Germany. Emerg Infect Dis. 2010, 16: 578-580. 10.3201/eid1603.091035.
Drexler JF, Corman VM, Müller MA, Maganga GD, Vallo P, Binger T, Gloza-Rausch F, Rasche A, Yordanov S, Seebens A, Oppong S, Adu Sarkodie Y, Pongombo C, Lukashev AN, Schmidt-Chanasit J, Stöcker A, Carneiro AJ, Erbar S, Maisner A, Fronhoffs F, Buettner R, Kalko EK, Kruppa T, Franke CR, Kallies R, Yandoko ER, Herrler G, Reusken C, Hassanin A, Krüger DH, Matthee S, Ulrich RG, Leroy EM, Drosten C: Bats host major mammalian paramyxoviruses. Nat Commun. 2012, 3: 396-
DeFrees SL, Wilson DE: Eidolon helvum. Mamm Species. 1988, 312: 1-5.
Mickleburgh SP, Hutson AM, Racey PA: Old World fruit bats. An action plan for their conservation. 1992, Gland, Switzerland: IUCN
Jones C: Comparative ecology of three pteropid bats in Rio Muni, West Africa. J Zool. 1972, 167: 353-370.
van Cleef BAGL, Monnet DL, Voss A, Krziwanek K, Allerberger F, Struelens M, Zemlickova H, Skov RL, Vuopio-Varkila J, Cuny C, Friedrich AW, Spiliopoulou I, Pászti J, Hardardottir H, Rossney A, Pan A, Pantosti A, Borg M, Grundmann H, Mueller-Premru M, Olsson-Liljequist B, Widmer A, Harbath S, Schweiger A, Unal S, Kluytmans JA: Livestock-associated methicillin-resistant Staphylococcus aureus in humans, Europe. Emerg Infect Dis. 2011, 17: 502-505. 10.3201/eid1703.101036.
van der Mee-Marquet N, François P, Domelier-Valentin AS, Coulomb F, Decreux C, Hombrock-Allet C, Lehiani O, Neveu C, Ratovohery D, Schrenzel J, Roland Q, Bloodstream Infection Study Group of Réseau des Hygiénistes du Centre (RHC): Emergence of unusual bloodstream infections associated with pig-borne like Staphylococcus aureus ST398 in France. Clin Infect Dis. 2011, 52: 152-153. 10.1093/cid/ciq053.
Mediavilla JR, Chen L, Uhlemann AC, Hanson BM, Rosenthal M, Stanak K, Koll B, Fries BC, Armellino D, Schilling ME, Weiss D, Smith TC, Lowy FD, Kreiswirth BN: Methicillin-susceptible Staphylococcus aureus ST398, New York and New Jersey, USA. Emerg Infect Dis. 2012, 18: 700-702. 10.3201/eid1804.111419.
Fayenuwo JO, Halstead LB: Breeding cycle of straw-colored fruit bat, Eidolon helvum at Ile-Ife, Nigeria. J Mammal. 1974, 55: 453-454. 10.2307/1379016.
Okon EE: Fruit bats at Ife: their roosting and food preferences (Ife Fruit Bat project No. 2). Nig Field. 1975, 39: 33-40.
Simonová M, Fotta M, Lauková A: Characteristics of Staphylococcus aureus isolated from rabbits. Folia Microbiol (Praha). 2007, 52: 291-296. 10.1007/BF02931312.
Sherein IA, Ahmed FY, Omaima HE: Staphylococcus aureus - A cause of fatal toxic shock syndrome in Egyptian horses (first record). Nature and Science. 2009, 7: 79-87.
Baba K, Ishihara K, Ozawa M, Tamura Y, Asai T: Isolation of methicillin-resistant Staphylococcus aureus (MRSA) from swine in Japan. Int J Antimicrob Agents. 2010, 36: 352-354. 10.1016/j.ijantimicag.2010.06.040.
Weese JS, Hannon SJ, Booker CW, Gow S, Avery BP, Reid-Smith RJ: The Prevalence of Methicillin-Resistant Staphylococcus aureus Colonization in Feedlot Cattle. Zoonoses Public Health. 2012, 59: 144-147. 10.1111/j.1863-2378.2011.01428.x.
Fitzgerald JR: Livestock-associated Staphylococcus aureus: origin, evolution and public health threat. Trends Microbiol. 2012, 20: 192-198. 10.1016/j.tim.2012.01.006.
Smith EM, Green LE, Medley GF, Bird HE, Fox LK, Schukken YH, Kruze JV, Bradley AJ, Zadoks RN, Dowson CG: Multilocus sequence typing of intercontinental bovine Staphylococcus aureus isolates. J Clin Microbiol. 2005, 43: 4737-4743. 10.1128/JCM.43.9.4737-4743.2005.
Lowder BV, Guinane CM, Ben Zakour NL, Weinert LA, Conway-Morris A, Cartwright RA, Simpson AJ, Rambaut A, Nübel U, Fitzgerald JR: Recent human-to-poultry host jump, adaptation, and pandemic spread of Staphylococcus aureus. Proc Natl Acad Sci U S A. 2009, 106: 19545-19550. 10.1073/pnas.0909285106.
Cuny C, Friedrich A, Kozytska S, Layer F, Nübel U, Ohlsen K, Strommenger B, Walther B, Wieler L, Witte W: Emergence of methicillin-resistant Staphylococcus aureus (MRSA) in different animal species. Int J Med Microbiol. 2010, 300: 109-117. 10.1016/j.ijmm.2009.11.002.
Guinane CM, Ben Zakour NL, Tormo-Mas MA, Weinert LA, Lowder BV, Cartwright RA, Smyth DS, Smyth CJ, Lindsay JA, Gould KA, Witney A, Hinds J, Bollback JP, Rambaut A, Pendadés JR, Fitzgerald JR: Evolutionary genomics of Staphylococcus aureus reveals insight into the origin and molecular basis of ruminant host adaptation. Genome Biol Evol. 2010, 2: 454-466. 10.1093/gbe/evq031.
Ng JWS, Holt DC, Lilliebridge RA, Stephens AJ, Huygens F, Tong SYC, Currie BJ, Giffard PM: Phylogenetically Distinct Staphylococcus aureus lineage prevalent among indigenous communities in Northern Australia. J Clin Microbiol. 2009, 47: 2295-2300. 10.1128/JCM.00122-09.
Monecke S, Kanig H, Rudolph W, Müller E, Coombs G, Hotzel H, Slickers P, Ehricht R: Characterisation of Australian MRSA Strains ST75- and ST883-MRSA-IV and Analysis of Their Accessory Gene Regulator Locus. PLoS One. 2010, 5: e14025-10.1371/journal.pone.0014025.
Ruimy R, Armand-Lefevre L, Barbier F, Ruppé E, Cocojaru R, Mesli Y, Maiga A, Benkalfat M, Benchouk S, Hassaine H, Dufourcq JB, Nareth C, Sarthou JL, Andremont A, Feil EJ: Comparisons between geographically diverse samples of carried Staphylococcus aureus. J Bacteriol. 2009, 191: 5577-5583. 10.1128/JB.00493-09.
Schaumburg F, Alabi AS, Köck R, Mellmann A, Kremsner PG, Boesch C, Becker K, Leendertz FH, Peters G: Highly divergent Staphylococcus aureus isolates from African non-human primates. Env Microbiol Rep. 2012, 4: 141-146. 10.1111/j.1758-2229.2011.00316.x.
Watanabe S, Ito T, Sasaki T, Li S, Uchiyama I, Kishii K, Kikuchi K, Skov RL, Hiramatsu K: Genetic diversity of staphylocoagulase genes (coa): insight into the evolution of variable chromosomal virulence factors in Staphylococcus aureus. PLoS One. 2009, 27: e5714-
Ben Ayed S, Boutiba-Ben Boubaker I, Ennigrou S, Ben Redjeb S: Accessory gene regulator (agr) typing of Staphylococcus aureus isolated from human infections. Arch Inst Pasteur Tunis. 2008, 85: 3-8.
Peerayeh SN, Azimian A, Nejad QB, Kashi M: Prevalence of agr specificity groups among Staphylococcus aureus isolates from University Hospitals in Tehran. LabMedicine. 2009, 40: 27-29.
Hirose M, Kobayashi N, Ghosh S, Paul SK, Shen T, Urushibara N, Kawaguchiya M, Shinagawa M, Watanabe N: Identification of Staphylocoagulase Genotypes I-X and Discrimination of Type IV and V Subtypes by Multiplex PCR Assay for Clinical Isolates of Staphylococcus aureus. Jpn J Infect Dis. 2010, 63: 257-263.
van den Berg S, van Wamel WJB, Snijders SV, Ouwerling B, de Vogel CP, Boelens HA, Willems RJL, Huijsdens XW, Verreck FAW, Kondova I, Heidt PJ, Verbrugh HA, van Belkum A: Rhesus Macaques (Maca mulatta) are natural hosts of specific Staphylococcus aureus lineages. PLoS One. 2011, 6: e26170-10.1371/journal.pone.0026170.
Sasaki T, Tsubakishita S, Tanaka Y, Sakusabe A, Ohtsuka M, Hirotaki S, Kawakami T, Fukata T, Hiramatsu K: Multiplex-PCR method for species identification of coagulase-positive staphylococci. J Clin Microbiol. 2010, 48: 765-769. 10.1128/JCM.01232-09.
Kwok AYC, Chow AW: Phylogenetic study of Staphylococcus and Macrococcus species based on partial hsp60 gene sequences. Int J Sys Evol Microbiol. 2003, 53: 87-92. 10.1099/ijs.0.02210-0.
Clinical and Laboratory Standards Institute (CLSI): CLSI document M31-A3. Performance Standards for Antimicrobial Disk and Dilution Susceptibility Tests for Bacteria Isolated From Animals; Approved Standard—Third Edition. 2009, 65-72.
Skov R, Frimodt-Møller N, Espersen F: Correlation of MIC methods and tentative interpretive criteria for disk diffusion susceptibility testing using NCCLS methodology for fusidic acid. Diagn Microbiol Infect Dis. 2001, 40: 111-116. 10.1016/S0732-8893(01)00262-0.
Udo EE, Farook VS, Mokadas EM, Jacob LE, Sanyal : Molecular fingerprinting of mupirocin-resistant Staphylococcus aureus from a Burn unit. Int J Infect Dis. 1999, 3: 82-87. 10.1016/S1201-9712(99)90014-0.
Fiebelkorn KR, Crawford SA, McElmeel ML, Jorgensen H: Practical disk diffusion method for detection of inducible clindamycin resistance in Staphylococcus aureus and coagulase-negative staphylococci. J Clin Microbiol. 2003, 41: 4740-4744. 10.1128/JCM.41.10.4740-4744.2003.
Lina G, Piemont Y, Godail-Gamot F, Bes M, Peter MO, Gauduchon V, Vadenesch F, Etienne J: Involvement of Panton-Valentine leukocidin-producing Staphylococcus aureus in primary skin infections and pneumonia. Clin Infect Dis. 1999, 29: 1128-1132. 10.1086/313461.
Shopsin B, Mathema B, Alcabes P, Said-Salim B, Lina G, Matsuka A, Martinez J, Kreiswirth BN: Prevalence of agr specificity groups among Staphylococcus aureus strains colonizing children and their guardians. J Clin Microbiol. 2003, 41: 456-459. 10.1128/JCM.41.1.456-459.2003.
Sakai F, Takemoto A, Watanabe S, Aoyama K, Ohkubo T, Yanahira S, Igarashi H, Kozaki S, Hiramatsu K, Ito T: Multiplex PCRs for assignment of Staphylocoagulase Types and Subtypes of Type VI Staphylocoagulase. J Microbiol Meth. 2008, 75: 312-317. 10.1016/j.mimet.2008.07.003.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance and Maximum Parsimony Methods. Mol Biol Evol. 2010, 28: 2731-2739.
Enright MC, Day NP, Davies CE, Peacock SJ, Spratt BG: Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J Clin Microbiol. 2000, 38: 1008-1015.
Suzuki H, Lefébure T, Bitar PP, Stanhope MJ: Comparative genomic analysis of the genus Staphylococcus including Staphylococcus aureus and its newly described sister species Staphylococcus simiae. BMC Genomics. 2012, 13: 38-10.1186/1471-2164-13-38.
Acknowledgments
We acknowledge the comments and suggestions of Professor Iruka Okeke in the preparation of the manuscript, and the kind assistance of Professor Johnson Lin, Dr. Stella Smith and Dr. Solayide Shittu.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
AS, OA, TS conceived the study, BA conducted the sample collection, preliminary identification and susceptibility testing of the isolates; TS carried out the molecular characterization. All authors read and approved the final version of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Akobi, B., Aboderin, O., Sasaki, T. et al. Characterization of Staphylococcus aureus isolates from faecal samples of the Straw-Coloured Fruit Bat (Eidolon helvum) in Obafemi Awolowo University (OAU), Nigeria. BMC Microbiol 12, 279 (2012). https://doi.org/10.1186/1471-2180-12-279
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-12-279