
Abstract
Background
Microsatellites or single sequence repeats (SSRs) are a powerful choice of marker in the study of Phytophthora population biology, epidemiology, ecology, genetics and evolution. A strategy was tested in which the publicly available unigene datasets extracted from genome sequences of P. infestans, P. sojae and P. ramorum were mined for candidate SSR markers that could be applied to a wide range of Phytophthora species.
Results
A first approach, aimed at the identification of polymorphic SSR loci common to many Phytophthora species, yielded 171 reliable sequences containing 211 SSRs. Microsatellites were identified from 16 target species representing the breadth of diversity across the genus. Repeat number ranged from 3 to 16 with most having seven repeats or less and four being the most commonly found. Trinucleotide repeats such as (AAG)n, (AGG)n and (AGC)n were the most common followed by pentanucleotide, tetranucleotide and dinucleotide repeats. A second approach was aimed at the identification of useful loci common to a restricted number of species more closely related to P. sojae (P. alni, P. cambivora, P. europaea and P. fragariae). This analysis yielded 10 trinucleotide and 2 tetranucleotide SSRs which were repeated 4, 5 or 6 times.
Conclusion
Key studies on inter- and intra-specific variation of selected microsatellites remain. Despite the screening of conserved gene coding regions, the sequence diversity between species was high and the identification of useful SSR loci applicable to anything other than the most closely related pairs of Phytophthora species was challenging. That said, many novel SSR loci for species other than the three 'source species' (P. infestans, P. sojae and P. ramorum) are reported, offering great potential for the investigation of Phytophthora populations. In addition to the presence of microsatellites, many of the amplified regions may represent useful molecular marker regions for other studies as they are highly variable and easily amplifiable from different Phytophthora species.
Similar content being viewed by others


Background
The genus Phytophthora, with other Oomycetes, fall within the kingdom Stramenopila, which also includes golden-brown algae, diatoms, and brown algae such as kelp [1–4]. This genus stands out among the plant pathogens since a significant number of the 80 or so described species continue to prove a threat to ecosystem stability and plant productivity on a global scale [5–8]. Despite the importance of Phytophthora species, studies of their molecular diversity have been limited by the power of the genetic markers and difficulties in comparing results among laboratories. Accurate studies based on the analysis of mitochondrial and nuclear DNA have resulted in a consensus of the phylogenetic relationships within the genus with a grouping into 10 genetically related clades now accepted [2, 3, 9]. However, these studies were based on genes commonly conserved within a species and therefore unsuitable to characterize intraspecific variability. Other approaches to study intraspecific variability among Phytophthora species including RAPD-PCR and AFLP have proved valuable within a particular study but comparing results from one laboratory to another has always proved challenging with such fingerprinting tools [10–13]. Although microsatellites or simple sequence repeats (SSRs) have been recognised as one of the most powerful choices of markers for molecular ecology they have only relatively recently been exploited in the study of Phytophthora populations. SSRs are tandemly repeated motifs of one to six bases which occur frequently and randomly in all eukaryotic genomes although their frequency varies significantly among different organisms [14]. They exhibit a high degree of length polymorphism among related organisms due to stepwise mutations affecting the number of repeat units and leading to polymorphism [14, 15]. Dinucleotide repeats account for the majority of microsatellites for many species whereas trinucleotide and hexanucleotide repeats are the most likely repeat classes to appear in coding regions because they do not cause a frameshift [16, 17]. Major advantages SSRs include: (i) multiple SSR alleles may be detected at a single locus using a simple PCR-based screen, (ii) SSRs are evenly distributed across the genome, (iii) they are co-dominant, (iv) very small quantities of DNA are required for screening, (v) analysis may be semi-automated, and (vi) results are objective compared to random amplification methods [18].
Microsatellites have been used to investigate genetic structure and reproductive biology of Oomycetes species including Plasmopara viticola, P. cinnamomi, P. infestans, and P. ramorum [19–21, 23–25]. However, a major limitation to their wider exploitation is the need for prior species-specific marker isolation that requires knowledge of the DNA sequence of the SSR flanking regions to which specific primers have to be designed. Such regions are usually conserved within a species but the likelihood of primers successfully working between species decreases with increasing genetic distance and, in practice, primers are usually developed anew for each species [25, 26]. Common methods for the discovery of SSR loci are based on constructing genomic DNA libraries enriched for SSR sequences. These methods were utilised for P. cinnamomi and P. ramorum, however they are time-consuming, and the specific sequencing of DNA libraries required is expensive [20, 25]. Many commercial and academic laboratories specialise in microsatellite isolation services and can provide a set of polymorphic microsatellite loci for a new species in 3–6 months for a cost of approximately USD 1,500 per locus, or USD 10,000 for 10–15 loci [14].
The availability of entire genome sequences for an increasing number of species including P. infestans http://www.broad.mit.edu/, P. ramorum and P. sojae http://genome.jgi-psf.org/ have proved novel opportunities to identify and evaluate potential SSR markers identified by computational tools (Abajian, 1994, http://espressosoftware.com/pages/sputnik.jsp) [27, 28]. This approach has been utilised to identify SSRs for the study of European and USA populations of P. ramorum and for monitoring the genetic variation in populations of P. infestans across Europe and worldwide [23, 24, 29, 30].
Recently, Garnica et al. used an in silico approach to survey and compare simple sequence repeats (SSRs) in transcript sequences from the genomes of P. sojae, P. ramorum and P. infestans [27]. They also evaluated in silico transferability of SSRs among the Phytophthora species and found that a proportion (7.5%) of primers could, in theory, be transferred between at least two of the three species. In the present study SSRs from P. infestans P. sojae and P. ramorum were analysed to identify useful loci common to many Phytophthora species (Approach 1) or to a restricted number of species closely related to P. sojae (Approach 2). Selected loci were amplified and sequenced from 16 (Approach 1) and 5 (Approach 2) different Phytophthora species and a comprehensive SSRs dataset was created.
Results
Approach 1 – SSRs for many Phytophthora species
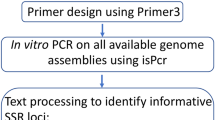
The aim of this approach was to identify loci containing SSRs common to a large number of Phytophthora species (Fig. 1A). The method was validated using 16 different species (Table 1) representing the breadth of diversity across the genus [2, 3].

Schematic representation of two different approaches utilised to identify SSRs in a broad range of Phytophthora species (A) or in Phytophthora spp. Clade 7 (B).
Analysis of sequences from P. infestans, P. sojae and P. ramorum genome projects and scanning for homologous SSRs
Predicted gene datasets from P. infestans http://www.broad.mit.edu/P. sojae and P. ramorum http://genome.jgi-psf.org/ were scanned for the presence of microsatellites defined as short tandem repeat motifs (SSRs) of 2–6 bp. Both perfect and compound SSRs were selected with a minimal acceptable length of 10 bp (dinucelotide motifs) and 12 bp (tri- and tetranucletide motifs). SSRs with a minimum of three repeats were included in the analyses of penta-nucleotide repeats. This search yielded 9333 sequences containing SSRs (1465 from P. infestans, 5348 from P. sojae and 2520 from P. ramorum). The relative abundance of SSRs was 103, 183 and 114 per Mb of predicted gene sequence for P. infestans, P. sojae and P. ramorum, respectively.
Selected regions were compared by BLAST analysis to identify homologous regions flanking SSRs in at least two of the three species (P. sojae, P. ramorum and P. infestans). This analysis identified 4135 SSRs from P. infestans (688), P. ramorum (1470), and P. sojae (1977). A very limited number of loci containing SSRs were common to the three species; most loci were common to P. ramorum and P. sojae (81.6%), P. infestans and P. ramorum (7%) or P. infestans and P. sojae (11%). In most of the cases, homologous loci contained the same SSR motif in different Phytophthoras, however the number of repeats was consistently higher in the 'source' species than the other two.
Among the selected loci, the number of SSR repeats ranged from 3 to 13, from 3 to 12 and from 3 to 17 in P. infestans, P. ramorum and P. sojae respectively. Most SSRs showed seven repeats or less (98.2% P. infestans, 97.4% P. ramorum, 94.4% P. sojae), with a repeat number of four being the most common in all species.
Selection and amplification of target regions containing SSRs
The 4135 homologous regions previously identified were manually analysed to select those with the highest number of repeats and flanked by the most conserved sequences on both sides. The latter condition was necessary to design primers suitable for as many species as possible. Based on this analysis 6, 7 and 12 target regions were identified across the genome of P. infestans, P. ramorum and P. sojae respectively. These regions, containing 8, 17 and 33 SSRs respectively, were selected (Table 2) for amplification from 16 different species of Phytophthora representing the breadth of diversity in the genus (Table 1). To this aim, a total number of 62 different degenerate primers (12 for P. infestans, 18 for P. ramorum, and 32 for P. sojae) were designed (Table 2). When target regions contained two or more SSRs and/or were too long to be amplified by a single amplification, a pool of different primers was designed (Table 2). Considerable effort was made to obtain successful amplification from as many species as possible. This involved screening of several primer pairs for each genomic region and for each Phytophthora species (Table 2) and adjustment of annealing temperatures and MgCl2 concentration for PCR reactions (Tables 3, 4, 5).
The resultant primers enabled the amplification of 271 single PCR bands of the expected size (Fig. 2). In the remaining primer-species combinations, 193 amplifications did not produce any product or produced complex profiles (two or more PCR fragments) impeding direct sequencing (Fig. 2). Some primer combinations failed to amplify a product from any of the Phytophthora species whereas other combinations amplified single bands from all or most Phytophthora species (Tables 3, 4, 5).

Amplification results obtained with 16 Phytophthora species (Table 1) using primers designed against P. sojae, P. ramorum and P. infestans genomes using Approach 1 (Fig. 1A). NA represents primer-species combinations in which amplification reactions did not produce any product or produced complex profiles (two or more PCR fragments) preventing direct sequencing. NS represents primer-species combinations in which amplification reactions produced single PCR bands, however direct sequencing did not yield reliable sequences. SQ represents primer-species combinations in which reliable sequences were obtained.
Sequencing of single PCR bands and scanning for SSRs
All single PCR bands (271) were purified to remove excess primers and nucleotides and sequenced in both directions using the same primers used for the amplification. When forward and/or reverse sequences were not identical, amplification, purification and sequencing were repeated twice and all unreliable sequences were discarded. Finally, 171 sequences were obtained with primers designed against P. sojae (70), P. ramorum (50) and P. infestans (51) genomes (Fig. 2) and scanned to identify SSRs by means of sputnik. Sequenced regions contained a total number of 211 SSRs distributed across the genome of the 16 target species with those of clade 7 (P. alni, P. cambivora, P. europaea, P. fragariae and P. sojae) and clade 8 (P. lateralis and P. ramorum) more highly represented (Fig. 3; Tables 3, 4, 5). A single microsatellite was identified in P. inundata. All SSRs identified in P. infestans were amplified with primers designed against its own genome (Fig. 3). Identified SSRs ranged in the number of repeats from 4 to 16, from 3 to 16 and from 4 to 14 in P. sojae, P. ramorum and P. infestans respectively (Tables 3, 4, 5). A single repeat of 24 was found in an SCRI isolate of P. ramorum (Table 4). Most SSRs were of seven repeats or less (88.9% P. infestans, 82.8% P. ramorum, 76.8 P. sojae), with a repeat number of four being the most common in all species (Fig. 4). Overall, the most common motifs were (AAG)n, (AGG)n and (AGC)n representing 40.9%, 23.3% and 17.6% respectively of the total number of identified SSRs (Fig. 5). Trinucleotide repeats were the most common (94.7%) followed by pentanucleotide (2.4%), tetranucleotide (1.9%) and dinucleotide (1.0%) repeats.

Number of SSRs identified for each of the 16 Phytophthora species using primers designed against P. sojae, P. ramorum and P. infestans genomes (Approach 1, Fig. 1A).


To evaluate intraspecific variability a few selected target regions amplified by primers S23F-S25R, S21F-S22R, I9-I10 and I5-6 (Table 2) were examined and sequenced from additional isolates of P. alni, P. cambivora, P. cinnamomi, P. pseudosyringae and P. ilicis (Table 1). The analysed target regions did not show intraspecific variability among analysed isolates of P. alni subsp. alni, P. pseudosyringae or P. ilicis, whereas P. cambivora and P. cinnamomi isolates were polymorphic in all the tested primer combinations. As an example, the target region amplified with primers I9-I10 from P. cinnamomi was characterised by 12, 14 and 18 repeated motifs (AGG) in three tested isolates (Table 6).
Approach 2 – Identification of SSRs in Phytophthora spp. clade 7
The aim of this approach was to focus the search for SSR loci to a restricted range of four clade 7 Phytophthora species (P. alni, P. cambivora, P. europaea and P. fragariae) phylogenetically related to P. sojae (Fig 1B) [9].
Identification of target regions
This approach was based on a detailed list of SSRs identified in the genome of P. sojae and provided by Dr. Niklaus Grunwald at the Agricultural Research Service, U.S. Department of Agriculture, Corvallis, Oregon. Among the list, sixty genomic regions (500–1000 bp) were manually selected on the basis of having the longest SSRs in exons (20), introns (20) and non coding regions (20). The selected regions (Table 7) were screened using BLAST against the entire genomes of P. ramorum and P. infestans to search for homology irrespective of the SSR regions. None of these regions aligned with sequences from the P. infestans genome whereas 18 of the 60 regions were sufficiently conserved to match homologous genes in P. ramorum (6 were localised in exons and 12 in introns). Surprisingly none of these 18 regions contained SSRs in P. ramorum, however it was hypothesised that microsatellites could be present in homologous regions of other Phytophthora species more closely related to P. sojae. To verify this hypothesis, thirty-six primers (18 pairs) were designed in the conserved flanking regions and used to amplify the target regions from P. alni, P. cambivora, P. europaea, P. fragariae and an SCRI isolate of P. sojae (Table 7). Degenerate primers were designed when necessary.
Amplification, sequencing and SSR scoring
Most primer-species combinations produced single PCR bands of the expected size (Table 8). Purification and direct sequencing of these PCR fragments produced 54 reliable sequences which were analysed as previously described for Approach 1. Twelve different microsatellites were identified: 2 in P. europaea, 3 in P. fragariae and P. alni and 4 in P. cambivora (Table 8). Among these, 10 were trinucleotides and 2 were tetranucleotides repeated 4, 5 or 6 times. All regions sequenced from the SCRI isolate of P. sojae contained the predicted/expected SSR (Table 8).
Discussion
The present study was undertaken to develop a method to rapidly identify loci containing SSRs and to create a pool of microsatellite markers for species of the genus Phytophthora taking advantage of publicly available sequences for P. sojae, P. ramorum and P. infestans. Recently, Garnica et al. explored the transferability of microsatellites across P. sojae, P. ramorum and P. infestans via an in silico virtual PCR approach [27]. In the present study, such an analysis on the same three species was conducted but also followed up with a comprehensive screening and validation process on multiple species to provide a practical evaluation of the procedure as a means of accelerating the search for new SSR markers in the genus Phytophthora.
The first approach was aimed at the identification of informative SSR loci common to many Phytophthora species. This approach was based on the hypothesis that among the large number of microsatellites distributed across the genome of species of the genus there may be a proportion in genes common to many species with sufficient sequence conservation in flanking regions to allow the design and use of universal SSR primers. Our search of the predicted gene sets yielded approximately 10% fewer SSRs and a corresponding lower abundance of SSRs per Mb of sequence than that of Garnica et al [27]. Preliminary analyses revealed a very limited number of loci containing SSRs that were common to the three Phytophthora species tested. The majority of the identified loci (81.6%) were common to P. sojae and P. ramorum only which is consistent with their closer phylogenetic relationship in clades 7 and 8 than to P. infestans in clade 1 [9, 2, 3]. Similarly, Garnica et al. found in their in silico analysis that 7.5% of their primers were, in theory, transferable between at least two species (mainly P. ramorum and P. sojae) and only 1.0% transferable between the three species [27]. Among the selected sequences satisfying the above conditions, the number of repeats ranged from 3 to 17 and most SSRs showed seven repeats or less, with a repeat number of four being the most common in all species. The abundance of different repeat motifs differed slightly between species however, on average, (AAG)n, (AGG)n and (AGC)n were the most abundant triplets in all three Phytophthoras (Fig. 5). These results differ from those reported by Garnica et al. in which (AGC)n, (ACG)n and (AGG)n were the most abundant triplets amongst all the screened EST sequences [27]. It should, however be considered that unlike the study of Garnica our data are confined to SSR sequences for which it was possible to identify a homologue in at least one of the other two species. Therefore it could be hypothesised that motifs (AAG)n and (AGG)n are more abundant in more conserved genes. The dominance of trinucleotide SSRs compared to dinucleotide SSRs was not surprising considering that trinucleotides are abundant in coding regions of all higher eukaryotic genomes [31–33]. Dinucleotide repeats, in contrast, are characterised by higher mutation rates which may explain their abundance in introns and non-coding regions and lower frequency in coding regions, which cannot tolerate frame-shift mutations [34, 35].
Primers designed in the present study with the first approach were tested against a panel of 16 different Phytophthora species representing the breadth of diversity across the genus to amplify P. sojae, P. ramorum and P. infestans target regions containing 33, 17 and 8 SSRs respectively. Overall, these primers enabled the sequencing of 171 target regions which contained 211 SSRs ranging in repeat number from 3 to 16. Most of these SSRs showed seven repeats or less with four the most common repeat number and (AAG)n, (AGG)n and (AGC)n the most common motifs. Trinucleotide repeats were dominant followed by pentanucleotide, tetranucleotide and dinucleotide repeats. This data indicate that such an approach can be useful to identify cross-specific SSR loci in the genus Phytophthora. As further genome sequences become available, for example, P. capsici http://www.jgi.doe.gov/sequencing/why/CSP2006/Pcapsici.html, the process can be refined to specific subsets of the genus. The mutation rates and, consequently, the practical utility of the identified SSRs in the study of the specific Phytophthora species need to be examined further. Undoubtedly, a risk of this approach is that the selection is biased towards more conserved sequences which may subsequently have a lower mutation rate that reduces their utility as polymorphic markers. Furthermore, the fact that P. infestans SSRs were all identified using primers designed on its own genome (Fig. 3) may indicate that this approach is less appropriate for distant relatives considering that, as stated above, P. infestans is phylogenetically distant from P. sojae and P. ramorum. However, the identification of intraspecific polymorphisms in some selected SSRs is encouraging and demonstrates that at least some of the selected SSRs are valuable for immediate practical applications (Table 6). This is consistent with the reported applicability of EST-SSRs across closely related taxa in other organisms as well as Phytophthora [23, 36–38]. In the present study, the focus on the breadth of species (16) prevented the analyses of a wider number of target regions. However, the same method could be easily applied to the study of more regions from one or a few species.
The application of the first method enabled the identification of novel SSRs from all the 16 target species with those of clade 7 and 8 more highly represented (Fig. 3). A higher proportion of SSRs from species of the clade 7 and 8 was expected considering that P. sojae and P. ramorum belong to these two clades [2, 3]. In light of this fact, a second approach to identify a greater number of polymorphic SSRs from within a more limited range of clade 7 taxa more closely related to P. sojae was investigated. Sixty P. sojae SSR candidates were compared by BLAST analysis against the complete genome sequence of the other two species yielding 18 SSR candidates which could be aligned with homologous regions in P. ramorum. However in none of these 18 candidates (6 exons and 12 introns) was the SSR maintained in P. ramorum. In four of the more closely related species (P. alni, P. cambivora, P. europaea and P. fragariae), however, some of the SSR regions were conserved (Table 8). In this study the focus was on discovery of SSRs in invasive forest Phytophthora species within the clade 7a, perhaps a higher success rate in marker discovery would have followed a search amongst the closest related species in clade 7b (P. sinensis, P. melonis, P. cajanae and P. vignae) [2]. Although a few SSR markers with potential were discovered using this approach, it was not a highly efficient means of identifying new polymorphic SSR loci and highlights the lack of conservation of SSR loci, even amongst coding regions within a single ITS clade of Phytophthora. Some degree of cross-species amplification has been observed between SSRs in P. infestans with other Clade 1c taxa and it is therefore likely that a wider application of this method concentrated on the closest relatives would be more productive [23].
Conclusion
The present study has tested two different methods to generate SSR markers that can be utilised across a broad range of Phytophthora species. The final number of identified loci for any single species may not be sufficient to run a complete population genetics analysis and key studies on the inter- and intraspecific variation remain. A comprehensive dataset of candidate SSRs from a range of species has been created (Table 3, 4, 5). The detailed groundwork needed to amplify these regions from such a diverse collection of species and target regions has been completed which moves beyond the previous in silico approach to improve our understanding of the range and sequence conservation of SSR loci amongst species [27]. In general, the level of interspecific SSR sequence conservation, even amongst more closely related species within a single clade, was low and the method may not be the most efficient means of identifying novel SSR loci. Apart from their application as molecular markers, determining the abundance and density of SSRs in Oomycetes may help understand whether these sequences have any functional and evolutionary significance [17]. Furthermore, irrespective of the microsatellites, some of the amplified regions represent valuable marker regions for a number of applications [39]. A single optimal target gene for all Phytophthora species and assay requirements is unlikely to exist, therefore the continued identification and characterization of new target genes offers new opportunities for detection and phylogenetic studies [3, 40, 41].
Methods
Phytophthora isolates and DNA extractions
Twenty-eight isolates (16 Phytophthora species) sourced from the SCRI culture collection were used in this study (Table 1). Isolates and species were selected to represent taxa most relevant to European forestry that also represented the breadth of Phytophthora diversity defined according to clades based on ITS sequence analysis [2]. Isolates were stored on oatmeal agar at 5°C and grown on French bean agar for routine stock cultures.
Total DNA was extracted from pure cultures of Phytophthora according to Schena and Cooke, diluted to 10 ng/μl and maintained at 5°C for routine amplifications and at -20°C for long term storage [42].
Analysis of sequences from P. infestans, P. sojae and P. ramorum genome projects and scanning for homologous SSRs
The predicted protein datasets of P. infestans (from the NCGR XGI database that was available prior to the Broad genome sequencing project) and P. ramorum and P. sojae http://genome.jgi-psf.org/ were screened for SSR loci using Sputnik (Chris Abaijan http://espressosoftware.com/pages/sputnik.jsp). Pairwise BLAST analysis using the default parameters was used to select loci conserved in different species combinations [43]. Manual screening of these loci on the basis of SSR and flanking region DNA sequence conservation yielded a short-list for further analysis.
Primer design and amplification conditions
All primers (Table 2 and 7) were designed with the Primer3 Software set up to generate a Tm of 60°C ± 2, a GC% between 20 and 80% and a length of 18–26 bp [44]. Primers were purchased from Eurogentec ltd. (Belgium). Considerable effort was made to obtain successful amplification of single PCR bands from as many species as possible. This involved adjustment of MgCl2 concentration (0.7, 1.0 or 1.7 mM) and annealing temperatures (55 or 58°C) for PCR reactions (Table 3, 4, 5). Furthermore in some circumstances alternative primers were designed and tested to amplify the target regions from as many taxa as possible (Table 2). PCR reactions were performed in a total volume of 15 μl containing 10 ng of genomic DNA, 1.5 μl of 10× Reaction Buffer (Promega Corporation, WI, USA), 100 μM dNTPs, 0.7, 1 or 1.7 mM MgCl2, 15 μg BSA, 2 unit of Taq polymerase (Taq DNA polymerase, Promega Corporation) and 1 μM of primers. PCR amplification conditions consisted of: 1 cycle of 95°C for 2 min; 40 cycles of 94°C for 30 s, 55 or 58°C for 30 s, 72°C for 60 s; and a final cycle of 72°C for 5 min.
DNA sequencing
The best primers and amplification conditions were identified for all primer-species combinations and target DNA was re-amplified in a total volume of 50 μl to provide sufficient amplicon for direct sequencing. Single PCR bands were purified with the MinElute PCR Purification Kit (Qiagen Ltd. West Sussex, UK) to remove excess primers and nucleotides. Sequencing was carried out with the same primers utilized for the amplification in a dye-terminator cycle-sequencing reaction (FS sequencing kit, Applied Biosystems, Warrington, UK) and run on an ABI373 automated sequencer (Applied Biosystems). All selected PCR fragments were sequenced using both the forward and the reverse primers.
Sequence analysis and SSRs scanning
The "Sequence Navigator" software (Applied Biosystems) was utilised to evaluate reliability of sequences and to compare forward and reverse sequences to create a consensus sequence. Non-reliable sequences in which both forward and reverse sequences contained doubtful bases were discarded. All sequences obtained in the present study were also parsed to a web version of SPUTNIK http://cbi.labri.fr/outils/Pise/sputnik.html, which uses a recursive algorithm to search for repeated patterns of nucleotides of length between 2 and 5.
References
Baldauf SL, Roger AJ, Wenk-Siefert I, Doolittle WF: A Kingdom-level phylogeny of eukaryotes based on combined protein data. Science. 2000, 290: 972-977.
Cooke DEL, Drenth A, Duncan JM, Wagels G, Brasier CM: A molecular phylogeny of Phytophthora and related Oomycetes. Fungal Genet Biol. 2000, 30: 17-32.
Kroon LPNM, Bakker FT, Bosch van den GBM, Bonants PJM, Flier WG: Phylogenetic analysis of Phytophthora species based on mitochondrial and nuclear DNA sequences. Fungal Genet Biol. 2004, 41: 766-782.
Tyler BM, Tripathy S, Zhang X, Dehal P, Jiang RHY, Aerts A, Arredondo F, Baxter L, Damasceno CMB, Dickerman A, Dorrance AE, Dou D, Dubchak I, Garbelotto M, Gijzen M, Gordon S, Govers F, Grunwald N, Huang W, Ivors K, Jones RW, Kamoun S, Krampis K, Lamour K, Lee MK, McDonald WH, Medina M, Meijer HJG, Nordberg E, MacLean DJ, Ospina-Giraldo MD, Morris P, Phuntumart V, Putnam N, Rash S, Rose JKC, Sakihama Y, Salamov AA, Savidor A, Scheuring C, Smith B, Sobral BWS, Terry A, Torto-Alalibo T, Win J, Xu Z, Zhang H, Grigoriev I, Rokhsar D, Boore J: Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science. 2006, 313: 1261-1265.
Erwin DC, Ribeiro OK: Phytophthora diseases worldwide. 1996, APS Press, St Paul, MN, USA
Jurskis V: Eucalypt decline in Australia, and a general concept of tree decline and dieback. Forest Ecol Manag. 2005, 215: 1-20.
Ristaino JB, Gumpertz ML: New frontiers in the study of dispersal and spatial analysis of epidemics caused by species in the genus Phytophthora. Annu Rev Phytopathol. 2000, 38: 541-576.
Rizzo DM, Garbelotto M, Hansen EM: Phytophthora ramorum: integrative research and management of an emerging pathogen in California and Oregon forests. Annu Rev Phytopathol. 2005, 43: 309-335.
Blair JE, Coffey MD, Park S, Geiser DM, Kang S: A multi-locus phylogeny for Phytophthora utilizing markers derived from complete genome sequences. Fungal Genet Biol. 2008, 45: 266-277.
De Merlier D, Chandelier A, Debruxelles N, Noldus M, Laurent F, Dufays E, Claessens H, Cavelier M: Characterization of alder Phytophthora isolates from Wallonia and development of SCAR primers for their specific detection. J Phytopathol. 2005, 153: 99-107.
Cooke DEL, Young V, Birch PRJ, Toth R, Gourlay F, Day JP, Carnegie S, Duncan JM: Phenotypic and genotypic diversity of Phytophthora infestans populations in Scotland (1995–97). Plant Pathol. 2003, 52: 181-92.
Ivors KL, Hayden K, Bonants PJM, Rizzo DM, Garbelotto M: AFLP and phylogenetic analyses of North American and European populations of Phytophthora ramorum. Mycol Res. 2004, 108: 378-392.
Cooke DEL, Lees AK: Markers, old and new, for examining Phytophthora infestans diversity. Plant Pathol. 2004, 53: 692-704.
Selkoe KA, Toonen RJ: Microsatellites for ecologists: a practical guide to using and evaluating microsatellite markers. Ecol Lett. 2006, 9: 615-629.
Mascheretti S, Croucher PJP, Vettraino A, Prospero S, Garbelotto M: Reconstruction of the Sudden Oak Death epidemic in California through microsatellite analysis of the pathogen Phytophthora ramorum. Mol Ecol. 2008, 17: 2755-2768.
Li YC, Korol AB, Fahima T, Beiles A, Nevo E: Microsatellites: genomic distribution, putative functions, and mutational mechanisms: A review. Mol Ecol. 2002, 11: 2453-2465.
Toth G, Gaspari Z, Jurka J: Microsatellites in different eukaryotic genomes: survey and analysis. Genome Res. 2000, 10: 967-981.
Chistiakov DA, Hellemans B, Volckaert FAM: Microsatellites and their genomic distribution, evolution, function and applications: A review with special reference to fish genetics. Aquaculture. 2006, 255: 1-29.
Gobbin D, Pertot I, Gessler C: Identification of microsatellites markers for Plasmopara viticola and establishment of high-throughput method for SSR analysis. Eur J Plant Pathol. 2003, 109: 153-164.
Dobrowolski MP, Tommerup IC, Blakeman HD, O'Brien PA: Non-mendelian inheritance revealed in a genetic analysis of sexual progeny of Phytophthora cinnamomi with microsatellite markers. Fungal Genet Biol. 2002, 35: 197-212.
Dobrowolski MP, Tommerup IC, Shearer BL, O'Brien PA: Three clonal lineages of Phytophthora cinnamomi in Australia revealed by microsatellites. Phytopathology. 2003, 93: 695-704.
Knapova G, Gisi U: Phenotypic and genotypic structure of Phytophthora infestans populations on potato and tomato in France and Switzerland. Plant Pathol. 2002, 51: 641-653.
Lees AK, Wattier R, Shaw DS, Sullivan L, Williams NA, Cooke DEL: Novel microsatellite markers for the analysis of Phytophthora infestans populations. Plant Pathol. 2006, 55: 311-319.
Ivors K, Garbelotto M, Vries ID, Ruyter-Spira C, Te Hekkert B, Rosenzweig N, Bonants P: Microsatellite markers identify three lineages of Phytophthora ramorum in US nurseries, yet single lineages in US forest and European nursery populations. Mol Ecol. 2006, 15: 1493-505.
Prospero S, Black JA, Winton ML: Isolation and characterization of microsatellite markers in Phytophthora ramorum, the causal agent of sudden oak death. Mol Ecol. 2004, 4: 672-674.
Glenn TC, Schable NA: Isolating microsatellite DNA loci. Molecular evolution: producing the biochemical data, part B. Edited by: Zimmer EA, Roalson E. 2005, Academic Press, San Diego, USA, 202-222.
Garnica DP, Pinzón AM, Quesada-Ocampo LM, Bernal AJ, Barreto E, Grünwald NJ, Restrepo S: Survey and analysis of microsatellites from transcript sequences in Phytophthora species: frequency, distribution, and potential as markers for the genus. BMC Genomics. 2006, 7: 245-
Robinson AJ, Love CG, Batley J, Barker G, Edwards D: Simple sequence repeat marker loci discovery using SSR primer. Bioinformatics. 2004, 20: 1475-1476.
Prospero S, Hansen EM, Grünwald NJ, Winton LM: Population dynamics of the sudden oak death pathogen Phytophthora ramorum in Oregon from 2001 to 2004. Mol Ecol. 2007, 16: 2958-2973.
Widmark AK, Andersson B, Cassel-Lundhagen A, Sandström M, Yuen JE: Phytophthora infestans in a single field in southwest Sweden early in spring: symptoms, spatial distribution and genotypic variation. Plant Pathol. 2007, 56: 573-579.
Borstnik B, Pumpernik D: Tandem repeats in protein coding regions of primate genes. Genome Res. 2002, 12: 909-915.
Metzgar D, Bytof J, Wills C: Selection against frameshift mutations limits microsatellite expansion in coding DNA. Genome Res. 2000, 10: 72-80.
Subramanian S, Madgula VM, George R, Mishra RK, Pandit MW, Kumar CS, Singh L: Triplet repeats in human genome: distribution and their association with genes and other genomic regions. Bioinformatics. 2003, 19: 549-552.
Karaoglu H, Lee CMY, Meyer W: Survey of simple sequence repeats in completed fungal genomes. Mol Biol Evol. 2004, 22: 639-649.
Katti MV, Ranjekar PK, Gupta VS: Differential distribution of simple sequence repeats in eukaryotic genome sequences. Mol Biol Evol. 2001, 18: 1161-1167.
Cordeiro GM, Casu R, McIntyre CL, Manners JM, Henry RJ: Microsatellite markers from sugarcane (Saccharum spp.) ESTs cross transferable to Erianthus and Sorghum. Plant Sci. 2001, 160: 1115-1123.
Woodhead M, Russell J, Squirrell J, Hollingsworth PM, Cardle L, Ramsay L, Gibby M, Powell W: Development of EST-SSRs from the Alpine lady-fern, Athyrium distentifolium. Mol Ecol Notes. 2003, 3: 287-290.
Zhan A, Bao ZM, Wang XL, Hu JJ: Microsatellite markers derived from bay scallop Argopecten irradians expressed sequence tags. Fisheries Science. 2005, 71: 1341-1346.
Cooke DEL, Schena L, Cacciola SO: Tools to detect, identify and monitor Phytophthora species in natural ecosystems. J Plant Pathol. 2007, 89: 13-28.
Schena L, Hughes KJD, Cooke DEL: Detection and quantification of P. ramorum, P. kernoviae, P. citricola and P. quercina in symptomatic leaves by multiplex real-time PCR. Mol Plant Pathol. 2006, 7: 365-379.
Schena L, Duncan JM, Cooke DEL: Development and application of a PCR based "molecular tool box" for the detection of Phytophthora species damaging forests and natural ecosystems. Plant Pathol. 2008, 57: 64-75.
Schena L, Cooke DEL: Assessing the potential of regions of the nuclear and mitochondrial genome to develop a "molecular tool box" for the detection and characterization of Phytophthora species. J Microbiol Methods. 2006, 67 (1): 70-85.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215: 403-410.
Rozen S, Skaletsky HJ: Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000, 132: 365-386.
Acknowledgements
This work was funded by the European Union with a Marie Curie Intra-European Fellowship (Project: MEIFCT-2003-502327) and by the Scottish Government Rural and Environment Research and Analysis Directorate (RERAD). We thank C.M. Brasier and others for providing isolates, N. Williams for invaluable help with the culture collection and Nik Grunwald for providing a list of P. sojae SSR markers. Cultures are held under SASA License No. PH/11/2008.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
LS conceived the study, carried out the molecular analyses, optimized the described method, performed data acquisition, data analysis and data interpretation and wrote the manuscript.
LM performed analysis of sequences from genome projects and scanned for homologous SSRs.
DELC coordinated, supervised and contributed to the design of the study, provided intellectual input to optimize the method and revised the manuscript critically.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Schena, L., Cardle, L. & Cooke, D.E. Use of genome sequence data in the design and testing of SSR markers for Phytophthora species. BMC Genomics 9, 620 (2008). https://doi.org/10.1186/1471-2164-9-620
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-9-620