
Abstract
Background
Myostatin (MSTN) is a member of the transforming growth factor-β superfamily that negatively regulates growth of skeletal muscle tissue. The gene encoding for the MSTN peptide is a consolidate candidate for the enhancement of productivity in terrestrial livestock. This gene potentially represents an important target for growth improvement of cultured finfish.
Results
Here we report molecular characterization, tissue expression and sequence variability of the barramundi (Lates calcarifer) MSTN-1 gene. The barramundi MSTN-1 was encoded by three exons 379, 371 and 381 bp in length and translated into a 376-amino acid peptide. Intron 1 and 2 were 412 and 819 bp in length and presented typical GT...AG splicing sites. The upstream region contained cis-regulatory elements such as TATA-box and E-boxes. A first assessment of sequence variability suggested that higher mutation rates are found in the 5' flanking region with several SNP's present in this species. A putative micro RNA target site has also been observed in the 3'UTR (untranslated region) and is highly conserved across teleost fish. The deduced amino acid sequence was conserved across vertebrates and exhibited characteristic conserved putative functional residues including a cleavage motif of proteolysis (RXXR), nine cysteines and two glycosilation sites. A qualitative analysis of the barramundi MSTN-1 expression pattern revealed that, in adult fish, transcripts are differentially expressed in various tissues other than skeletal muscles including gill, heart, kidney, intestine, liver, spleen, eye, gonad and brain.
Conclusion
Our findings provide valuable insights such as sequence variation and genomic information which will aid the further investigation of the barramundi MSTN-1 gene in association with growth. The finding for the first time in finfish MSTN of a miRNA target site in the 3'UTR provides an opportunity for the identification of regulatory mutations on the expression of this gene.
Similar content being viewed by others


Background
The transforming growth factor-β (TGF-β) superfamily encompasses a number of peptides, such as inhibins and activins, which share a similar structure and a relatively conserved amino acid sequence. Transforming growth factors are involved in important biological functions, including cell growth and differentiation [1]. Myostatin (MSTN), formerly known as growth differentiation factor-8, is a member of the TGF-β superfamily recently isolated from murine muscle tissue by McPherron et al. [2]. The biological activity model proposed for MSTN is experimentally supported by gene inactivation and revealed a negative correlation between MSTN expression and growth/number of muscle fibres [2]. The lack of evident non-specific defects in null-MSTN mice suggests that mammalian MSTN physiological functions are essentially confined to skeletal muscle. To corroborate this hypothesis, the MSTN gene is found expressed almost exclusively in skeletal muscle and heart of higher vertebrates [2, 3]. Alternatively, piscine MSTN transcripts have been ubiquitously detected in tissues such as muscle, gill, brain, kidney and gonad, highlighting that in lower vertebrates MSTN is possibly involved in several developmental and physiological functions [4, 5]. However, MSTN activity has been successfully inhibited in model fish species via N-terminal pro-peptide over-expression, morpholino, or double strand RNA interference, resulting in some degree of hypertrophic and/or hyperplasic muscle mass increase. This suggests that regulation of skeletal growth has remained the principal developmental role of MSTN across all vertebrates [6–8].
The MSTN gene has been cloned and characterized in a large number of high value commercial fish species such as atlantic salmon [9], shi drum [10], rainbow trout [11], gilthead sea bream [12], white bass [13], Mozambique tilapia [13], catfish spp. [14–16], European sea bass [17], striped bass [18], white perch [18], orange spotted grouper [19], Japanese sea perch [20] and croceine croaker [21]. In fish, this gene consists of three exons and two introns. Exon 1, encoding for the N-terminal signal sequence for secretion, contains the highest inter-specific variability, while exons 2 and 3 are highly conserved across species and are translated into the pro-peptide and C-terminal bioactive dimer [15, 21]. Alternative forms of MSTN have been independently isolated in zebrafish, gilthead sea bream, fugu and salmonid spp. [9, 11, 22, 23]. Different rates of identity shared between alternative MSTN isoforms suggests that at least two events of duplication occurred in finfish. A first event, which separated MSTN-1 and MSTN-2, occurred early during teleost evolution, while a second event, likely due to tetraploidization, occurred in salmonids (MSTN1a-2a; MSTN1b-2b) [24]. According to Rodgers et al. (2007), the new nomenclature proposed for the MSTN gene family has been adopted in this study.
The desire to select fast growing strains for the rapid improvement of livestock production in selective breeding programs has encouraged the identification of genes with a putative role in the enhancement of growth. Several authors have attempted to establish a quantitative correlation between production trait variability and genetic markers. For example, a single nucleotide polymorphism (SNP) occurring at intron 3 of the chicken growth hormone gene was associated with a 16.8% enhancement of body weight [25], and the "long allele" of a microsatellite described at the cattle g rowth hormone receptor promoter corresponded to a 6.9% production improvement compared to the "short allele" [26]. Therefore, targeting the improvement of a trait under quantitative genetic control with a single-gene approach (marker assisted selection) ensures more predictable and consistent outcomes compared to widely used traditional breeding techniques, reflecting an increasing demand for additional genes to be characterized in commercial animal production species.
Because of the unquestionable involvement of MSTN in differentiation and proliferation of muscle fibres, MSTN gene is considered a candidate for livestock growth improvement. However, while several authors have studied phenotypic implications of naturally occurring mutations in terrestrial livestock [27–31], only a few studies have described polymorphisms in the piscine MSTN gene. Maccatrozzo et al. [10] detected variation in a Simple Sequence Repeat (SSR) present at the 3'UTR of shi drum (Umbrina cirrosa), whereas Kocabas et al. [15] described several synonymous and non-synonymous mutations in the catfish MSTN gene, including a length polymorphism of a CAG-microsatellite present at exon 1. Surprisingly, neither microsatellites in non-coding regions (probable source of variation in traits under quantitative genetic control such as growth [32]), nor variation at the exons have been investigated in association with growth traits in finfish thus far.
Barramundi (Lates calcarifer) is a high-value aquaculture fish endemic and widely farmed in south-east Asia and Australia and with increasing aquaculture interest in Europe and North America [33, 34]. On account of the increasing worldwide aquaculture importance of this species, a growing number of farmers have expressed interest in maximising production through selective breeding. Ongoing experiments carried out in our research group have revealed extremely large size variability in F1 hatchery juveniles grown under identical conditions (Smith-Keune et al., unpublished data). The possibility that a certain amount of the observed variability might be explained by means of additive effects at the MSTN gene encouraged us to commence this investigation. The aims of this study, therefore, were to characterize the MSTN-1 gene in barramundi and to understand the gene's tissue-specific expression pattern. The project also aimed to identify possible regions of sequence variability among barramundi individuals that could be tested for their correlation with phenotypic growth in future studies.
Results
Sequence characterization and analysis
PCR amplifications from barramundi tissues resulted in six fragments of the following sizes: 925 bp (Myo-pro/Myo-L8), 384 bp (Myo-up1/Jcu-R), 765 bp (Myo-up2/Myo-L1), 1068 bp (Myo-up3/Myo-L3), 749 bp (Myo-up5/Myo-L5) and 831 bp (Myo-up8/Myo-utr). The relative position of each primer is shown in Fig. 1. The whole consensus sequence (4484 bp) (Fig. 2), including 5' and 3' flanking region, was obtained by merging overlapping PCR fragments and has been deposited in Genbank [35] (accession number EF672685). Gene structure of the putative barramundi MSTN-1 gene (LcMSTN-1) was deduced by cross-comparing gDNA consensus sequence, the entire cDNA sequence from a single clone and the gilthead sea bream MSTN-1 gene [12]. Three exons were shown to be encoded by this gene in barramundi. Exon 1, exon 2 and exon 3 were 379, 371, 381 bp in length, whereas the enclosed introns were 412 and 810 bp, respectively. Distinctive sites were recognized: i) a typical start codon (ATG) in exon 1; ii) a stop codon (TGA) in exon 3; iii) distinctive splicing sites (GT...AG) at the exon-intron boundaries; iv) a putative TATA box 137 bp upstream from the ATG start codon (28 bp upstream the putative start transcription site); v) two putative E-boxes (CANNTG motif), 811 and 398 bp upstream from the ATG starting codon. Four single nucleotide polymorphisms were also identified in the 5' flanking region among barramundi from the seven locations: i) a T → G transversion (position 280); ii) a C → G transition (position 306); iii) a C → T transition (position 490); iv) a A → G transition (position 612). BLASTN search results showed that nucleotide sequence produced significant alignments (above 90%) with MSTN-1 gene sequences in Genbank, displaying extremely high identity with most perciformes. Within the gene sequence, higher rates of similarity were shared among coding regions, in particular exons 2 and 3.

Schematization of the barramundi MSTN-1 gene. For clarity, the MSTN gene and deduced protein have not been represented in their real proportions. Primers sequences are presented in Table 1.

Genomic sequence of the barramundi MSTN-1 gene. Non-coding regions are represented italicized. TATA-box and E-boxes are grey-highlighted. Coding regions are shown in normal capital letters and deduced amino acid translation is reported in bolded single-letter code. Putative proteolytic and glycosilation sites are boxed and conserved cysteines underlined. Star represents STOP codon. This figure shows the upper quartile, for the full image please see Additional file 1.
The translated amino acid sequence (376 aa) was used as a query for a BLASTP search, showing significant similarities with most vertebrate MSTN genes, including chicken and human (Fig. 3). Protein alignment between species representative of various classes of vertebrates showed that the amino acid sequence obtained in the current study coded for a protein with homologous structure to that observed in other vertebrate MSTN containing a pro-peptide domain (residues 41–258) and a bioactive domain (residues 282–376). One putative conserved proteolytic RXXR motif for the releasing of the mature peptide (matching with RARR, residues 264–267) was identified along with two putative conserved glycosylation sites (N) in the propeptide domain (residues 73 and 223) (Fig. 2). Finally, as for all vertebrate MSTN proteins, 13 conserved cysteines were found, nine of which were in the bioactive domain, a characteristic shared by all the TGF-β superfamily.

Alignment of the barramundi MSTN-1 amino acid sequence with representative species of vertebrates. Numbers on the right indicate amino acid positions. References and accession numbers are provided in appendix.
Characterization of micro RNA target sites
Micro RNA (miRNA) target site analyses carried out on the barramundi 3'UTR genomic sequence revealed one putative site interacting with dre-let-7a, dre-let-7b and dre-let-7c, members of the let-7 miRNA family originally isolated in zebrafish (Fig. 4) [36]. A similar analysis of target sites in other teleost species indicated that all perciform MSTN-1 putatively interact with some members of the let-7 miRNA family. In particular, interaction of dre-let-7c was common to most species, with the only exception being gilthead seabream (data not shown). Atlantic salmon also displayed (with weak support value) a putative target site for dre-let-7d in a different 3'UTR region than the perciform counterpart, whereas no sites of interaction were found in zebrafish. An alignment of putative perciform interaction sites highlights 100% identity in the sequence complementary to the 5' critical region of the let-7 family (known as "seed") (5'-UGAGGUAG-3') (Fig. 5).

Graphic representation of barramundi MSTN-1 (3'UTR) MiRNA target sites. Double dot indicates G-U wobble bonds.

Alignment of 3'UTR putative target site. Bracket indicates the conserved site, matching with the critical 5' end of miRNA. Numbers refers to the nucleotide position relative to the barramundi MSTN-1. Abbreviations for species are provided in appendix.
Phylogenetic analyses
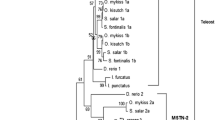
Phylogenetic analyses of the MSTN gene and related sequences indicates that LcMSTN-1 clusters with high support (100%) with that of Nile perch MSTN-1 gene in the perciform clade. Phylogeny of MSTN coding regions shows a clear separation between teleost orders (siluriformes, salmoniformes, perciformes) and the second MSTN isoform, which are all grouped together with extremely high support (Fig. 6). The resulting tree also suggests that the sequence isolated in this study is the LcMSTN-1 isoform. The overall bootstrap values are reasonably high providing strong statistical support for the main structure of the tree, which is also supported by the known phylogenies of the represented species. However, the perciform branch was not fully resolved based purely on phylogeny of MSTN coding regions. Increased resolution of perciformes MSTN-1 evolutionary relationships arises from phylogenetic analyses achieved when the whole gene sequence was analysed (coding and intron regions) (Fig. 7).

Phylogenetic tree of teleosts MSTN -1 and -2 coding regions. A maximum parsimony analysis was carried out using HsMSTN, MmMSTN and GgMSTN as outgroups. Numbers at the tree nodes represent percentage bootstrap support after 1000 replicates. Species abbreviations are shown in appendix. Tree topology and statistical support were similar for the NJ analysis (not shown).

Phylogenetic tree of perciformes whole MSTN-1 gene sequence. No outgroup was used for the whole gene analysis. Numbers at the tree nodes represent percentage bootstrap support after 1000 replicates. Species abbreviations are shown in appendix. Tree topology and statistical support were similar for the NJ analysis (not shown).
MSTN gene qualitative expression pattern
To further characterize the LcMSTN-1 gene, reverse transcriptase PCR (RT-PCR) was used to detect expression in eye, gill, heart, brain, muscle, gonad, intestine, spleen, kidney and liver (Fig. 8). The amplification products were identical in length and of the expected size. Cloning and sequencing the resulting 265 bp bands revealed that the fragments belonged to the LcMSTN-1 coding region. Transcripts were widely represented in each analyzed tissue by bands of variable intensities, suggesting that a possible differential expression pattern might occur among tissues. After 30 cycles, LcMSTN-1 transcripts were clearly visible in muscle and eye, intermediately detected in liver and kidney and weakly observed in brain, spleen and intestine. However, after 35 cycles, LcMSTN-1 transcripts were present in each tissue with very weak intensity in gills and heart. Amplification of RT control reactions showed no amplification products (data not shown).

Tissue-specific qualitative expression of the barramundi MSTN-1 gene. Reverse transcriptase PCR was conducted for 30 cycles (30×) and 35 cycles (35×). Lanes are represented as follows: Marker; E, eye; G, gill; H, heart; B, brain; M, muscle; Go, gonad; I, intestine; S, spleen; K, kidney; L, liver; C, negative control. As a positive control of the RT-PCR reaction step, a 94 bp fragment was successfully amplified in 18S rRNA.
Discussion
In the present investigation we isolated and characterized the MSTN-1 gene from barramundi. The LcMSTN-1 contains three exons and two introns, as for all known vertebrate MSTN, and encodes for a 376 amino acid long peptide. Like other members of the TGF-β superfamily, MSTN is synthesized as a precursor peptide, which includes an N-terminal signal sequence, pro-peptide and bioactive domain [2]. Results of the BLASTP query show that Lc MSTN-1 protein structure is consistent with that previously reported by other authors [15, 20]. In accordance with past studies, the peptide sequence is highly conserved in the C-terminal bioactive domain, where nine cysteine residues are present [1, 2, 20]. The nine conserved cysteines are involved in the formation of interchain disulphide-bonds, essential for the dimerization of the two MSTN subunits [37]. Two putative glycosylation sites were also identified in the pro-peptide domain. Glycosylation sites have also been demonstrated to be actively involved in the regulation of TGF-β superfamily bioactivity [38, 39]. Finally, the RXXR motif (matching with RARR in LcMSTN-1) identified at residues 264–267 represents the putative cleavage site to release the processed mature peptide. The putative functional and structural roles associated with the above mentioned sites are corroborated by the extremely high identity maintained throughout the evolution of vertebrates.
With regard to the nucleotide sequence, LcMSTN-1 shares high similarity with most known perciform MSTN-1, particularly in the coding region. The 5' flanking region contains putative regulatory sites including a TATA box, located 28 bp upstream of the transcription starting point, and two E-boxes. The TATA box surrounding sequence (± 30 bp) is highly conserved across teleost species, sharing in this region 100% identity with other available sequence from perciformes. The E-box 1 is also found conserved in the same group of species that share the identical TATA box region. E-boxes have been shown to interact with the helix-loop-helix proteins of myogenic regulatory factors enhancing the expression of the MSTN gene in vertebrates, therefore representing interesting target sites for the screening of regulatory mutations [40].
A first screening of sequences from different Australian populations highlights that LcMSTN-1 is variable in the 5' flanking region, where four SNPs have been identified. These point mutations, however, did not appear to result in changes in putative functional sites. A lack of major polymorphisms in the coding region suggests that functional or loss-of-function mutations might be rarer in fish than in higher vertebrates [28, 31]. However, the number of individuals targeted in this study did not provide enough elements for robust conclusions. As wild fish are continuously utilized in hatcheries for the production of barramundi larvae, it is essential in future studies to screen a larger number of individuals/populations and understand the spatial genetic variability of LcMSTN-1. A better understanding of LcMSTN-1 variability will allow selection of appropriate brood-stocks in an experimental MSTN-based selective breeding program. Finally, it is notable that repeated motifs of significant length were not found in non-coding regions. In particular, from sequence alignment emerges that intron 2 contains a (CA)n-SSR (Simple Sequence Repeat) which is observed to be extremely conserved across perciformes and salmonids, once again suggesting a possible functional role (i.e. gene expression regulation [32]). In barramundi the CA-microsatellite is reduced to five repeated units.
The central finding of this study is the identification of a miRNA target in the 3'UTR. MiRNA are small non-coding RNA (~22 nucleotide) that can actively regulate the expression of several genes at the post-transcriptional level by physical interaction with complementary mRNA sequences [41]. Sites presenting perfect complementarity at position 2–8 of the 5' end of the miRNA are likely to be targeted by the miRNA itself [42], but sites with one mismatch in the target sequence have also been considered as seeds [43]. Herein, a site located in the 3'UTR of LcMSTN-1 and highly conserved across perciformes represents a putative target for the let-7 miRNA family. To support this finding, members of the let-7 family have been predicted, via evolutionary conservation, to interfere with the TGF-β signalling pathway [43]. To highlight the importance of gene expression regulation at the post-transcriptional level, a recent study has described a polymorphism in the 3'UTR of the sheep MSTN gene that generates an illegitimate new target site for two miRNAs [44]. This point mutation was statistically associated with an increased hypertrophy of muscle fibres, therefore defining a new class of regulatory mutations. The homologous site of interaction, herein described for the first time in finfish, represents a possible valuable target for the identification of fast growing haplotypes. Further, miRNAs may also play a pivotal developmental role inhibiting the translation of MSTN in lower vertebrates in a tissue-specific manner.
It has been well established that the piscine MSTN gene evolved subject to different events of duplication and in some cases tissue-specific differential expression of MSTN- 1 and MSTN-2 was detected [9, 11, 22, 23, 45]. Loss of physiological function might have evolved in parallel to MSTN duplication events as, for example, in the case of salmonids where different MSTN isoforms are specifically expressed in distinct tissues [9, 11, 45]. However, it is possible that miRNAs have a crucial role in regulating the expression of piscine MSTN genes. The latter hypothesis is strongly supported by miRNA differential expression detected in teleost fishes [46] and might also explain the loss of the miRNA target site observed in salmonids compared to the sister clade of perciformes. Therefore, it would be useful in future studies to target the miRNA pathway as a source of variation for the identification of fast growing mutants and the improvement of finfish productivity. In fact, although mutations occurring at MSTN miRNA target sites have been shown to affect muscularity in higher vertebrates [44], the post-transcriptional regulatory pathway has not been targeted in finfish to date.
The phylogeny of the MSTN gene has been widely assessed by previous authors and is largely consistent with the current study [12, 20, 21, 47]. However, former investigations have been carried out on protein sequence of a limited number of species [12, 21]. Thanks to recent intensive submissions of piscine MSTN genes to public databases, we present the first attempt to assess the utility of MSTN introns as a phylogenetic nuclear marker. In accordance with the tree presented by Ye et al. [20], phylogeny of coding regions was unable to produce a fully resolved perciform clade. On the contrary, when coding regions were analyzed in conjunction with introns a better picture of perciformes evolutionary relationships emerged. In particular, largemouth sea bass, Japanese sea perch and orange spotted grouper were clustered together, with high bootstrap values supporting the monophyly of this clade. The resolution within the perciformes using the complete MSTN-1 gene is consistent with that of Smith et al [48] based on 4036 bp of mitochondrial and nuclear sequence, demonstrating that MSTN also provides strong phylogenetic signal and has utility as a marker for future broader piscine phylogeny studies.
The principal biological activity of MSTN is to negatively regulate growth of muscle fibres [2]. The suppression of hyperplasic and hypertrophic growth of somites triggered by MSTN has been experimentally demonstrated in vertebrates, where the inactivation of protein synthesis leads to an extraordinary increase of muscle mass both in fish and mammals [2, 6]. However, in mammals MSTN physiological activity seems to be restricted to skeletal muscle and cardiomyocytes and major loss-of-function natural mutations have been identified, causing giant phenotypes without any other detectable side effects [3, 28, 49]. On the contrary, previous investigations are contrasting with regard to MSTN expression in spleen, kidney, liver and intestine of teleost fishes. In particular, tissues that showed no MSTN transcript detection included kidney in Japanese sea perch [20], spleen in croceine croaker [21], gonad, liver and spleen in gilthead seabream [12] and adipose tissue, liver, spleen, intestine and kidney in shi drum [10]. In the present study, MSTN transcripts were expressed at some level in each target tissue examined. However, in gills and heart the expression was so weak that 35 amplification cycles were needed to visualize a PCR product. These results do demonstrate, however, that in adult barramundi at least, MSTN is widely expressed in many tissues at the same time.
Although one aim of the present investigation was simply to determine the qualitative tissue-specific expression of the MSTN gene and quantitative conclusion without verification from quantitative PCR might be hazardous, it was clearly evident that LcMSTN-1 transcripts were more highly expressed in barramundi skeletal muscle than in other tissues. This is a similar finding to other fish studies where MSTN expression has been shown to be highest in skeletal muscle, and sometimes, one or other tissues like brain, eye or liver [9, 10, 16, 20]. Unlike many other studies, however, we observed some level of simultaneous LcMSTN-1 expression in skeletal muscle, eye, gill, heart, brain, muscle, gonad, intestine, spleen, kidney and liver. The fact that LcMSTN-1 transcripts were not found to be simultaneously expressed in such a wide range of non-muscle tissues in other fish might have depended on several factors including methodology (i.e. number of amplification cycles – in tissues where the transcripts are very weak a difference of five cycles is significant), tissue preservation (RNA degrades relatively quickly) or age effects on expression (MSTN expression intensity is significantly correlated with developmental stages [15]).
Conclusion
The LcMSTN-1 gene was successfully isolated and characterized. The gene displays high sequence identity and equal organization both with other teleost fish and mammalian counterparts. Putative functional sites have been recognized in LcMSTN-1, either in the translated protein and nucleotide sequence. LcMSTN-1 transcripts are ubiquitously expressed in several tissues with greater intensity in skeletal muscles. The high similarity observed across teleost, mammalian and avian species indicates that the main biological function of MSTN (muscle growth suppressor) has been evolutionary conserved to some extent, although some physiological activities might have been lost/acquired during the evolution of vertebrates. However, the successful association between MSTN gene variation and growth traits observed in higher vertebrates corroborates the importance that the MSTN gene might have for the improvement of aquaculture finfish production and encourages further investigations. Furthermore, a putative miRNA site of interaction identified in this study and localized in the 3'UTR suggests a possible new direction to be undertaken for the improvement of finfish productivity.
Methods
Sample collection
Three one-year-old barramundi were collected from a local barramundi farm. Fish were euthanized by diluting 1 ml clove oil per litre of water. Brain, muscle, liver, intestine, heart, eye, spleen, gills, gonads, and kidney were dissected, immediately preserved in liquid nitrogen and subsequently stored at -80°C until further processing. Fin clips were also removed and preserved in 100% ethanol. Furthermore, fin clips from seven wild-caught barramundi were collected from seven separated Australian locations (Ashburton River (Western Australia – WA), De Grey River (WA), Roebuck Bay (WA), Ord River (WA), Daly River (Northern Territory), Gulf of Carpentaria (Queensland – QLD), Boyle River (QLD)) to provide an opportunity for maximum levels of genetic variability and preserved in 100% ethanol.
Cloning and characterization of the MSTN gene
Total genomic DNA (gDNA) was extracted from barramundi fin clips by digestion in CTAB buffer with 20 mg/ml proteinase K for 1 hour at 60°C, followed by a phenol:chloroform:isoamyl alcohol purification protocol [50]. Genomic DNA was quantified by comparison to DNA concentration standards after 0.8% agarose gel electrophoresis using the ImageJ 1.33 software package (Wayne Rasband, National Institutes of Health, MA) and re-suspended in ddH2O to a concentration of 10 ng/μl.
Twelve specific oligonucleotides were designed based on conserved regions of known MSTN-1 sequences in perciformes (Table 1). Four pairs of primers, Myo-up1/Jcu-R, Myo-up2/Myo-L1, Myo-up3/Myo-L3, Myo-up5/Myo-L5 were designed. For the amplification of 5' and 3' UTRs (untranslated regions) two pairs of primers, Myo-pro/Myo-L8 and Myo-up8/Myo-utr (Table 1), were designed. PCR reactions were carried out in a final volume of 50 μl using the following final concentration of reagents: 1 × of 10× PCR buffer (Qiagen), 2 mM of MgCl2, 0.4 mM of dNTP mix, 0.4 μM of each primer, 0.75 units of Taq polymerase (Qiagen) and 10–20 ng of gDNA. The amplification cycle, performed in a MJ research thermocycler, consisted of 2 min pre-denaturation at 94°C followed by 32 cycles of denaturation at 94°C for 45 sec, annealing at specific temperature (Table 1) for 45 sec and elongation at 72°C for 1 min, followed by 10 min final extension at 72°C. The PCR fragments of expected size obtained from these amplifications were separated on 1.5% agarose gels, purified and cloned into a pGEM-T Easy Vector System (Promega, Madison, WI). Clones containing the insert of expected size were isolated and sequenced in both directions using T7 and SP6 universal primers. Fragment sequences were analysed and spliced in to a single consensus sequence using Sequencher version 4.2 (GeneCode Corp) and visually analysed for polymorphisms. In addition, the whole cDNA was sequenced in both directions from three single clones and compared to the gDNA sequence as a control.
Genomic sequence: nucleotide analysis
The resulting LcMSTN-1 consensus nucleotide and amino acid sequences were first compared with those available from the Genbank public database using BLASTN and BLASTP, respectively [51]. The intron-exon boundaries were deduced by alignment with cDNA sequence amplified using the Myo-up1/Myo-L3 and Myo-up3/Myo-L5 primer pairs (Table 1). Translated amino acid sequences were aligned for analysis of putative conserved functional residues by ClustalW using MEGA 3.1 [52]. Putative full-transcripts sequences, starting and stop codon and other functional sites where deduced by comparison with known MSTN sequences.
Micro RNA target sites identification
To find potential miRNA target sites, a further analysis of non-coding regions was carried out using the miRanda software package [53], using default settings and SHUFFLE mode ON (1000 random shuffled analysis). The entire collection of Danio rerio miRNA available from a public non-coding RNA database [52, 54–56] was used as a query and run against LcMSTN-1 3'UTR. Output parameters considered for the scope of this investigation were: i) score; ii) energy and iii) z-value (index of rudimental statistical test for the reliability of results) [53]. Threshold score and energy were set at 100 and -19 kcal/mol respectively. MiRNA showing a putative interaction with LcMSTN-1 3'UTR were then tested on representative teleosts species using default thresholds and parameters. Finally, presumed target sites of interactions were visually confirmed by screening sequence site conservation between relatively close species.
Phylogenetic analysis
Phylogenetic analysis of evolutionary relationship between LcMSTN-1 and other vertebrates was performed using two optimality criteria. Firstly, a maximum parsimony analysis with tree-bisection-reconnection (TBR) branch-swapping and gaps treated as missing characters was undertaken in PAUP version 4.08 [57]. Secondly, a neighbour-joining (NJ) tree was constructed using the Jukes-Cantor model of sequence evolution in MEGA 3.1 [52]. Support for both types of trees was obtained by bootstrapping data (1000 replicates). Sequences were rooted using Homo sapiens, Mus musculus and Gallus gallus as outgroups.
Expression analysis: RNA extraction, complementary DNA synthesis and Reverse Transcriptase PCR
Total RNA was extracted from preserved barramundi tissues with Trizol reagent (Invitrogen, Carlsbad, CA). RNA quality and DNA contamination were verified by 1.5% agarose gel electrophoresis. RNA concentration and purity was checked on a spectrophotometer using 230/260 and 260/280 nm wavelengths. Nucleic acid samples were diluted in ddH2O to a concentration of 100 ng/μl. First strand complementary DNA (cDNA) was synthesized from the same amount of total RNA (1 μg) for each sample using SuperScript III Reverse Transcriptase enzyme (SS-III) (Invitrogen, Carlsbad, CA). A mixture of (T)25 oligos and random hexamers were used as primers for the reverse transcriptase reaction. A further reaction was carried out without the SS-III (-RT) enzyme as a control. 2 μl of the resulting cDNA and -RT control were used as templates for the RT-PCR. RT-PCR reactions were prepared in a final volume of 25 μl with primers Myo-up3 and Myo-L3, designed to encompass intron 2 in order to eliminate potential positive results due to genomic contamination. Concentrations of reagents were maintained consistent with the ones previously used for gDNA amplifications. To confirm the specificity of the amplification, fragments of expected size (approximately 270 bp) were cloned and sequenced. In RT-PCR, 18S rRNA (18S-F and 18S-R primers, Table 1) were used as a control to test the quality of the reverse transcribed RNA. A 55 to 48°C step-down program was used for the 18S rRNA amplification reaction. The step-down program consisted of: 94°C × 3 min (pre-denaturation); 4× (94°C × 30 sec + 55°C × 40 sec + 72°C × 1 min) + 4× (94°C × 30 sec + 50°C × 40 sec + 72°C × 1 min) + 20× (94°C × 30 sec + 48°C × 40 sec + 72°C × 1 min); 72°C × 10 min (final extension).
Appendix
List of abbreviations and Genbank accession number of fish species used in alignments and phylogenetic analyses (see Table 2).
References
Massague J: The Transforming Growth-Factor-Beta Family. An Rev Cell Biol. 1990, 6: 597-641.
McPherron AC, Lawler AM, Lee SJ: Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997, 387 (6628): 83-90. 10.1038/387083a0.
Sharma M, Kambadur R, Matthews KG, Somers WG, Devlin GP, Conaglen JV, Fowke PJ, Bass JJ: Myostatin, a transforming growth factor-&bgr; superfamily member, is expressed in heart muscle and is upregulated in cardiomyocytes after infarct. J Cell Physiol. 1999, 180 (1): 1-9. 10.1002/(SICI)1097-4652(199907)180:1<1::AID-JCP1>3.0.CO;2-V.
Radaelli G, Rowlerson A, Mascarello F, Patruno M, Funkenstein B: Myostatin precursor is present in several tissues in teleost fish: a comparative immunolocalization study. Cell Tissue Res. 2003, V311 (2): 239-250.
Roberts SB, Goetz FW: Myostatin protein and RNA transcript levels in adult and developing brook trout. Mol Cell Endocrinol. 2003, 210 (1–2): 9-20. 10.1016/j.mce.2003.09.002.
Acosta J, Carpio Y, Borroto I, Gonzalez O, Estrada MP: Myostatin gene silenced by RNAi show a zebrafish giant phenotype. J Biotechnol. 2005, 119 (4): 324-331. 10.1016/j.jbiotec.2005.04.023.
Amali AA, Lin CJF, Chen YH, Wang WL, Gong HY, Lee CY, Ko YL, Lu JK, Her GM, Chen TT, Wu JL: Up-regulation of muscle-specific transcription factors during embryonic somitogenesis of zebrafish (Danio rerio) by knock-down of myostatin-1. Dev Dynam. 2004, 229 (4): 847-856. 10.1002/dvdy.10454.
Xu C, Wu G, Zohar Y, Du SJ: Analysis of myostatin gene structure, expression and function in zebrafish. J Exp Biol. 2003, 206 (22): 4067-4079. 10.1242/jeb.00635.
Ostbye T-K, Galloway TF, Nielsen C, Gabestad I, Bardal T, Andersen O: The two myostatin genes of Atlantic salmon (Salmo salar) are expressed in a variety of tissues. Eur J Biochem. 2001, 268 (20): 5249-5257. 10.1046/j.0014-2956.2001.02456.x.
Maccatrozzo L, Bargelloni L, Patarnello P, Radaelli G, Mascarello F, Patarnello T: Characterization of the myostatin gene and a linked microsatellite marker in shi drum (Umbrina cirrosa, Sciaenidae). Aquaculture. 2002, 205 (1–2): 49-60. 10.1016/S0044-8486(01)00659-7.
Rescan P-Y, Jutel I, Ralliere C: Two myostatin genes are differentially expressed in myotomal muscles of the trout (Oncorhynchus mykiss). J Exp Biol. 2001, 204 (20): 3523-3529.
Maccatrozzo L, Bargelloni L, Radaelli G, Mascarello F, Patarnello T: Characterization of the myostatin gene in the gilthead seabream (Sparus aurata): Sequence, genomic structure, and expression pattern. Mar Biotechnol. 2001, 3 (3): 224-230. 10.1007/s101260000064.
Rodgers BD, Weber GM, Sullivan CV, Levine MA: Isolation and characterization of myostatin complementary deoxyribonucleic acid clones from two commercially important fish: Oreochromis mossambicus and Morone chrysops. Endocrinology. 2001, 142 (4): 1412-1418. 10.1210/en.142.4.1412.
Gregory DJ, Waldbieser GC, Bosworth BG: Cloning and characterization of myogenic regulatory genes in three Ictalurid species. Anim Genet. 2004, 35 (6): 425-430. 10.1111/j.1365-2052.2004.01193.x.
Kocabas AM, Kucuktas H, Dunham RA, Liu Z: Molecular characterization and differential expression of the myostatin gene in channel catfish (Ictalurus punctatus). Biochimica et Biophysica Acta (BBA) – Gene Structure and Expression. 2002, 1575 (1–3): 99-107. 10.1016/S0167-4781(02)00289-0.
Pan J, Wang X, Song W, Chen J, Li C, Zhao Q: Molecular cloning and expression pattern of myostatin gene in yellow catfish (Pelteobagrus fulvidraco). DNA Sequence. 2007, 18 (4): 279-287. 10.1080/10425170701243492.
Terova G, Bernardini G, Binelli G, Gornati R, Saroglia M: cDNA encoding sequences for myostatin and FGF6 in sea bass (Dicentrarchus labrax, L.) and the effect of fasting and refeeding on their abundance levels. Dom Anim Endocrinol. 2006, 30 (4): 304-319. 10.1016/j.domaniend.2005.08.003.
Rodgers BD, Weber GM: Sequence conservation among fish myostatin orthologues and the characterization of two additional cDNA clones from Morone saxatilis and Morone americana. Comp Biochem Physiol B. 2001, 129 (2–3): 597-603. 10.1016/S1096-4959(01)00350-5.
Ko C-F, Chiou T-T, Chen T, Wu J-L, Chen J-C, Lu J-K: Molecular Cloning of Myostatin Gene and Characterization of Tissue-Specific and Developmental Stage-Specific Expression of the Gene in Orange Spotted Grouper, Epinephelus coioides. Marine Biotechnol. 2007, 9 (1): 20-32. 10.1007/s10126-006-6059-8.
Ye H-Q, Chen S-L, Sha Z-X, Liu Y: Molecular Cloning and Expression Analysis of the Myostatin Gene in Sea Perch (Lateolabrax japonicus). Mar Biotechnol. 2007, 9 (2): 262-272. 10.1007/s10126-006-6093-6.
Xue L, Qian K, Qian H, Li L, Yang Q, Li M: Molecular Cloning and Characterization of the Myostatin Gene in Croceine Croaker, Pseudosciaena crocea. Mol Biol Rep. 2006, V33 (2): 129-135. 10.1007/s11033-006-0015-6.
Biga PR, Roberts SB, Iliev DB, McCauley LAR, Moon JS, Collodi P, Goetz FW: The isolation, characterization, and expression of a novel GDF11 gene and a second myostatin form in zebrafish, Danio rerio. Comp Biochem Physiol B. 2005, 141 (2): 218-230. 10.1016/j.cbpc.2005.03.004.
Maccatrozzo L, Bargelloni L, Cardazzo B, Rizzo G, Patarnello T: A novel second myostatin gene is present in teleost fish. FEBS Letters. 2001, 509 (1): 36-40. 10.1016/S0014-5793(01)03124-6.
Rodgers BD, Roalson EH, Weber GM, Roberts SB, Goetz FW: A proposed nomenclature consensus for the myostatin gene family. Am J Physiol Endocrinol Metab. 2007, 292 (2): E371-372. 10.1152/ajpendo.00395.2006.
Nie Q, Sun B, Zhang D, Luo C, Ishag NA, Lei M, Yang G, Zhang X: High Diversity of the Chicken Growth Hormone Gene and Effects on Growth and Carcass Traits. J Hered. 2005, 96 (6): 698-703. 10.1093/jhered/esi114.
Hale CS, Herring WO, Shibuya H, Lucy MC, Lubahn DB, Keisler DH, Johnson GS: Decreased growth in Angus steers with a short TG-microsatellite allele in the P1 promoter of the growth hormone receptor gene. J Anim Sci. 2000, 78 (8): 2099-2104.
Li MH, Adamowicz T, Switonski M, Ammosov I, Ivanova Z, Kiselyova T, Popov R, Kantanen J: Analysis of population differentiation in North Eurasian cattle (Bos taurus) using single nucleotide polymorphisms in three genes associated with production traits. Anim Genet. 2006, 37 (4): 390-392. 10.1111/j.1365-2052.2006.01479.x.
McPherron AC, Lee SJ: Double muscling in cattle due to mutations in the myostatin gene. PNAS. 1997, 94 (23): 12457-12461. 10.1073/pnas.94.23.12457.
Gu ZL, Zhu DH, Li N, Li H, Deng XM, Wu CX: The single nucleotide polymorphisms of the chicken myostatin gene are associated with skeletal muscle and adipose growth. Sci China Ser C. 2004, 47 (1): 25-30. 10.1360/02yc0201.
Jiang YL, Li N, Fan XZ, Xiao LR, Xiang RL, Hu XX, Du LX, Wu CX: Associations of T -> A mutation in the promoter region of myostatin gene with birth weight in Yorkshire pigs. AJAS. 2002, 15 (11): 1543-1545.
Kambadur R, Sharma M, Smith TPL, Bass JJ: Mutations in myostatin (GDF8) in double-muscled Belgian blue and Piedmontese cattle. Gen Res. 1997, 7 (9): 910-916.
Kashi Y, King D, Soller M: Simple sequence repeats as a source of quantitative genetic variation. Trends Genet. 1997, 13 (2): 74-78. 10.1016/S0168-9525(97)01008-1.
O'Sullivan D: Australian barra for New York. Austasia Aquaculture. 2004, 18: 40-43.
Tucker JW, Russell DJ, Rimmer M: Barramundi culture: A success story for aquaculture in Asia and Australia. World aquaculture. 2002, 33 (3): 67-72.
GenBank. [http://www.ncbi.nlm.nih.gov/Genbank]
Chen PY, Manninga H, Slanchev K, Chien M, Russo JJ, Ju J, Sheridan R, John B, Marks DS, Gaidatzis D, Sander C, Zavolan M, Tuschl T: The developmental miRNA profiles of zebrafish as determined by small RNA cloning. Genes Dev. 2005, 19 (11): 1288-1293. 10.1101/gad.1310605.
Daopin S, Piez KA, Ogawa Y, Davies DR: Crystal-Structure of Transforming Growth-Factor-Beta-2 – an Unusual Fold for the Superfamily. Science. 1992, 257 (5068): 369-373. 10.1126/science.1631557.
Brunner AM, Lioubin MN, Marquardt H, Malacko AR, Wang WC, Shapiro RA, Neubauer M, Cook J, Madisen L, Purchio AF: Site-Directed Mutagenesis of Glycosylation Sites in the Transforming Growth Factor-Beta-1 (Tgf-Beta-1) and Tgf-Beta-2 (414) Precursors and of Cysteine Residues within Mature Tgf-Beta-1 – Effects on Secretion and Bioactivity. Mol Endocrinol. 1992, 6 (10): 1691-1700. 10.1210/me.6.10.1691.
Sha X, Yang L, Gentry LE: Identification and Analysis of Discrete Functional Domains in the Pro Region of Pre-Pro-Transforming Growth Factor-Beta-1. J Cell Biol. 1991, 114 (4): 827-839. 10.1083/jcb.114.4.827.
Spiller MP, Kambadur R, Jeanplong F, Thomas M, Martyn JK, Bass JJ, Sharma M: The myostatin gene is a downstream target gene of basic helix-loop-helix transcription factor MyoD. Mol Cell Biol. 2002, 22 (20): 7066-7082. 10.1128/MCB.22.20.7066-7082.2002.
Bartel DP, Chen CZ: Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet. 2004, 5 (5): 396-400. 10.1038/nrg1328.
Lewis BP, Burge CB, Bartel DP: Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005, 120 (1): 15-20. 10.1016/j.cell.2004.12.035.
Gaidatzis D, van Nimwegen E, Hausser J, Zavolan M: Inference of miRNA targets using evolutionary conservation and pathway analysis. Bmc Bioinformatics. 2007, 8: 69-10.1186/1471-2105-8-69.
Clop A, Marcq F, Takeda H, Pirottin D, Tordoir X, Bibe B, Bouix J, Caiment F, Elsen JM, Eychenne F, Larzul C, Laville E, Meish F, Milenkovic D, Tobin J, Charlier C, Georges M: A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet. 2006, 38 (7): 813-818. 10.1038/ng1810.
Roberts SB, Goetz FW: Differential skeletal muscle expression of myostatin across teleost species, and the isolation of multiple myostatin isoforms. FEBS Letters. 2001, 491 (3): 212-216. 10.1016/S0014-5793(01)02196-2.
Kloosterman WP, Steiner FA, Berezikov E, de Bruijn E, van de Belt J, Verheul M, Cuppen E, Plasterk RHA: Cloning and expression of new microRNAs from zebrafish. Nucl Acids Res. 2006, 34 (9): 2558-2569. 10.1093/nar/gkl278.
Kerr T, Roalson EH, Rodgers BD: Phylogenetic analysis of the myostatin gene sub-family and the differential expression of a novel member in zebrafish. Evol Devel. 2005, 7 (5): 390-400. 10.1111/j.1525-142X.2005.05044.x.
Smith WL, Craig MT: Casting the percomorph net widely: The importance of broad taxonomic sampling in the search for the placement of serranid and percid fishes. Copeia. 2007 (1): 35-55. 10.1643/0045-8511(2007)7[35:CTPNWT]2.0.CO;2.
Schuelke M, Wagner KR, Stolz LE, Hubner C, Riebel T, Komen W, Braun T, Tobin JF, Lee S-J: Myostatin Mutation Associated with Gross Muscle Hypertrophy in a Child. N Engl J Med. 2004, 350 (26): 2682-2688. 10.1056/NEJMoa040933.
Sambrook J, Fritsch E, Maniatus T: Molecular cloning: a laboratory manual. 1989, Cold 12 Spring Harbour Laboratory Press, New York, 2
BLAST (Basic Local Alignment Search Tool). [http://www.ncbi.nlm.nih.gov/BLAST]
Kumar S, Tamura K, Nei M: MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform. 2004, 5: 150-163. 10.1093/bib/5.2.150.
Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS: MicroRNA targets in Drosophila. Gen Biol. 2004, 5 (1):
Non coding RNA database. [http://biobases.ibch.poznan.pl/ncRNA]
Griffiths-Jones S: The microRNA Registry. Nucl Acids Res. 2004, 32 (suppl_1): D109-111. 10.1093/nar/gkh023.
Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ: miRBase: microRNA sequences, targets and gene nomenclature. Nucl Acids Res. 2006, 34 (suppl_1): D140-144. 10.1093/nar/gkj112.
Swofford DL: PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4. 2000, Sinauer Associates, Sunderland, Massachusetts
Acknowledgements
We are grateful to Dr. Serena Aceto for contributing in the analysis of data. This work was funded in part by the JCU Finfish and Emerging Aquaculture Species Research Advancement Program Grant awarded to DRJ.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
CDS and DRJ conceived the intellectual design of the project and co-wrote the manuscript. Laboratory work was undertook by CDS. CSK and BSE formed part of the supervisory team and provided intellectual input into experimental design, statistical analyzes and laboratory procedures. All authors read and approved the final manuscript.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
De Santis, C., Evans, B.S., Smith-Keune, C. et al. Molecular characterization, tissue expression and sequence variability of the barramundi (Lates calcarifer) myostatin gene. BMC Genomics 9, 82 (2008). https://doi.org/10.1186/1471-2164-9-82
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-9-82