
Abstract
Background
The effects of acetic acid, a common food preservative, on the bacteriophage-encoded enterotoxin A (SEA) expression and production in Staphylococcus aureus was investigated in pH-controlled batch cultures carried out at pH 7.0, 6.5, 6.0, 5.5, 5.0, and 4.5. Also, genomic analysis of S. aureus strains carrying sea was performed to map differences within the gene and in the temperate phage carrying sea.
Results
The sea expression profile was similar from pH 7.0 to 5.5, with the relative expression peaking in the transition between exponential and stationary growth phase and falling during stationary phase. The levels of sea mRNA were below the detection limit at pH 5.0 and 4.5, confirmed by very low SEA levels at these pH values. The level of relative sea expression at pH 6.0 and 5.5 were nine and four times higher, respectively, in the transitional phase than in the exponential growth phase, compared to pH 7.0 and pH 6.5, where only a slight increase in relative expression in the transitional phase was observed. Furthermore, the increase in sea expression levels at pH 6.0 and 5.5 were observed to be linked to increased intracellular sea gene copy numbers and extracellular sea- containing phage copy numbers. The extracellular SEA levels increased over time, with highest levels produced at pH 6.0 in the four growth phases investigated. Using mitomycin C, it was verified that SEA was at least partially produced as a consequence of prophage induction of the sea-phage in the three S. aureus strains tested. Finally, genetic analysis of six S. aureus strains carrying the sea gene showed specific sea phage-groups and two versions of the sea gene that may explain the different sea expression and production levels observed in this study.
Conclusions
Our findings suggest that the increased sea expression in S. aureus caused by acetic acid induced the sea-encoding prophage, linking SEA production to the lifecycle of the phage.
Similar content being viewed by others

Background
Staphylococcal enterotoxins (SEs) are extracellular proteins, produced mainly by Staphylococcus aureus, causing food intoxication when ingested. Staphylococcal food poisoning (SFP) was the fourth most common causative agent in food-borne illness within the EU in 2008 [1]. It is associated with food, generally rich in protein, which requires extensive manual handling, often in combination with inadequate heating and/or inappropriate storage of the food [2, 3]. To date, 21 staphylococcal enterotoxins or enterotoxin-like proteins (SEA-SEE, SEG-SEV), excluding variants, have been identified. These SE genes are widely disseminated by several mobile genetic elements leading to variations in the SE expression behavior among enterotoxigenic staphylococci [2–5]. The expression of a number of the enterotoxins including SEB, SEC, and SED is to some extent known to involve regulatory systems such as the accessory gene regulator (Agr), the staphylococcal accessory regulator (Sar) and the repressor of toxin (Rot) [6]. However, we still have limited information about SEA, the toxin considered to be mainly responsible for staphylococcal food poisoning outbreaks [7–11]. The SEA gene is carried in the bacterial genome by a polymorphic family of temperate bacteriophages [12–14]. Recent studies of S. aureus strain MSSA476 have shown that mitomycin C (MC) induction of ΦSa3ms, resulted in increased transcription of enterotoxins SEA, SEG, and SEK and the fibrinolytic enzyme staphylokinase (Sak) carried by the prophage [14]. Although, it is still unclear if the increased transcription of these virulence determinants lead to increased amounts of SE proteins. Furthermore, identification of the environmental parameters that control the expression of SEA in food, and the mechanism by which these signals are transduced to bring about changes in gene expression, are very limited. This knowledge is crucial for understanding the potential of S. aureus to cause food poisoning.
Acetic acid is a weak organic acid often used in the food industry as a preservative due to its antagonistic effect on bacterial pathogens [15]. Weak acids have the ability to pass through the cell membrane in the undissociated form. Once inside the cell, the acid dissociates in the more alkaline interior, lowering the intracellular pH of the cell. A decrease in intracellular pH can lead to the damage of macromolecules (e.g. proteins and DNA) and the cell membrane, and have a negative effect on cell maintenance [16, 17]. Also, the anion of the acid is accumulated intracellularly, increasing turgor pressure [18]. Acetic acid has been found to be more inhibitory to the growth of S. aureus than lactic acid, citric acid, phosphoric acid and hydrochloric acid, respectively [19]. Also, acetic acid has been found to almost completely inhibit SEA formation in brain heart infusion (BHI) broth when added gradually over time [20].
In the present study, the effects of acetic acid on S. aureus growth, sea expression and SEA production were investigated in four growth phases. Furthermore, the relationship between SEA production and the lifecycle of the phage carrying the toxin gene was determined. Finally, genomic analysis of S. aureus strains carrying sea was performed to map differences within the gene and in the temperate phage carrying sea.
Results
Effects of acetic acid on sea expression and SEA production in S. aureus Mu50
Batch cultures of S. aureus Mu50, harboring the sea-containing Φ42-like prophage ΦMu50A [21], were carried out at controlled pH levels of 7.0, 6.5, 6.0, 5.5, 5.0, and 4.5 (Figure 1A). Acetic acid was used to set the pH to investigate the effects of acetic acid on growth, relative sea expression and extracellular SEA levels during all stages of growth. The maximal growth rate of S. aureus Mu50 was highest at pH 7.0 and decreased with decreasing pH (Figure 1A). Batch cultivations performed at lower pH values showed that pH 5.0 was highly growth-inhibitory, with only a modest increase in optical density, OD, and viable cells in the late stationary growth phase, and that pH 4.5 was too toxic; < 1% of the starting inoculum was viable after 24 h. The relative sea expression pattern was similar at all tested pH levels that allowed expression analysis (Figure 1B); the highest relative levels of sea mRNA were found in the transitional phase and fell during the stationary growth phase. Small increases in sea expression were found in the transitional phase at pH 7.0 and 6.5. However, relative sea expression in the transitional phase at pH 6.0 (n = 2) and 5.5 (n = 3) were high, nine and four times higher, respectively, than in the exponential growth phase. At pH 5.5, extended sea mRNA expression was observed with the peak associated with the transitional phase. However, sea mRNA was not possible to detect at pH 5.0 or 4.5.

Growth and relative sea levels of S. aureus Mu50 when grown at different pH levels. (A) Growth curves determined by OD measurements at 620 nm at pH 7.0, 6.5, 6.0, 5.5, 5.0, and 4.5. (B) Relative expression (RE) of sea at pH 7.0, 6.5, 6.0, and 5.5. Solid and dashed lines represent growth and RE, respectively. For pH 6.0 and 5.5, the mean and standard deviations of independent batch cultures; two and three, respectively, is displayed.
Extracellular SEA was detected in all cultivations of S. aureus Mu50 and the levels increased over time at tested pH levels allowing growth (Figure 2). The SEA levels increased from pH 7.0 to 6.0 and decreased significantly at lower pH levels, i.e. pH 5.5, 5.0 and 4.5. The specific extracellular SEA concentrations (i.e. the extracellular SEA concentrations divided by the value of the OD at that point in time) correlating the SEA production to growth, showed the same trend. The specific SEA concentrations were 100, 450, 510, 210, 40, and 870 ng per ml and OD unit for pH 7.0, 6.5, 6.0, 5.5, 5.0, and 4.5, respectively. The specific SEA concentration at pH 4.5 is misleading since the culture was not growing.

SEA levels, growth rate and sea expression of S. aureus Mu50 at different pH levels. Extracellular SEA levels in the mid-exponential, the transitional, the early stationary, and late stationary growth phase; maximal growth rate (μmax), and relative sea levels (RE) in the transitional phase. At pH 4.5 the SEA values are after 10, 24 and 30 h of growth, shown in the figure as transitional, early stationary and late stationary phase samples, respectively. The values at pH 6.0 and 5.5 are the average and standard deviations of two and three independent batch cultures, respectively.
Phage-associated sea expression
Samples of bacterial cells and culture supernatants from S. aureus Mu50 were collected to determine the trends of the relative sea gene copy number (and thus the replicative form of the sea- carrying phage) and relative phage copy number in the four growth phases at different pH values (Figure 3). The relative sea gene copy number was low throughout the cultivations at pH 7.0 and 6.5. The sea gene copy number peaked at pH 5.5, being twelve times higher than at pH 7.0 in the mid-exponential growth phase, and a trend of the sea gene copy number decreasing over time was observed at this pH. The sea gene copy number increased over time at pH 5.0 and 4.5, showing markedly higher levels at the last sampling point. The phage copy number increased over time at all pH levels, with a peak at pH 5.5. In the late stationary growth phase, the phage copy number was 13 times higher at pH 5.5 than at pH 7.0.

Change in sea gene copy number and sea -carrying phage copy number of S. aureus Mu50. The relative sea gene copy numbers and phage copy numbers in the mid-exponential, the transitional, the early stationary, and the late stationary growth phase of S. aureus Mu50 at different pH levels; black symbols are the relative sea gene copy numbers and white symbols are the relative phage copy numbers. At pH 4.5, the SEA values are after 10, 24 and 30 h of growth, shown in the figure as transitional, early stationary and late stationary phase samples, respectively. For pH 6.0 and 5.5, representative values of several independent batch cultures are shown.
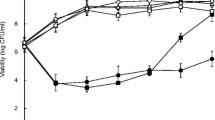
To investigate if the extracellular SEA levels were affected by prophage induction, 0.5 μg/ml or 5.0 μg/ml MC was added to exponentially growing S. aureus strains Mu50, SA17, and SA45 (Figure 4). The number of viable cells of strain Mu50 after three hours of growth following MC addition was reduced by two log units in cultures containing 0.5 μg/ml MC and five log units in cultures containing 5.0 μg/ml MC, compared with control cultures containing no MC. For both strains SA17 and SA45 the viable cell counts were reduced by one and four log units in cultures containing 0.5 μg/ml and 5.0 μg/ml MC, respectively (data not shown). The specific extracellular SEA levels, i.e. the extracellular SEA concentration per colony-forming unit, CFU, of S. aureus strains Mu50, SA17, and SA45, increased with MC concentration compared to the control cultures, being ten, 50, and 20 times higher at 0.5 μg/ml MC, and 3000, 40 000, and 6000 times higher at 5.0 μg/ml MC for Mu50, SA17, and SA45, respectively. Viable phage particles, defined as plaque forming units, were observed for strains SA17 and SA45 after MC treatment but not for Mu50 using S. aureus RN450 as recipient strain (for Mu50, S. aureus RN4220 was also tested) (data not shown).

Specific extracellular SEA levels of S. aureus Mu50, SA17, and SA45 after mitomycin C treatment.
Effects of acetic acid on sea expression and SEA production in S. aureus SA45
To determine if the response to acetic acid was specific to strain Mu50 or a more general S. aureus response, a strain isolated from ham involved in a food poisoning outbreak, S. aureus SA45, was used to replicate the batch cultivations at pH 7.0 and pH 5.5 (Figure 5 A and B). S. aureus SA45 had higher maximal growth rate than S. aureus Mu50, but the cultures never reached the same maximum OD as Mu50. The relative sea expression pattern of S. aureus SA45 was the same as for S. aureus Mu50, with the highest relative sea levels found in the transitional phase. The sea mRNA levels and extracellular SEA amounts were very similar for both strains at pH 7.0. However, at pH 5.5 the relative sea expression in the transitional phase was 38% higher in S. aureus Mu50 compared to in S. aureus SA45 and the final extracellular SEA concentration in the S. aureus Mu50 cultures was 61% higher than in S. aureus SA45 cultures on average.

Growth, SEA levels, and sea mRNA levels of S. aureus SA45 grown at two pH levels. (A) Growth curves determined by OD measurements at 620 nm and extracellular SEA levels at pH 7.0 and pH 5.5. (B) Relative expression (RE) of sea at pH 7.0 and pH 5.5. Solid, dotted and dashed lines represents growth, SEA levels and RE, respectively. Values are the mean and standard deviations of two independent batch cultures.
Genetic diversity of sea
Nucleotide sequence analysis of sea and prophage regions immediately upstream and downstream of the gene was performed on the whole-genome sequenced S. aureus strains MRSA252 [22], MSSA476 [22], Mu3, Mu50 [21], MW2 [23], and Newman [24] to determine genetic differences that may explain the different sea expression and SEA production profiles observed at pH 5.5 with S. aureus Mu50 and SA45. Sequence alignment of the coding region of sea revealed two main groups of sea-carrying phages. Within a group the sea sequences showed 100% sequence similarity and between the two groups the sequence similarity was 98%. Prophages ΦMu3, ΦMu50A, ΦSa3ms, and ΦSa3mw clustered together in a sea-group designated sea1, while Φ252B and ΦNM3 formed a sea group, designated sea2.
All six phages shared a homologous region of 3.2 kb downstream of the sea gene containing the sak gene. Thereafter, the nucleotide sequences diverged, forming three subgroups of sea phages. The same grouping of phages was observed immediately upstream of the translational start site of sea (Figure 6). An analogous phage grouping was recently reported when comparing the integrase (int) nucleotide sequences of these bacteriophages [25]. To improve the resolution of phylogenetic analysis of these bacteriophages based on int genes, we repeated the int gene grouping (data not shown). The ΦMu3A/ΦMu50A branch was found to be closer to the Φ252B/ΦNM3 branch than to the ΦSa3ms/ΦSa3mw branch. This is in direct contrast to what was found for the sea gene.

Gene map of the sea virulence region of S. aureus. Gene map of the sea gene and regions immediately upstream and downstream of the gene in six different S. aureus strains. The map is based on nucleotide sequence analysis of the strains. Solid lines are sequences within the sea-carrying prophage. Dotted lines represent sequences outside the prophage region. The letters a-h indicates were PCR amplicons are located within the region; numbers 1-2 indicate transcription start sites [14].
In order to identify the phage- and sea-group of SA45, eight different regions were targeted by PCR (see Table 1 and Figure 6). This analysis showed that SA45 carries the sea1-version of the sea gene and belongs to the same subgroup as ΦSa3mw.
Discussion
The genetic diversity analysis of the prophage region encoding SEA showed two main groups of genes, sea 1 and sea 2 . To our knowledge this has not been observed before. Furthermore, Figure 6 shows that the sea 1 and sea 2 genes are associated with specific bacteriophages which could be further grouped based on sequence similarities within regions upstream and downstream of the sea gene. Borst and Betley divided enterotoxin-A-producing S. aureus into high-SEA producing and low-SEA producing strains [13]. The variation in SEA production was associated with differences in the prophage region immediately upstream of sea. The six strains analyzed here could be divided in three groups based on sequence differences in the sea-virulence region. However, a different grouping than for the sea gene was observed upon comparing the int gene of these phages. The int gene, being part of the core genome, is essential for the phage's lifecycle unlike the sea gene, and is therefore reflecting the evolutionary relationship among these phages. Nucleotide sequence analysis of S. aureus Mu50 and SA45 showed that they belong to different groups based on variations in the nucleotide sequences within the sea-virulence region. This division may explain the differences observed between the two strains regarding sea expression and SEA levels at pH 5.5.
The sea expression was highest in the transition from the exponential to the stationary growth phase in both S. aureus Mu50 and SA45 at all pH levels that allowed expression analysis, as established previously [26, 27]. A boost in sea expression was observed in the transitional phase in S. aureus Mu50 at pH 6.0 and pH 5.5, which was also found in strain SA45. The same expression pattern has been found for the prophage-encoded Panton-Valentine leukocidin (PVL, luk-PV) of S. aureus[28]. Maximal expression of luk-PV in the late exponential growth phase was followed by a rapid decline post-exponentially. Our observation could partially be explained by the induction of the prophage carrying the toxin gene. The sea-phage copy numbers of S. aureus Mu50 at pH 6.0 remained constant during the first part of cultivation. In the late stationary growth phase, however, the number had increased four times (average increase of two biological replicates) compared to levels in early stationary growth phase. The phage copy numbers might have increased further if growth was allowed to continue. An acetic-acid induced intracellular drop in pH, leading to oxidative stress [29] would activate the SOS response system inducing the prophage [30]. Sumby and Waldor showed that upon prophage induction in S. aureus, the phage DNA was replicated, resulting in an increase in sea gene copy number, and that a second prophage-regulated sea promoter was also activated, resulting in increased sea expression [14]. Similar enhanced transcription of phage-encoded virulence genes upon prophage induction has also been observed for PVL in S. aureus and the Shiga toxins in E. coli[28, 31].
Mitomycin C, a well-known prophage inducer, was used in this study. The more MC added, the more SEA was produced per CFU for all three strains tested, supporting the association between prophage induction and SEA production. However, the expected boost in extracellular SEA levels accompanying the increased sea mRNA levels and sea gene copy levels observed at pH 5.5 was not found. This could be because of the pronounced phage production at pH 5.5 seen as a rapid increase in extracellular sea-phage copy number (Figure 3). The window for phage-encoded SEA-biosynthesis prior to phage-release could be too narrow in the bacteria at this pH level. The relative phage copy number generally increased over time at all pH levels investigated. At pH 5.5, the relative phage copy number was increasing dramatically over time, suggesting that substantial prophage induction had occurred. The sea gene copy number, however, was decreasing over time at pH 5.5. This could be due to cell lysis occurring upon prophage induction at this pH. At pH 5.0 and 4.5, a big increase in relative sea gene copy number was observed between the two last sampling points. This suggests that the prophage has been induced and the replicative form of the phage DNA is produced. However, at these low pH values, no great increase in SEA or phage copies were observed, suggesting protein synthesis was impaired.
In addition, the reason why the sea expression of S. aureus Mu50 at pH 5.5 was not as high as at pH 6.0, despite the fact that an even higher level of acetic acid was added, could also be because the acetic acid concentration here was on the limit of that tolerated by the organism. The growth rate of the culture at pH 5.5 was almost half of that at pH 6.0. The expression pattern at pH 5.5 was different from the patterns at the higher pH levels studied, in that it lacked the sharp expression peak in the transitional phase. At pH levels below 6.0, low amounts of SEA were produced. This supports the theory that pH 5.5 is close to the limiting pH of the bacterium. The SEA levels remained constant at pH 5.0 and pH 4.5 during the cultivation of Mu50, with a final SEA concentration of 62 ng/ml for both pH levels, indicating that no SEA production occured ≤ pH 5.0. This observation is supported by Barber and Deibel [32]. Using hydrochloric acid, they found that the lowest pH values that supported SEA biosynthesis in buffered BHI medium incubated aerobically was 4.9. SFP can be caused by as little as 20-100 ng of enterotoxin [33]. Levels higher than 100 ng/ml were detected at pH levels 7.0-5.5 in the mid-exponential growth phase.
Conclusions
This study has shown that the food preservative acetic acid increases sea gene expression in S. aureus. At pH 6.0 and 5.5, maximal sea expression was observed. At pH 6.0 there was a marked shift in growth rate and phage production peaked at pH 5.5. These findings suggest prophage induction. At pH 5.0 and 4.5, the sea gene copy numbers increased dramatically during late stages of cultivation, but SEA levels and phage copy numbers were low indicating that protein synthesis was affected. It is our hypothesis that the acetic acid lowers the intracellular pH of S. aureus, activating the temperate phage and, as a consequence, boosts the sea expression. Our results support the theory proposed by other research groups that prophages not only facilitate the dissemination of virulence genes, but also take part in the regulation of the expression of the genes.
Methods
Culture conditions
The S. aureus strains used in this study were Mu50 (LGC Promochem, London, UK), MW2 (donated by Dr. T. Baba, Juntendo University, Tokyo, Japan), Newman (donated by Dr. H. Ingmer, Copenhagen University, Copenhagen, Denmark), RN4220 (Culture Collection University of Göteborg, Göteborg, Sweden), RN450 (donated by Dr. J. R. Penadés, Instituto Valenciano de Investigaciones Agrarias, Castellón, Spain), SA17 and SA45 (donated by the Swedish Institute for Food and Biotechnology, SIK, Göteborg, Sweden). All cultivations were performed in BHI (Difco Laboratories; BD Diagnostic Systems, Le Point de Claix, France) broth (with agitation) or agar at 37°C. S. aureus was transferred from glycerol stock to broth for overnight cultivation prior to the experiments. Broth (300 ml) was inoculated with a sufficient volume of S. aureus overnight culture to give an OD value at 620 nm (OD620) of 0.1 at the start of cultivation. Batch cultivations were then performed at different pH levels (pH 7.0, 6.5, 6.0, 5.5, 5.0, and 4.5) using in-house fermentors. The pH was set using acetic acid (Merck; Darmstadt, Germany), pH 7.0: 5.1 mM; pH 6.5: 12 mM; pH 6.0: 18 mM; pH 5.5: 28 mM; pH 5.0: 43 mM and pH 4.5: 93 mM final concentration of acetic acid, and maintained by adding sodium hydroxide (Merck) by automatic titration. The study was designed using several sampling points over time to visualize trends and all samples were analyzed three times. Where trend deviations were observed, cultivations were repeated to confirm the results.
The OD620 was measured to follow growth. All OD measurements were performed using a U-1800 spectrophotometer (Hitachi High Technologies Inc., Pleasanton, CA). Samples for quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis and enzyme-linked immunosorbent assay (ELISA) analysis, and intracellular-DNA and extracellular-DNA extractions were taken in the mid-exponential growth phase, in the transitional phase, i.e. between the exponential and stationary phases of growth, in the early stationary phase of growth, and in the late stationary phase of growth. At pH 5.0, samples were taken after 12, 27, 36 and 49 h of growth. At pH 4.5, samples were taken after 10, 24, and 30 h of growth.
Viable counts were determined in the late stationary growth phase to confirm OD620 measurements, except at pH 4.5, where viable counts were determined on each sampling occasion. Serial decimal dilutions of the bacterial cultures in physiological saline (Merck) solution were performed. The dilutions were plated on agar, incubated overnight and the CFU per ml was calculated.
Primer and probe design
The forward primer, ESA-1, specific to sea was identified from the literature [34], and the reverse primer was designed in-house using LightCycler Probe Design© software ver. 1.0 (Roche Diagnostics GmbH, Mannheim, Germany) (Table 2). Primers for the reference gene rrn were designed as the reverse primer of the sea gene. All primers were purchased from MWG Biotech AG (Ebersberg, Germany). Hybridization probes specific to sea and rrn were also designed using the LightCycler Probe Design© software and purchased from TIB Molbiol GmbH (Berlin, Germany). The probes work in pairs. A donor probe labeled with fluorescein at the 3" end transmits the signal to an acceptor probe labeled with LCRed640/LCRed705 at the 5" end and the 3" hydroxy group is phosphorylated.
sea expression analysis
Total RNA was extracted using phenol and chloroform as described by Lövenklev et al. [35], except that the RNA was re-suspended in 100 μl RNA storage solution (Applied Biosystems, Foster City, CA). First-strand cDNA was synthesized in two separate reverse-transcription assays using reverse primers specific to SEA and the reference gene 16S rRNA, as described previously [36], with 0.1 μg RNA in the reference gene assay and 0.5 μg RNA in the toxin gene assay.
Real-time PCR amplification was carried out on a LightCycler™ 1.0 instrument (Roche Diagnostics GmbH). The total volume of PCR mixture was 20 μl including 4 μl of template cDNA. The sea PCR mixture consisted of 1 × PCR buffer, 3.25 mM MgCl2, 0.2 mM each of dATP, dTTP, dCTP, and dGTP, 0.5 μM each of the forward and reverse primers, 0.05 U Tth DNA polymerase, and 0.3 μM of each hybridization probe. The rrn PCR mixture was the same as the sea PCR mixture, except that 0.15 μM of each hybridization probe was added. All reagents except the primers and probes were obtained from Roche Diagnostics GmbH. The water used was autoclaved ultrapure water. In order to detect the amplification of possible contaminants, a negative control consisting of water instead of DNA was added to the PCR. Genomic DNA was used as a positive control. The following PCR protocol was used: initial denaturation at 95°C for 1 min, followed by 45 cycles of denaturation at 95°C for 0 s (i.e., no hold at 95°C), primer annealing at 46°C (sea) or 48°C (rrn) for 5 s, and extension at 72°C for 25 s, with a single fluorescence measurement at the end of the extension step. The crossing point cycle for each transcript was determined using the second derivative maximum mathematical model in the LightCycler™ software (ver. 3.5) (Roche Diagnostics GmbH), and the amplification efficiency in the exponential phase was calculated using the equation of Klein et al. [37]. The sea gene assay was linear at 1.0 × 10-6 to 6.3× 10-8 g/ml RNA. The threshold cycle number of the reference gene varied <1.3 cycles in between samples. The efficiency was 0.96 ± 0.066 and 1.1 ± 0.075, respectively for the sea and the rrn assays.
The relative expression of the sea gene was calculated by relating the toxin gene expression to the constant expression of a reference gene, the 16S rRNA gene [38]. To determine the amplification efficiency and the log-linear range of amplification for each real-time PCR assay, the total RNA was serially diluted. The dilutions were reverse transcribed and amplified in the LightCycler™ instrument three times to obtain standard curves. Samples were also amplified three times. Equal amounts of total RNA from each sample were reverse transcribed to quantify the transcript levels of sea. The relative expression was calculated from the amplification efficiencies of each PCR assay and the crossing point deviation of the unknown sample versus a calibration sample, as described previously [38], see equation 1.

Phage induction analysis
Cell-DNA was extracted using a protocol described by Walsh et al. [39] modified to include a 20% Chelex® (BioRad Laboratories; Hercules, USA) solution instead of 5%. 20 ng of DNA was added to the sea real-time PCR assay, see above.
Phage DNA was purified using zinc chloride as previously described by Santos [40] without previous DNase or RNase treatments. 200 ng of DNA was added to the sea real-time PCR assay, see above.
Induction of the bacteriophage using MC (Duchefa Biochemie, Haarlem, the Netherlands) was performed according to Resch et al. [41]. S. aureus overnight culture (0.2 ml) was added to 30 ml of fresh broth in 250 ml Erlenmeyer flasks. When cultures were in the mid-exponential phase of growth, MC was added to a final concentration of 0.5 μg/ml or 5 μg/ml, followed by continued incubation for 3 h. SEA concentrations, viable cell counts, and viable virus particles were determined. Cultures without addition of MC were used as controls.
The phage plaque assay was performed as described by France and Markham [42]. Supernatants from S. aureus cultures were spotted onto agar and the plates were then incubated at least overnight. S. aureus RN450 and RN4220 were used as receiver strains.
The relative sea gene copy number was calculated using equation 1. The relative phage copy number was calculated using the nominator part of equation 1.
ELISA
A modified protocol was developed for ELISA analysis of SEA using affinity-purified sheep polyclonal antibodies based on Poli et al. [43]. A microtiter plate (Immulon® 2HB polystyrene, Flat Bottom Microtiter® Plates, 96 wells solid; Thermo Electron Corporation; Waltham, MA) was coated with 100 μl/well of a solution containing 2 μg/ml SEA affinity-purified antibody (Toxin Technology, Inc.; Sarasota, FL) in coating buffer (0.1 M sodium carbonate, pH 9.6, Merck) and left at 37°C overnight. All sites were blocked with 185 μl blocking buffer (SuperBlock Blocking Buffer in PBS, pH 7.4, Pierce, Rockford, IL) for one hour at 37°C and at least one hour at 4°C. The plate was washed four times with washing buffer (0.05% Tween 20, BioRad Inc., in 10 mM PBS, Sigma-Aldrich, St Louis, MO). Standards or culture supernatants were loaded onto the plate (100 μl/well) at appropriate dilutions and incubated for 90 min at 37°C. As SEA standard, highly purified SEA staphylococcal enterotoxin from Toxin Technology Inc. (Sarasota, FL), was used. The plate was washed and the biotinylated antibody (Toxin Technology, Inc.), diluted 2000 × in assay buffer (50 mM PBS, 0.01% bovine serum albumin, Sigma-Aldrich, 0.1% Tween 20, 0.01% Thimerosal, Sigma-Aldrich, 1% milk powder, Semper, Sundbyberg, Sweden) was added (100 μl/well). The plate was incubated for one hour at 37°C and washed. NeutrAvidin™-linked alkaline phosphatase (ImmunoPure NeutrAvidin™, alkaline phosphatase conjugated, 0.9 mg/ml, Pierce; diluted 1000 × in assay buffer, no milk powder included) was added (100 μl/well), and the plate was incubated for 30 minutes at 37°C. The plate was washed and substrate (SIGMAFAST™ p-nitrophenyl phosphate tablets N2770, Sigma-Aldrich) was added (100 μl/well). The color was allowed to develop for 45 min in darkness and the optical density was determined using a microplate reader with a filter at 405 nm (Multiskan Ascent, Thermo Electron Corporation). Absorbance values (mean of triplicate wells) were plotted against toxin concentrations, and values were determined from linear regression. The detection limit was at 0.31 ng/ml of SEA.
Nucleotide sequence analysis
The sea nucleotide sequences of six S. aureus strains (MRSA252 [GenBank: BX571856], MSSA476 [GenBank: BX571857], Mu3 [GenBank: AP009324], Mu50 [GenBank: BA000017], MW2 [GenBank: BA000033], and Newman [GenBank: AP009351]) were retrieved from GenBank (http://www.ncbi.nlm.nih.gov/Genbank/index.html April 2009) and pairwise aligned using BioEdit v. 7.0.9.0 (Ibis Biosciences; Carlsbad, CA). DNA sequences (8 kb) upstream and downstream of the sea gene were also compared. The sea genes of all six strains have previously been annotated.
Conventional PCR
Primers were designed to confirm the results of the nucleotide sequence analysis of sea and regions adjacent to the gene (Table 1). Two primer pairs were designed to distinguish between the two groups of nucleotide sequences, sea1 and sea2. Six primer pairs were designed to validate sequence differences found between strains in regions upstream and downstream of the sea gene. All primers were ordered from MWG Biotech AG. Genomic DNA from S. aureus Mu50, MW2, Newman, and SA45 was used as template. The total volume of PCR mixture was 50 μl including 200 ng template DNA. The PCR mixture consisted of 1 × PCR buffer, 2 mM MgCl2, 0.2 mM each of dATP, dTTP, dCTP, and dGTP, 0.2 μM each of forward and reverse primer and 2 U Tth DNA polymerase. All reagents except primers were obtained from Roche Diagnostics GmbH. The water used was autoclaved ultrapure water. In order to detect the amplification of possible contaminants, a negative control consisting of water instead of DNA was added to the PCR. The following PCR protocol was used: initial denaturation at 94°C for 4 min, followed by 30 cycles of denaturation at 94°C for 30 s, primer annealing at 47-55°C (see Table 1) for 30 s, and extension at 72°C for 1 min, with a final extension step at 72°C for 5 min. All amplifications were carried out using the Gene Amp 9700 thermal cycler (Perkin-Elmer Cetus; Norwalk, CT). The PCR products were visualized using 0.8% agarose (Bio-Rad Laboratories, Hercules, CA) gel electrophoresis according to Sambrook and Russell [44].
References
EFSA: The Community Summary Report on Trends and Sources of Zoonoses, Zoonotic Agents and food-borne outbreaks in the European Union in 2008. The EFSA journal. 2010, 1496:
Smyth CJ, Smyth DS, Kennedy J, Twohig J, Bolton DJ: Staphylococcus aureus: from man or animal - an enterotoxin iceberg?. EU-RAIN, 3-4 December, 2004, Padua, Italy. Edited by: Maunsell B, Sheridan J, Bolton DJ. 2004, Teagasc - The National Food Centre, 85-102.
Le Loir Y, Baron F, Gautier M: Staphylococcus aureus and food poisoning. Genet Mol Res. 2003, 2 (1): 63-76.
Thomas DY, Jarraud S, Lemercier B, Cozon G, Echasserieau K, Etienne J, Gougeon ML, Lina G, Vandenesch F: Staphylococcal enterotoxin-like toxins U2 and V, two new staphylococcal superantigens arising from recombination within the enterotoxin gene cluster. Infect Immun. 2006, 74 (8): 4724-4734. 10.1128/IAI.00132-06.
Ono HK, Omoe K, Imanishi K, Iwakabe Y, Hu DL, Kato H, Saito N, Nakane A, Uchiyama T, Shinagawa K: Identification and characterization of two novel staphylococcal enterotoxins, types S and T. Infect Immun. 2008, 76 (11): 4999-5005. 10.1128/IAI.00045-08.
Bronner S, Monteil H, Prévost G: Regulation of virulence determinants in Staphylococcus aureus: complexity and applications. FEMS Microbiol Rev. 2004, 28 (2): 183-200. 10.1016/j.femsre.2003.09.003.
Cha JO, Lee JK, Jung YH, Yoo JI, Park YK, Kim BS, Lee YS: Molecular analysis of Staphylococcus aureus isolates associated with staphylococcal food poisoning in South Korea. J Appl Microbiol. 2006, 101 (4): 864-871. 10.1111/j.1365-2672.2006.02957.x.
Kérouanton A, Hennekinne JA, Letertre C, Petit L, Chesneau O, Brisabois A, De Buyser ML: Characterization of Staphylococcus aureus strains associated with food poisoning outbreaks in France. Int J Food Microbiol. 2007, 115 (3): 369-375. 10.1016/j.ijfoodmicro.2006.10.050.
Wieneke AA, Roberts D, Gilbert RJ: Staphylococcal food poisoning in the United Kingdom, 1969-90. Epidemiol Infect. 1993, 110 (3): 519-531. 10.1017/S0950268800050949.
Casman EP: Staphylococcal food poisoning. Health Lab Sci. 1967, 4 (4): 199-206.
Payne DN, Wood JM: The incidence of enterotoxin production in strains of Staphylococcus aureus isolated from foods. J Appl Bacteriol. 1974, 37 (3): 319-325.
Betley MJ, Mekalanos JJ: Staphylococcal enterotoxin A is encoded by phage. Science. 1985, 229 (4709): 185-187. 10.1126/science.3160112.
Borst DW, Betley MJ: Phage-associated differences in staphylococcal enterotoxin A gene (sea) expression correlate with sea allele class. Infect Immun. 1994, 62 (1): 113-118.
Sumby P, Waldor MK: Transcription of the toxin genes present within the Staphylococcal phage phiSa3ms is intimately linked with the phage's life cycle. J Bacteriol. 2003, 185 (23): 6841-6851. 10.1128/JB.185.23.6841-6851.2003.
Smittle RB: Microbiological safety of mayonnaise, salad dressings, and sauces produced in the United States: a review. J Food Prot. 2000, 63 (8): 1144-1153.
Cotter PD, Hill C: Surviving the acid test: responses of gram-positive bacteria to low pH. Microbiol Mol Biol Rev. 2003, 67 (3): 429-453. 10.1128/MMBR.67.3.429-453.2003.
Clements MO, Foster SJ: Stress resistance in Staphylococcus aureus. Trends Microbiol. 1999, 7 (11): 458-462. 10.1016/S0966-842X(99)01607-8.
Foster JW: When protons attack: microbial strategies of acid adaptation. Curr Opin Microbiol. 1999, 2 (2): 170-174. 10.1016/S1369-5274(99)80030-7.
Minor TE, Marth EH: Growth of Staphylococcus aureus in acidified pasteurized milk. J Milk Food Tech. 1970, 33: 516-520.
Domenech A, Hernandez FJ, Orden JA, Goyache J, Lopez B, Suarez G, Gomez-Lucia E: Effect of six organic acids on staphylococcal growth and enterotoxin production. Z Lebensm Unters Forsch. 1992, 194 (2): 124-128. 10.1007/BF01190181.
Kuroda M, Ohta T, Uchiyama I, Baba T, Yuzawa H, Kobayashi I, Cui L, Oguchi A, Aoki K, Nagai Y, Lian J, Ito T, Kanamori M, Matsumaru H, Maruyama A, Murakami H, Hosoyama A, Mizutani-Ui Y, Takahashi NK, Sawano T, Inoue R, Kaito C, Sekimizu K, Hirakawa H, Kuhara S, Goto S, Yabuzaki J, Kanehisa M, Yamashita A, Oshima K, Furuya K, Yoshino C, Shiba T, Hattori M, Ogasawara N, Hayashi H, Hiramatsu K: Whole genome sequencing of methicillin-resistant Staphylococcus aureus. Lancet. 2001, 357 (9264): 1225-1240. 10.1016/S0140-6736(00)04403-2.
Holden MT, Feil EJ, Lindsay JA, Peacock SJ, Day NP, Enright MC, Foster TJ, Moore CE, Hurst L, Atkin R, Barron A, Bason N, Bentley SD, Chillingworth C, Chillingworth T, Churcher C, Clark L, Corton C, Cronin A, Doggett J, Dowd L, Feltwell T, Hance Z, Harris B, Hauser H, Holroyd S, Jagels K, James KD, Lennard N, Line A, Mayes R, Moule S, Mungall K, Ormond D, Quail MA, Rabbinowitsch E, Rutherford K, Sanders M, Sharp S, Simmonds M, Stevens K, Whitehead S, Barrell BG, Spratt BG, Parkhill J: Complete genomes of two clinical Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug resistance. Proc Natl Acad Sci USA. 2004, 101 (26): 9786-9791. 10.1073/pnas.0402521101.
Baba T, Takeuchi F, Kuroda M, Yuzawa H, Aoki K, Oguchi A, Nagai Y, Iwama N, Asano K, Naimi T, Kuroda H, Cui L, Yamamoto K, Hiramatsu K: Genome and virulence determinants of high virulence community-acquired MRSA. Lancet. 2002, 359 (9320): 1819-1827. 10.1016/S0140-6736(02)08713-5.
Baba T, Bae T, Schneewind O, Takeuchi F, Hiramatsu K: Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: polymorphism and evolution of two major pathogenicity islands. J Bacteriol. 2008, 190 (1): 300-310. 10.1128/JB.01000-07.
Goerke C, Pantucek R, Holtfreter S, Schulte B, Zink M, Grumann D, Broker BM, Doskar J, Wolz C: Diversity of prophages in dominant Staphylococcus aureus clonal lineages. J Bacteriol. 2009, 191 (11): 3462-3468. 10.1128/JB.01804-08.
Borst DW, Betley MJ: Mutations in the promoter spacer region and early transcribed region increase expression of staphylococcal enterotoxin A. Infection and Immunity. 1993, 61: 5421-5425.
Wallin-Carlquist N, Marta D, Borch E, Rådström P: Prolonged expression and production of Staphylococcus aureus enterotoxin A in processed pork meat. Int J Food Microbiol.
Wirtz C, Witte W, Wolz C, Goerke C: Transcription of the phage-encoded Panton-Valentine leukocidin of Staphylococcus aureus is dependent on the phage life-cycle and on the host background. Microbiology. 2009, 155: 3491-3499. 10.1099/mic.0.032466-0.
Clements MO, Watson SP, Foster SJ: Characterization of the major superoxide dismutase of Staphylococcus aureus and its role in starvation survival, stress resistance, and pathogenicity. J Bacteriol. 1999, 181 (13): 3898-3903.
Selva L, Viana D, Regev-Yochay G, Trzcinski K, Corpa JM, Lasa I, Novick RP, Penadés JR: Killing niche competitors by remote-control bacteriophage induction. Proc Natl Acad Sci USA. 2009, 106 (4): 1234-1238. 10.1073/pnas.0809600106.
Wagner PL, Neely MN, Zhang X, Acheson DW, Waldor MK, Friedman DI: Role for a phage promoter in Shiga toxin 2 expression from a pathogenic Escherichia coli strain. J Bacteriol. 2001, 183 (6): 2081-2085. 10.1128/JB.183.6.2081-2085.2001.
Barber LE, Deibel RH: Effect of pH and oxygen tension on staphylococcal growth and enterotoxin formation in fermented sausage. Appl Microbiol. 1972, 24 (6): 891-898.
Asao T, Kumeda Y, Kawai T, Shibata T, Oda H, Haruki K, Nakazawa H, Kozaki S: An extensive outbreak of staphylococcal food poisoning due to low-fat milk in Japan: estimation of enterotoxin A in the incriminated milk and powdered skim milk. Epidemiol Infect. 2003, 130 (1): 33-40. 10.1017/S0950268802007951.
Rosec JP, Gigaud O: Staphylococcal enterotoxin genes of classical and new types detected by PCR in France. International Journal of Food Microbiology. 2002, 77 (1-2): 61-70. 10.1016/S0168-1605(02)00044-2.
Lövenklev M, Holst E, Borch E, Rådström P: Relative neurotoxin gene expression in Clostridium botulinum type B, determined using quantitative reverse transcription-PCR. Appl Environ Microbiol. 2004, 70 (5): 2919-2927. 10.1128/AEM.70.5.2919-2927.2004.
Artin I, Carter AT, Holst E, Lövenklev M, Mason DR, Peck MW, Rådström P: Effects of carbon dioxide on neurotoxin gene expression in nonproteolytic Clostridium botulinum Type E. Appl Environ Microbiol. 2008, 74 (8): 2391-2397. 10.1128/AEM.02587-07.
Klein D, Janda P, Steinborn R, Müller M, Salmons B, Günzburg WH: Proviral load determination of different feline immunodeficiency virus isolates using real-time polymerase chain reaction: influence of mismatches on quantification. Electrophoresis. 1999, 20 (2): 291-299. 10.1002/(SICI)1522-2683(19990201)20:2<291::AID-ELPS291>3.0.CO;2-R.
Pfaffl MW: A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Research. 2001, 29 (9): e45-10.1093/nar/29.9.e45.
Walsh PS, Metzger DA, Higuchi R: Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques. 1991, 10 (4): 506-513.
Santos MA: An improved method for the small scale preparation of bacteriophage DNA based on phage precipitation by zinc chloride. Nucleic Acids Res. 1991, 19 (19): 5442-10.1093/nar/19.19.5442.
Resch A, Fehrenbacher B, Eisele K, Schaller M, Götz F: Phage release from biofilm and planktonic Staphylococcus aureus cells. FEMS Microbiol Lett. 2005, 252 (1): 89-96. 10.1016/j.femsle.2005.08.048.
France DR, Markham NP: Epidemiological aspects of Proteus infections with particular reference to phage typing. J Clin Pathol. 1968, 21 (1): 97-102. 10.1136/jcp.21.1.97.
Poli MA, Rivera VR, Neal D: Sensitive and specific colorimetric ELISAs for Staphylococcus aureus enterotoxins A and B in urine and buffer. Toxicon. 2002, 40 (12): 1723-1726. 10.1016/S0041-0101(02)00202-7.
Sambrook J, Russell D: Molecular cloning: a laboratory manual. 2001, Cold Spring Harbor: Cold Spring Harbor Laboratory Press, 3
Acknowledgements
This work was supported by grants from the Swedish Research Council for Environment, Agricultural Sciences, and Spatial Planning (FORMAS) and by PathogenCombat, part of the European Commission's 6th Framework Programme. Dora Marta was awarded a grant by the Swedish Institute within the Guest Scholarship Programme 2008/2009.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
NWC participated in designing the study, in carrying out the cultivations, the expression analysis and phage induction analysis, and in drafting the manuscript. RC participated in designing the study, and in carrying out the cultivations, the expression analysis, the phage induction analysis, the ELISA, and the nucleotide sequence analysis. DM participated in carrying out the cultivations, the expression analysis, phage induction analysis and the ELISA. AS participated in the phage induction analysis. JS and PR participated in designing the study and drafting the manuscript. All authors read and approved the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wallin-Carlquist, N., Cao, R., Márta, D. et al. Acetic acid increases the phage-encoded enterotoxin A expression in Staphylococcus aureus. BMC Microbiol 10, 147 (2010). https://doi.org/10.1186/1471-2180-10-147
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-10-147