
Abstract
Background
Mycobacteria can be quickly and simply identified by PCR restriction-enzyme analysis (PRA), but misidentification can occur because of similarities in band sizes that are critical for discriminating among species. Capillary electrophoresis can provide computer-aided band discrimination. The aim of this research was to develop an algorithm for identifying mycobacteria by combined rpo B duplex PRA (DPRA) and hsp65 PRA with capillary electrophoresis.
Results
Three hundred and seventy-six acid-fast bacillus smear-positive BACTEC cultures, including 200 Mycobacterium tuberculosis complexes (MTC) and 176 non-tuberculous mycobacteria (NTM) were analyzed. With combined hsp65 and rpo B DPRA, the accuracy rate was 100% (200 isolates) for the MTC and 91.4% (161 isolates) for the NTM. Among the discordant results (8.6%) for the NTM, one isolate of Mycobacterial species and an isolate of M. flavescens were found as new sub-types in hsp65 PRA.
Conclusions
This effective and novel identification algorithm using combined rpo B DPRA and hsp65 PRA with capillary electrophoresis can rapidly identify mycobacteria and find new sub-types in hsp65 PRA. In addition, it is complementary to 16 S rDNA sequencing.
Similar content being viewed by others

Background
Detection and identification of mycobacteria in clinical specimens is a key issue in the therapy of pulmonary diseases because misidentification can lead to inappropriate treatment. Traditionally, mycobacterial species are identified based on their growth rate, presence or absence of pigmentation, and using biochemical assays of the isolates recovered from specimens. The biochemical assays are time-consuming and labor-intensive, usually taking 1 to 2 months to complete, and assays for non-tuberculous mycobacteria (NTM) species can have poor reproducibility and provide ambiguous results[1, 2].
By contrast, molecular identification, notably PCR-restriction enzyme analysis (PRA), is rapid and simple. The hsp65 PRA method, developed by Telenti et al. in 1993, is a popular DNA-based method for mycobacteria identification[3]. Using hsp65 PRA, Wong et al.[4] reported 100% sensitivity and specificity in identifying Mycobacterium tuberculosis complexes but only 74.5% sensitivity in identifying NTM species. This misidentification may occur because of similarities in band sizes that are critical for species discrimination[3]. An additional contributing factor is a lack of knowledge of all existing PRA profiles, especially among species that are very heterogeneous, such as M. gordonae, M. scrofulaceum, and M. terrae complexes. Recently, capillary electrophoresis (CE) with computer analysis[5–9] has provided more precise band discrimination than analysis by the naked eye.
Previously, we developed an algorithm for mycobacterial species identification from acid-fast bacillus (AFB) smear-positive BACTEC tubes by combining the rpo B duplex PRA (DPRA)[10] and the key phenotypic characters of mycobacteria recovered from the tubes[11]. Using rpo B DPRA, we differentiated Mycobacterium tuberculosis complexes (MTC) from NTM with 235 base pair (bp) and 136 bp PCR amplicons in AFB smear-positive BACTEC cultures. The 136 bp rpo B duplex PCR amplicon was further digested with Msp I and Hae III (rpo B DPRA) to divide the NTM species into eight easily distinguishable groups (A–H) as described by Kim et al.[10]. Using two phenotypic characters (growth rate and photoreactivity on pigment production) and two simple biochemical assays (nitrate reduction test and Tween 80 hydrolysis test)[11], the mycobacterial species were identified. However, the sub-culture and biochemical tests for this algorithm took three weeks.
In the present study, we developed a rapid and effective algorithm for identification of mycobacteria by combined rpo B DPRA and hsp65 PRA with CE.
Results
Mycobacteria identification
There were 376 AFB smear-positive BACTEC culture tubes (positive BACTEC cultures), including 200 MTC and 176 NTM-containing BACTEC cultures. A further 20 bacteria were MGIT positive but AFB culture smear negative, and these were classed as contaminated and excluded from subsequent evaluation.
By rpo B duplex PCR, all of the 200 MTC-containing BACTEC cultures and the 176 NTM-containing BACTEC cultures showed 235-bp and 136-bp PCR amplicons specific for MTC and NTM, respectively. The species were identified according to the flow chart shown in Figure1.

An flow chart of the identification of Mcobacterial species from clinical specimens by combined rpo B duplex PCR(DCR) and hsp 65 PCR-Restriction Fragment Length Polymorpgism analysis(PRA).
Concordant results from rpo B DPRA and hsp65 PRA
Combining rpo B DPRA and hsp65 PRA with computer-aided CE gave an accuracy rate of 100% (200/200) for MTC and 91.4% (161/176) for NTM (Table1).
Discordant results from rpo B DPRA and hsp65 PRA
There were 15 isolates (8.6%) of NTM with discordant results with rpo B DPRA and hsp65 PRA (Table2). The two isolates, Mycobacterial species (A group) and M. flavescens (A group) identified by 16 S rDNA sequencing represented new patterns not available in the hsp 65 PRA databases and might be new sub-types in hsp65 PRA. For Mycobacterial species, 16 S rDNA sequencing did not confirm the identity of the isolate but conventional biochemical identification showed it was M. mucogenicum.
Development of a species identification algorithm
The results in Tables1 and2 and the mycobacterial identification flow chart (Figure1) were used to develop a species identification algorithm by combining rpo B duplex PCR[10] and hsp65 PRA[3] using the most common 74 patterns of 40 species in Table3. In this algorithm (Table3), we added M. gordonae types 3 and 4 to the F group. The accuracy rate for NTM by combining the two methods could reach 96.6% (170/176).
Discussion
Mycobacterial species identification is generally achieved by standard culture and biochemical methods[12]. Biochemical methods are labor-intensive and time-consuming, and not all are reproducible[2, 13]. Recently, PRA and DPRA have been developed for molecular identification of mycobacterial species using different regions of hsp65, 16 S rDNA, 16 S-23 S rDNA spacer, dnaJ, and rpo B as an amplification target[3, 14–17]. The most common method is hsp65 PRA, and 74 patterns for 40 species are available in the PRASITE database (http://app.chuv.ch/prasite/ index.html).
Previous studies[18, 19] have reported that hsp65 PRA is faster and more accurate for species identification than conventional (phenotypic or biochemical) testing. This is because more incorrect and ambiguous results are obtained with conventional methods. The results in our study (Tables1 and2) also support this finding. Incorrect and ambiguous results are caused by phenotypic homogeneity among different species and phenotypic variability within species[18]. With by hsp65 PRA, some sub-species, such as M. kansasii, can be identified and rapid-growing mycobacterium can be divided into M. abscessus and M.chelonae, M. fortuitum and M. smegmatis[20], whereas these identifications are difficult with conventional methods[21]. As found in our study (Tables1 and2), M. peregrinum was identified as M. fortuitum and M. avium subsp. avium and M. intracellulare were both identified as M. avium complex by the conventional biochemical method.
However, hsp65 PRA limitations have been reported in some articles[22, 23]. Failure to identify or incorrect identification of the species may occur because of similarities in band sizes critical for discriminating species, including difficult to distinguish M. tuberculosis complex (M. tuberculosis and M. bovis)[22], and closely related sub-species such as M. avium or M. gordonae, because of sequence heterogeneity[22]. In addition, technical problems can also cause misinterpretation or incorrect identification[23]. Patterns in PRA profiles are complex and difficult to interpret with the naked eye, especially when more detailed sub-types are included[21].
This study combined rpo B DPRA and hsp65 PRA to test both reference strains and clinical respiratory isolates. The mycobacterial identification flow chart (Figure1) can identify species to the sub-species level, and final species identification can be obtained instantly with concordant results from the two PRA. M. gordonae has a highly variable gene sequence with 10 sub-types in hsp65 PRA, and there are two groups (G and F) in rpoB DPRA. Most M. gordonae is in the G group, but M. gordonae types 3 and 4 by hsp65 PRA are in the F group (Tables1 and2).
In addition, there were different rpo B DPRA results (Table2) for M. simaie type 5 (G group but not E group), M. scrofulaceum type 1 (D group but not H group), and M. intracellulare type 3 (F group but not G group). The identities of all of these isolates were finally confirmed by 16 S rDNA sequencing. Variable numbers of restriction sites for Hae III in these species may derive from genetic sequence mutation. However, these species are included in the species identification algorithm even though they are uncommon isolates.
Using the mycobacteria identification flow chart (Figure1) and algorithm (Table3), M. avium-intracellulare complex (MAC) can be easily divided into M. avium spp. avium and M. intracellulare by both rpo B DPRA and hsp65 PRA. By contrast, this was not possible with the conventional method. Using the results in Table3, some NTM species with identical or similar hsp65 PRA can be clearly grouped by rpoB DPRA (Table4). Ambiguous results from hsp65 PRA alone are easier to interpret with combined rpo B DPRA and hsp65 PRA. However, M. intermedium type 1 and M. intracellulare type 3 with identical hsp65 PRA and rpo B DPRA (G group) could not be differentiated further by this species identification algorithm and required 16 S rDNA sequencing for confirmation.
Although 16 S rDNA sequencing is the standard method for mycobacterium species identification, it cannot differentiate some closely related rapid-growing mycobacterium species[24] or slow-growing M. kansasii and M. gastri that had identical 16 S rDNA sequences, but these can be differentiated by hsp65 PRA and rpo B DPRA. There are some reports[6, 25] of conflicting results from different methods for mycobacterial species identification, probably caused by a failure of one method to identify all test strains correctly. Combining methods for mycobacterial species identification can improve the accuracy rate, avoid ambiguous results, and save time.
Many CE-based studies[5–9] in PCR-RFLP analysis have investigated improving band size discrimination. In one study by Chang et al.[7], high-resolution CE gave more precise estimates of DNA fragment sizes than analysis by the naked eye, and CE could detect low molecular weight fragments (down to 12 bp). In our study, restriction fragments <50 bp were not selected for analysis to avoid confusion with primer and primer dimer bands. We found that more hsp 65 fragment differences than rpo B fragment (data not shown) may explain the size differences with highly variable sequence for species identification but difficult interpretation in hsp65 PRA.
Some sub-types of NTM species are relevant to clinical management, such as the M. kansasii and MAC. M. kansasii type 1 is the most common type associated with human disease[26–28] because of its high pathogenicity. However, M. kansasii types 3–7 are most often isolated from the environment and rarely from humans, and have no significant role in clinical management[26, 27]. MAC can be divided into M. avium subsp. avium and M. intracellulare because drug sensitivity test and clinical outcomes are different between these two sub-types[29, 30].
It is important to identify NTM to the sub-type level both for epidemiologic data and for differentiating potentially pathogenic sub-types[26, 27]. Combined rpo B DPRA and hsp65 PRA with capillary electrophoresis provides precise species identification and overcomes problems associated with discrimination by hsp65 PRA band sizes. This combined method takes around 2–3 days to complete in the laboratory once clinical isolates have been received. However, the identification algorithm has some limitations. First, it could not discriminate M. intermedium type 1 from M. intracellulare type 3, and second, not every hospital laboratory will be equipped with the appropriate equipment for this method.
Conclusion
In conclusion, the novel flow chart and identification algorithm obtained by combined rpo B DPRA and hsp65 PRA with capillary electrophoresis can easily differentiate MTC from NTM and identify mycobacterial species to the sub-type level, which is helpful for clinical management. The results are complementary to 16 S rDNA sequencing, and the effective algorithm provides rapid and accurate mycobacterial species identification.
Methods
Mycobacterial isolates
Fourteen mycobacterial reference strains including one MTC and 13 NTM strains and 376 clinical respiratory specimens, including sputum, broncho-alveolar lavage, and aspirated secretion from endotracheal tubes, were collected from January to July 2007 from Taichung Veterans General Hospital (Taichung, Taiwan). The respiratory specimens were digested by a N-acetyl-l-cysteine-NaOH decontamination procedure, centrifugal concentration, and sputum dissolving agents[31]. The processed specimens or the concentrated specimens were inoculated into MGIT culture tubes and incubated in the BACTEC 960 instrument at 37°C until a positive signal appeared. Positive BACTEC cultures were smeared on glass slides and Kinyoun staining was used to screen for AFB[31]. Mycobacteria in the positive BACTEC cultures were isolated and identified by conventional methods[12, 13] .
Identification of mycobacterial isolates by conventional methods
All 376 isolates from cultures in the AFB smear-positive BACTEC tubes were streaked on Löwenstein-Jensen (LJ) slants and incubated at 37°C under 5% CO2. Colonies on the LJ slants were used for species identification by conventional culture and biochemical methods[12, 13]. These methods included growth rates, photoreactivity for pigment production, morphology in microcolonies on LJ slants, and biochemical tests, including nitrate reduction, arylsulfatase, Tween 80 hydrolysis, urease, semiquantitative catalase, tolerance to 5% NaCl and niacin production.
Genomic DNA extraction
Mycobacterial DNA was extracted from positive BACTEC cultures using a DTB specimen processing kit (Becton Dickinson, Franklin Lakes, NJ) according to the manufacturer’s instructions[11].
rpoB DPCR and rpoB DPRA
The rpo B DPCR was performed using genomic DNA as template and primer pairs Tbc1 (5’-CGTACGGTCGGCGAGCTGATCCAA-3’)-TbcR5 (5’-CCACCAGTCGGCGCTTGTGGGTCAA-3’) and M5 (5’-GGAGCGGATGACCACCCAGGACGTC-3’)-RM3 (5’-CAGCGGGTT GTTCTGGTCCATGAAC-3’) as described by Kim et al.[10]. A 235 bp DNA PCR amplicon from MTC and a 136 bp DNA PCR amplicon from NTM were specifically amplified[10], and these two amplification products were analyzed by electrophoresis on a 2% agarose gel (Seakem LE agarose, Cambrex, East Rutherford, NJ).
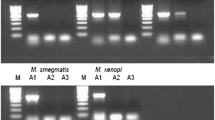
For rpo B DPRA, the 136-bp DNA PCR amplicon was further digested with Msp I and Hae III after DPRA, and analyzed by electrophoresis on a 3% agarose gel (NuSieve 3:1 agarose, Cambrex) or CE (eGene). The rpo B restriction fragment length polymorphism (RFLP) patterns were compared to eight groups described by Kim et al.[10]. Eight NTM reference strains (M. abscessus ATCC 19977, M. avium subsp. avium ATCC 25291, M. kansasii ATCC 12479, M. terrae ATCC 15755, M. szulgai ATCC 29716, M. intracellulare ATCC 13950, M. scrofulaceum ATCC 19981, M. xenopi ATCC 19250) from each rpo B group (A-H) were subjected to rpo B DPRA by CE (eGene).
hsp65 PCR and hsp65PRA
The hsp65 PCR was performed using genomic DNA as template and primer Tb11(5’-ACC AAC GAT GGT GTG TCC-3’) and Tb12 (5’-CTT GTC GAA CCG CAT ACC CT-3’) as described by Telenti et al.[3]. A 439-bp DNA hsp 65 PCR amplicon was specifically amplified from the extracted DNA, and the amplification product was analyzed by electrophoresis on a 2% agarose gel ( Seakem LE agarose, Cambrex).
For hsp65 PRA, the 439-bp DNA hsp65 PCR amplicon was further digested with BstE II and Hae III after completing hsp65 PCR, and analyzed by electrophoresis on a 3% agarose gel (NuSieve 3:1 agarose, Cambrex) or by CE (eGene). The sizes of the restriction fragment by hsp65 PRA were compared to those reported on the PRASITE database (http://app.chuv.ch/prasite/index.html). Thirteen ATCC NTM reference strains and one MTC reference strain were subjected to hsp65 PRA by CE (eGene).
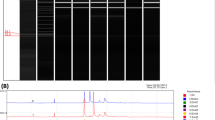
eGene CE
The restriction fragment sizes were estimated by the naked eye or by eGene CE according to the manufacturer’s instructions using size markers (100 bp to 3 kb) and alignment markers (15 bp to 3 kb).
Mycobacterial identification flow chart
The mycobacterial identification flow chart is shown in Figure1.
16 S rDNA sequencing
The 16 S rDNA sequencing of mycobacterial DNA as the reference standard method for mycobacterial species identification was carried out using primer pair 8FPL (5’AGTTTGATCCTGGCTCAG 3’) and 1492 (5’GGTTACCTTGTTACGACT T 3’) as described by Turenne et al.[32]. The species were identified by comparing the 16 S rDNA sequences with similar sequences from GenBank.
Abbreviations
- PRA:
-
Polymerase chain reaction restriction-enzyme analysis
- DPRA:
-
Duplex Polymerase chain Reaction restriction-enzyme Analysis
- MTC:
-
Mycobacterium Tuberculosis Complexes
- NTM:
-
Non-Tuberculous Mycobacteria
- CE:
-
Capillary Electrophoresis
- AFB:
-
Acid-Fast Bacillus
- MAC:
-
M. Avium-intracellulare Complex
- LJ:
-
Löwenstein-Jensen.
References
Collins CH, Grange JM, Yates MD: Tuberculosis bacteriology: organization and practice. 1997, Oxford; Boston: Butterworth-Heinemann, 2
Springer B, Stockman L, Teschner K, Roberts GD, Bottger EC: Two-laboratory collaborative study on identification of mycobacteria: molecular versus phenotypic methods. J Clin Microbiol. 1996, 34: 296-303.
Telenti A, Marchesi F, Balz M, Bally F, Bottger EC, Bodmer T: Rapid identification of mycobacteria to the species level by polymerase chain reaction and restriction enzyme analysis. J Clin Microbiol. 1993, 31: 175-178.
Wong DA, Yip PC, Tse DL, Tung VW, Cheung DT, Kam KM: Routine use of a simple low-cost genotypic assay for the identification of mycobacteria in a high throughput laboratory. Diagn Microbiol Infect Dis. 2003, 47: 421-426. 10.1016/S0732-8893(03)00133-0.
Chang PL, Hsieh WS, Chiang CL, Yen-Liberman B, Procop GW, Chang HT, Ho HT: Identification of individual DNA molecule of Mycobacterium tuberculosis by nested PCR-RFLP and capillary electrophoresis. Talanta. 2008, 77: 182-188. 10.1016/j.talanta.2008.06.004.
Sajduda A, Martin A, Portaels F, Palomino JC: hsp65 PCR-restriction analysis (PRA) with capillary electrophoresis in comparison to three other methods for identification of Mycobacterium species. J Microbiol Methods. 2010, 80: 190-197. 10.1016/j.mimet.2009.12.012.
Chang PL, Hsieh WS, Chiang CL, Tuohy MJ, Hall GS, Procop GW, Chang HT, Ho HT: The hsp65 gene patterns of less common Mycobacterium and Nocardia spp. by polymerase chain reaction-restriction fragment length polymorphism analysis with capillary electrophoresis. Diagn Microbiol Infect Dis. 2007, 58: 315-323. 10.1016/j.diagmicrobio.2007.02.004.
Yokoyama E, Kishida K, Uchimura M, Ichinohe S: Comparison between agarose gel electrophoresis and capillary electrophoresis for variable numbers of tandem repeat typing of Mycobacterium tuberculosis. J Microbiol Methods. 2006, 65: 425-431. 10.1016/j.mimet.2005.08.014.
Lindstedt BA, Vardund T, Aas L, Kapperud G: Multiple-locus variable-number tandem-repeats analysis of Salmonella enterica subsp. enterica serovar Typhimurium using PCR multiplexing and multicolor capillary electrophoresis. J Microbiol Methods. 2004, 59: 163-172. 10.1016/j.mimet.2004.06.014.
Kim BJ, Hong SK, Lee KH, Yun YJ, Kim EC, Park YG, Bai GH, Kook YH: Differential identification of Mycobacterium tuberculosis complex and nontuberculous mycobacteria by duplex PCR assay using the RNA polymerase gene (rpoB). J Clin Microbiol. 2004, 42: 1308-1312. 10.1128/JCM.42.3.1308-1312.2004.
Shen GH, Hung CH, Hu ST, Wu BD, Lin CF, Chen CH, Wu KM, Chen JH: Combining polymerase chain reaction restriction enzyme analysis with phenotypic characters for mycobacteria identification in Taiwan. Int J Tuberc Lung Dis. 2009, 13: 472-479.
Witebsky FG, Kruczak-Filipov P: Identification of mycobacteria by conventional methods. Clin Lab Med. 1996, 16: 569-601.
Vossler JL: Mycobacterium tuberculosis and other non-tuberculous mycobacteria. Text-book of diagnostic microbiology. Edited by: Mahon CRMG. 2000, Philadephia, PA, USA: W B Saunders, 667-707.
Domenech P, Menendez MC, Garcia MJ: Restriction fragment length polymorphisms of 16 S rRNA genes in the differentiation of fast-growing mycobacterial species. FEMS Microbiol Lett. 1994, 116: 19-24. 10.1111/j.1574-6968.1994.tb06669.x.
Lee H, Park HJ, Cho SN, Bai GH, Kim SJ: Species identification of mycobacteria by PCR-restriction fragment length polymorphism of the rpoB gene. J Clin Microbiol. 2000, 38: 2966-2971.
Roth A, Reischl U, Streubel A, Naumann L, Kroppenstedt RM, Habicht M, Fischer M, Mauch H: Novel Diagnostic Algorithm for Identification of Mycobacteria Using Genus-Specific Amplification of the 16 S-23S rRNA Gene Spacer and Restriction Endonucleases. J Clin Microbiol. 2000, 38: 1094-1104.
Takewaki S, Okuzumi K, Manabe I, Tanimura M, Miyamura K, Nakahara K, Yazaki Y, Ohkubo A, Nagai R: Nucleotide sequence comparison of the mycobacterial dnaJ gene and PCR-restriction fragment length polymorphism analysis for identification of mycobacterial species. Int J Syst Bacteriol. 1994, 44: 159-166. 10.1099/00207713-44-1-159.
Chimara E, Ferrazoli L, Ueky SY, Martins MC, Durham AM, Arbeit RD, Leao SC: Reliable identification of mycobacterial species by PCR-restriction enzyme analysis (PRA)-hsp65 in a reference laboratory and elaboration of a sequence-based extended algorithm of PRA-hsp65 patterns. BMC Microbiol. 2008, 8: 48-10.1186/1471-2180-8-48.
Kim BJ, Park JH, Lee SA, Kim H, Cha CY, Kook YH, Kim EC, Joo SI, Lee JS, Yim JJ: Differentiation of mycobacteria in sputa by duplex polymerase chain reaction for mycobacterial hsp65 gene. Diagn Microbiol Infect Dis. 2008, 62: 193-198. 10.1016/j.diagmicrobio.2008.05.013.
Brown-Elliott BA, Wallace RJ: Clinical and taxonomic status of pathogenic nonpigmented or late-pigmenting rapidly growing mycobacteria. Clin Microbiol Rev. 2002, 15: 716-746. 10.1128/CMR.15.4.716-746.2002.
Kim HJ, Mun HS, Kim H, Oh EJ, Ha Y, Bai GH, Park YG, Cha CY, Kook YH, Kim BJ: Differentiation of Mycobacterial species by hsp65 duplex PCR followed by duplex-PCR-based restriction analysis and direct sequencing. J Clin Microbiol. 2006, 44: 3855-3862. 10.1128/JCM.01214-06.
McNabb A, Eisler D, Adie K, Amos M, Rodrigues M, Stephens G, Black WA, Isaac-Renton J: Assessment of partial sequencing of the 65-kilodalton heat shock protein gene (hsp65) for routine identification of Mycobacterium species isolated from clinical sources. J Clin Microbiol. 2004, 42: 3000-3011. 10.1128/JCM.42.7.3000-3011.2004.
Leao SC, Bernardelli A, Cataldi A, Zumarraga M, Robledo J, Realpe T, Mejia GI, da Silva Telles MA, Chimara E, Velazco M, et al: Multicenter evaluation of mycobacteria identification by PCR restriction enzyme analysis in laboratories from Latin America and the Caribbean. J Microbiol Methods. 2005, 61: 193-199. 10.1016/j.mimet.2004.11.015.
Ringuet H, Akoua-Koffi C, Honore S, Varnerot A, Vincent V, Berche P, Gaillard JL, Pierre-Audigier C: hsp65 sequencing for identification of rapidly growing mycobacteria. J Clin Microbiol. 1999, 37: 852-857.
Häfner B, Haag H, Geiss H-K, Nolte O: Different molecular methods for the identification of rarely isolated non-tuberculous mycobacteria and description of new hsp65 restriction fragment length polymorphism patterns. Mol Cell Probes. 2004, 18: 59-65. 10.1016/j.mcp.2003.09.003.
da Silva Telles MA, Chimara E, Ferrazoli L, Riley LW: Mycobacterium kansasii: antibiotic susceptibility and PCR-restriction analysis of clinical isolates. J Med Microbiol. 2005, 54: 975-979. 10.1099/jmm.0.45965-0.
Taillard C, Greub G, Weber R, Pfyffer GE, Bodmer T, Zimmerli S, Frei R, Bassetti S, Rohner P, Piffaretti JC, et al: Clinical implications of Mycobacterium kansasii species heterogeneity: Swiss National Survey. J Clin Microbiol. 2003, 41: 1240-1244. 10.1128/JCM.41.3.1240-1244.2003.
Zhang Y, Mann LB, Wilson RW, Brown-Elliott BA, Vincent V, Iinuma Y, Wallace RJ: Molecular analysis of Mycobacterium kansasii isolates from the United States. J Clin Microbiol. 2004, 42: 119-125. 10.1128/JCM.42.1.119-125.2004.
Maekura R, Okuda Y, Hirotani A, Kitada S, Hiraga T, Yoshimura K, Yano I, Kobayashi K, Ito M: Clinical and prognostic importance of serotyping Mycobacterium avium-Mycobacterium intracellulare complex isolates in human immunodeficiency virus-negative patients. J Clin Microbiol. 2005, 43: 3150-3158. 10.1128/JCM.43.7.3150-3158.2005.
Yamori S, Tsukamura M: Comparison of prognosis of pulmonary diseases caused by Mycobacterium avium and by Mycobacterium intracellulare. Chest. 1992, 102: 89-90. 10.1378/chest.102.1.89.
Hanna BA: Diagnosis of tuberculosis by microbiologic techniques. 1996, Philadelphia, PA, USA: Little, Brown and Company
Turenne CY, Tschetter L, Wolfe J, Kabani A: Necessity of Quality-Controlled 16 S rRNA Gene Sequence Databases: Identifying Nontuberculous Mycobacterium Species. J Clin Microbiol. 2001, 39: 3637-3648. 10.1128/JCM.39.10.3638-3648.2001.
Acknowledgements
This work was supported by grants from the Center of Disease Control (Grant No. DOH95-DC-1106) and the National Science Foundation (Grant No. NSC-982A52) of Taiwan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The authors declare that they have no competing interests.
Authors’ contributions
CCH wrote the manuscript. CSC, JHC, STH participated in the study design, and analysis. GHS and WCH managed the project. JYH, JJL assisted in improving the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Huang, CC., Chen, JH., Hu, ST. et al. Combined rpo B duplex PCR and hsp65 PCR restriction fragment length polymorphism with capillary electrophoresis as an effective algorithm for identification of Mycobacterial species from clinical isolates. BMC Microbiol 12, 137 (2012). https://doi.org/10.1186/1471-2180-12-137
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-12-137