
Abstract
Background
A key problem in the sequence-based reconstruction of regulatory networks in bacteria is the lack of specificity in operator predictions. The problem is especially prominent in the identification of transcription factor (TF) specific binding sites. More in particular, homologous TFs are abundant and, as they are structurally very similar, it proves difficult to distinguish the related operators by automated means. This also holds for the LacI-family, a family of TFs that is well-studied and has many members that fulfill crucial roles in the control of carbohydrate catabolism in bacteria including catabolite repression. To overcome the specificity problem, a comprehensive footprinting approach was formulated to identify TF-specific operator motifs and was applied to the LacI-family of TFs in the model gram positive organism, Lactobacillus plantarum WCFS1. The main premise behind the approach is that only orthologous sequences that share orthologous genomic context will share equivalent regulatory sites.
Results
When the approach was applied to the 12 LacI-family TFs of the model species, a specific operator motif was identified for each of them. With the TF-specific operator motifs, potential binding sites were found on the genome and putative minimal regulons could be defined. Moreover, specific inducers could in most cases be linked to the TFs through phylogeny, thereby unveiling the biological role of these regulons. The operator predictions indicated that the LacI-family TFs can be separated into two subfamilies with clearly distinct operator motifs. They also established that the operator related to the 'global' regulator CcpA is not inherently distinct from that of other LacI-family members, only more degenerate. Analysis of the chromosomal position of the identified putative binding sites confirmed that the LacI-family TFs are mostly auto-regulatory and relate mainly to carbohydrate uptake and catabolism.
Conclusion
Our approach to identify specific operator motifs for different TF-family members is specific and in essence generic. The data infer that, although the specific operator motifs can be used to identify minimal regulons, experimental knowledge on TF activity especially is essential to determine complete regulons as well as to estimate the overlap between TF affinities.
Similar content being viewed by others

Background
Numerous studies have been devoted to the identification of Transcription Factor (TF)-binding sites or other regulatory elements in bacterial genomes. So far, most large-scale approaches relied heavily on statistics and the input of known binding motifs [1–7]. Unfortunately, purely statistical approaches are seriously hampered by the trade-off that exists between a high true-positive rate and a low false-negative rate of the prediction. Nonetheless, both rates can be considerably improved by taking advantage of additional data [2, 8] like, for instance, sequence data from related species [9–11], structural information [12] or transcriptome data [13, 14]. Another way to enhance the accuracy is phylogenetic footprinting which takes both 'phylogeny' and 'synteny' into account[8, 14–16].
We have recently developed a large-scale automated regulatory motif prediction method for prokaryotic genomes [17]. It was applied with success in the identification of a relatively large number of regulatory motifs in genomes of the Firmicutes, a phylum that comprises many well-studied families like the Bacillaceae, Clostridiaceae, Lactobacillaceae, Staphylococcaceae and Streptococcaceae. The identified motifs included several new motifs besides known ones. Nevertheless, in many cases the method appeared less suited to couple a specific TF or signal to the regulatory motif in a straightforward manner. For example, although the characteristic T-box motif was easily identified – the T-box is a regulatory element that responds to uncharged t-RNA [18] and is found in all Firmicutes – the amino acid specificity of that element was not retrieved for the individual instances automatically (Wels et al. unpublished results). Likewise, the 'CRE-like' motif that was retrieved is very similar to known operator motifs of various TFs of the LacI-family, suggesting that the recovered motif is not specific.
The LacI-family of TFs plays a crucial role in many bacterial species, and certainly in those of the phylum Firmicutes, as these TFs mediate preferences in the utilization of certain carbohydrates over others. The prioritization involves both repression (or activation) of catabolic genes (i) in the absence (or presence) of a related substrate and (ii) in the presence (or absence) of a preferred substrate [19–21]. The latter process is referred to as carbon catabolite repression (CCR) and its main mediator in Firmicutes species is CcpA [21–25]. CcpA operators were called CREs (CcpA-responsive elements [26]) and a CRE consensus motif was defined on basis of experiments in various Firmicutes species [21–23, 25, 27–30]. The consensus motif is very similar to, and sometimes coincides with, operators related to other TFs of the LacI-family [30–33]. Most family members, however, interact with only a few operators on the genome, like LacI of Escherichia coli, which represses specifically the lac-operon in the absence of lactose [34]. This raises the question how these bacteria coordinate 'local' (def: control of the expression of one or a few genes/operons) and 'global' (def: control of the expression of many genes/operons) regulatory effects using homologous TFs.
Thus, the lack in specificity of the current prediction methods is a key issue in case one wants to disentangle complex regulatory relationships, like between those of the TFs of the LacI-family and the operons involved in carbohydrate catabolism. Therefore, we have formulated a comprehensive sequence-based comparative approach for the prediction of TF-specific operators in bacteria. Specificity is ensured by building upon a proper phylogenetic classification of each family of TFs (whose members can for instance be found in reference databases [35–37]) and very strict criteria to define synteny.
The value of the approach was put to the test on the well-described LacI-family of TFs, and more specifically, to uncover the regulatory connections of the 12 LacI-family TFs in L. plantarum WCFS1. This species was chosen as a representative of the phylum Firmicutes, as it is an industrially and medically relevant model organism that is encountered in very different environmental niches, i.e. in association with plants, fermenting food and feed, and in the animal and human gastrointestinal tract [38, 39]. The approach proved successful and each LacI-family TF of L. plantarum was linked to a putative operator motif and thereby to a putative regulon. In addition, several principles that should govern LacI-family TF mediated 'local' and 'global' transcription regulation could be inferred from the results. Ample experimental and structural information was used to evaluate and support the predictions and inferences.
Results
I) A comprehensive approach to identify TF-specific operators
It has been observed consistently that orthologous protein sequences [40] are very likely to have molecular properties that are alike [41]. Similarly, synteny – conserved gene order – was found to be a strong indicator of functional equivalency [42]. Thus, genes that are orthologous and share 'gene context' can be assumed to be functionally more equivalent than orthologous genes that are not syntenous. Based on this premise we formulated a generic phylogenetic footprinting [43]/shadowing [44] approach for the identification of TF-specific operator sequences in bacteria (description in Methods). High specificity in the motif prediction was achieved by properly classifying orthologous TFs into groups that share gene context to yield putative Groups of Orthologous Functional Equivalents or GOOFEs. To develop the approach, we chose the well-described LacI-family of transcriptional regulators (PFAM PF00356), limiting the analysis to Firmicutes and focusing specifically on the model organism Lactobacillus plantarum WCFS1, which has a high number of LacI-family TFs for which we have ample experimental and transcriptome data for validation.
Collect homologs: LacI-family TFs in the genomes of L. plantarum WCFS1 and other Firmicutes
The search for LacI-family TF specific operators was initiated by collecting the LacI-family TF protein sequences from taxonomically related genomes and grouping them using the Neighbor Joining (NJ) algorithm (for relevant data see Additional files 1, 2, 3, 4). The resulting NJ-tree indicated a clear separation between two subfamilies of LacI-family TF homologs (top of Figure 1 and see Additional file 5). One subfamily represented the vast majority of LacI-family TF homologs including CcpA, whereas the other represented only 1 to 3 homologs per species. The latter subfamily contained one well-studied TF from E. coli, the evolved beta-galactosidase repressor or EbgR [45]. Henceforth, the two LacI-family TF subfamilies will be referred to as 'CcpA-like' and 'EbgR-like'. The number of LacI-family TF homologs ranged from 2 to 17 and the homolog composition was found highly variable between different species and also variable between strains (see Figure 1 and Additional file 1) and correlated roughly with genome size. For example, although all 6 LacI-family TFs of Pediococcus pentosaceus [46] were orthologous to a LacI-family TF in L. plantarum WCFS1, only 5 out of 9 where orthologous in Lactobacillus brevis [46], another close relative. Moreover, when the LacI-family TF content of various strains of L. plantarum was analyzed – the data were derived from a strain diversity analysis [47] – it appeared that apart from ccpA none of the individual LacI-family genes was present in all other strains. In fact, the master regulator CcpA was the only LacI-family member present in all sequenced Firmicutes genomes.

The number of LacI-family TF homologs and the presence of Lactobacillus plantarum orthologs in different Firmicutes. The organisms are organized on basis of their phylogeny (left; inferred from phosphoglycerate kinase amino acid sequence data [98]) and the TFs on basis of the NJ-tree of the L. plantarum LacI-family TFs (top). The presence of an ortholog to the L. plantarum proteins is indicated by open (different cluster in the NJ-tree) and closed circles (same cluster in the NJ-tree). The members of the various L. plantarum GOOFEs are colored. Some orthologs have been experimentally characterized and are indicated by '+'. remark: Although the PFAM HMM that is used to identify the LacI-domain represents only a small part of the DNA-binding domain, in most instances there was complete correspondence between the number of LacI-family TFs identified by us and the number listed by PFAM [96]. However, there were a few exceptions and in these cases the number given by PFAM appeared erroneous [see Additional file 1]. In some cases the PFAM database was just incomplete (e.g. Pediococcus pentosaceus and Leuconostoc mesenteroides). In other cases sequences were counted twice as a result of double Uniprot entries (e.g. for CcpA in L. plantarum). Other proteins were missing in the PFAM database because of mistakes in the ORF definition.
Determine synteny: Identification of TF-specific binding motifs
Inspection of the gene-neighborhood of the genes encoding LacI-family TF homologs in L. plantarum indicated that most of them are associated with genes encoding proteins that catabolize carbohydrates. Although the gene association appeared conserved in other genomes, it was mostly only true for a limited number of species. A TF-specific GOOFE was defined for each L. plantarum LacI-family TF on basis of context conservation and then the upstream regions preceding the conserved operons/genes were selected (see Methods for the precise procedure). Multiple sequence alignments, motif-finding methods, as well as visual inspection, were then used to identify potential GOOFE-specific LacI-family TF operator motifs for all 12 LacI-family TFs (see Additional file 6). A first comparison of the motifs, depicted in Figure 2, showed that the 'CcpA-like' and 'EbgR-like' LacI-family TF operators had characteristic yet distinct subfamily traits. The 'CcpA-like' operators carry a central CG nucleotide pair, whereas the 'EbgR-like' operators have only a single central C or G nucleotide. Moreover, within the subfamilies, the motifs appeared to be discretely different in at least one position.

Left panel: Sequence motifs of predicted LacI-family TF specific operators in L. plantarum. Right panel: The protein sequence motif of the DNA-binding region of the LacI-family TFs per GOOFE. The numbering of the protein residues deviates slightly from that in the various crystal structures. This relates to the fact that the alignment includes some gaps that are necessary to accommodate all the LacI protein sequences that have been compared by us. The visualization of the sequences was created using Weblogo [99]. remark: NMR studies have shown that the hinge helix plays an important role in kinking the DNA whilst forming an alpha-helix (helix 4) and thereby stabilizing the induced fit of the recognition helix within the major groove of the operator [33,81]. In fact, the 3D-structures of operator-bound CcpA and LacI implicate many residues of helix 3 and 4 in the contact of the TF with the operator [56,57]. Moreover, the 3D-structures indicate that in both CcpA and LacI the same residues are involved. The DNA-protein contacts are indicated with triangles. The blue triangles mark the residues interacting with the phosphate backbone and the red triangles mark the residues interacting directly with a nucleotide (the position of the nucleotide is indicated in a box). In the case of Lp_0188 (SacR), a well-conserved guanine and corresponding cytidine are found at positions -7 and 7 of the operator, respectively. This suggests that the operator recognized by Lp_0188 (SacR) and its orthologs, is two nucleotides wider than that recognized by other 'CcpA-like' LacI-TFs. The 'EbgR-like' LacI-family TFs carry a conserved insertion before helix 3 and seem to lack the characteristic conserved alanine and leucine (or methionine in the case of RbsR) at position 60 of the hinge helix in the 'CcpA-like' LacI-family TFs. The absence of these residues coincides perfectly with the absence of the central CG nucleotide pair in the predicted Lp_3470 (LacR), Lp_3479 (GalR) and Lp_3488 (RafR) operators.
Validation: Comparison of the predicted motifs with experimental functional data from literature
Compelling evidence that the recovered motifs were genuine and the approach was effective came from a comparison of the motif predictions with experimentally characterized operators. In all cases that could be checked, the prediction was in full agreement with the experimental findings. This was true for CcpA in Lactobacilli [48, 49], for the Lp_3470 ortholog LacR in Lactobacillus delbrueckii subsp. lactis [50], for the Lp_3479 ortholog GalR in Streptococcus thermophilus [51] and in Streptococcus mutans [52], as well as for MalR in Stretococcus pneumoniae [53], for MalI in E. coli [31] and for ExuR in Bacillus subtilis [54] (see Tables 1, 2 and 3).
Validation: Comparison of the predicted with 3-D structure information from literature
It also proved possible to use structural information on the binding of several LacI-family members to their respective operator [55–57] to validate predicted motifs. Differences in the conservation of certain amino acid residues in the DNA-binding domain of the TF were compared to the composition of the connected operator. Two clear correlations between protein sequence and operator sequence were found (see also the legend to Figure 2):
- Firstly, the structural data suggest that, in the case of CcpA and LacI, the conserved arginine located at position 24 is one of the few residues that hydrogen bonds directly with one of the nucleotide bases, a guanine at position -6 of the operator [56, 57]. In Lp_3661 (RbsR) and its orthologs, the arginine is replaced by a glutamine (or leucine) and correspondingly the otherwise 'conserved' guanine is replaced by a thymidine. In fact, such a replacement was observed for all other studied LacI-family TFs deviant at position 24 (see Additional file 7). These anomalous TFs include MalI from E. coli which was proven experimentally to indeed bind an operator with a thymidine at position -6 [31] (Table 2).
- Secondly, the 'EbgR-like' TFs (i.e. Lp_3470 (LacR), Lp_3479 (GalR) and Lp_3488 (RafR)) are expected to have distinct DNA-binding features. Members of this subfamily lack the conserved leucine residue (position 60 in Figure 2) that according to the 3D-structure of operator-bound CcpA [57] intercalates between the central CG base pairs that are characteristic for 'CcpA-like' operators [22]. Concordantly, the predicted 'EbgR-like' LacI-family TF operators lack the central CG nucleotide pair. Possibly, the conserved arginine at position 24 interacts with the conserved single C or G nucleotide in the operator (Table 3).
II) Identification of the biological role of a TF through comparative genomics
The biological role of a transcription factor is to activate or repress the transcription of certain genes in response to the presence of a signal (e.g. a nutrient or metabolite). In principle, once the sequence of a TF-specific operator is known, a genome-wide search for the related motif could be used to find putative TF-binding sites on the genome and to establish the regulated functionalities (regulon). The signal that triggers the transcriptional response can be obtained by linking the specific TF to an ortholog that has experimentally verified 'inducer' specificities. Finally, the transcriptional effect (i.e. activation or repression) of the binding of the TF can be deduced from the relative position of the putative binding site with respect to the promoter [25, 58].
Regulon predictions for the LacI-family TF homologs in L. plantarumWCFS1
The 12 predicted specific operator motifs were used to search the genome for potential TF-binding sites. For each of the identified specific motifs an initial list of 30 to 100 putative binding sites was retrieved and the list was reduced by application of a distance and similarity criterion to yield a few putative highly specific binding sites per TF (visualized in Figure 3; data in Additional file 8). Not surprisingly, the 'best hits' included those sites that were used to create the search motif in the first place. However, they also included multiple sites that were not used as input. For instance in L. plantarum, new Lp_0172 (MalR) operators were detected upstream of the operon comprising the gene lp_0172 (malR) and upstream of the neighboring operon. Furthermore, the notion that auto-regulation is an important feature connected to LacI-family TF mediated regulation [27, 49–51, 54] was confirmed within L. plantarum, by the identification of a specific binding site upstream of all LacI-family TFs with the exception of Lp_0173, Lp_3488 (RafR) and Lp_3661 (RbsR). It is generally accepted that auto-regulation provides stability to a transcriptional network [59–61].

The L. plantarum operons predicted to be controlled by (a) 'CcpA-like' LacI-family TFs and (b) 'EbgR-like' LacI-family TFs. The set of operons is restricted to those having a very high probability of being correctly predicted. The positions of putative operators are marked by triangles and the direction in which transcription is presumably regulated is indicated (< and >). The functional categories of the proteins encoded by the genes that are under the control of LacI-family TFs are color-coded as depicted in the inset. The functional annotations were taken from the in-house annotation database of L. plantarum WCFS1 ([38] and C. Francke unpublished results). See [Additional file 9] for a detailed description of the functional annotation.
As expected, most potential binding sites were identified upstream of operons that encoded functionalities related to the catabolism of particular carbohydrates. In L. plantarum WCFS1, 11 out of 12 LacI-family TFs were found to be associated with active carbohydrate transport systems (driven by protons: GPH family; ATP: ABC transport systems; or phosphoenolpyruvate: PhosphoTransferaseSystems). Furthermore, the size of the putative regulons varied slightly. For instance, the putative regulon of Lp_3625 encompassed only one operon, whereas that of Lp_0172 encompassed five operons (Figure 3). Although the putative regulon of CcpA was the largest, it was still limited in size, which is slightly in contrast with the global role of CcpA [21, 24, 25]. The precise composition and functionality of most of the predicted regulons is discussed in some detail in Additional file 9.
The molecular function of LacI-family TFs and the connection with the predicted biological role
The functional similarity between homologs can be derived from a proper phylogeny of all homologous sequences [41, 62]. However, the low bootstrap support for the 'early' branches in the NJ-tree of all LacI homologs made it impossible to deduce functional similarities between the members of different clusters of orthologous sequences (Methods and see Additional file 4). It was observed by us (Francke et al. unpublished results) and others [63] that generating a NJ-tree of intra-species homologs of specific functional domains is extremely helpful to overcome this problem. To obtain putative links with experimental functional data, orthologous sequences linked with experimentally verified 'inducer' specificities can be added. The branching pattern within such a NJ-tree for the LacI-family TFs of L. plantarum (Figure 4 and see Additional file 5) and the bootstrap support for that pattern, suggested clear similarities in the encoded affinity for certain inducer substrates. It must be noted here that 'inducer' does not necessarily mean that the binding of the TF to the DNA is promoted by the particular molecule. In fact, in many cases the interaction with the 'inducer' causes a release of the LacI-family TF from the DNA and thereby a relief from repression (as shown experimentally for MalR of E. faecalis [64]; SacR in L. plantarum [65]; GalR in S. thermophilus [51]; and RbsR in L. sakei [66]).

A reduced NJ-tree of the inducer-binding domain for the LacI-family TF homologs of L. plantarum. The sequences of LacI-family TF homologs with known inducer from other organisms were added for comparison [21, 34, 45, 50-53, 64, 67-69, 84, 100-109]. Orthology is indicated by color-coding. The numbers accompanying the clusters in the NJ-tree represent the bootstrap support for the individual divisions (out of 1000).
The identification of various additional sites whose presence should be expected (i.e. related to autoregulation or regulation of genomically associated operons) supported the view that the approach yielded genuine TF-specific binding sites. Other support for the validity of the identified sites and regulons was provided by a comparison of the functionalities encoded by the regulons and the molecules that induced the activity (or better: the in-activity) of the related TFs. Figures 3 and 4 show that in almost all cases a straightforward metabolic link existed between the predicted regulated functionality and the assigned 'inducer' of the TF. For example in L. plantarum, Lp_0188 (SacR) is predicted to respond to sucrose or oligofructose, a prediction that was derived from experimental evidence obtained for orthologous TFs [67–69]. Concordantly, its putative operators are found upstream of two operons that harbor the genes encoding an active oligofructose/sucrose uptake system [65] and enzymes that catalyze the conversion of the phosphorylated oligosaccharide into phosphorylated disaccharide and the phosphorylated disaccharide into glucose-6-P and fructose.
Some of the predicted regulatory connections could be substantiated directly by published transcription data for L. plantarum or related species. On the other hand, the predictions also could often not be extrapolated in a straightforward way. For instance, similar to the prediction for L. plantarum, the expression of the ribose utilization operon (rbsUDK) in L. sakei was shown to be controlled by RbsR and induced by ribose [66]. Unfortunately, the induction of other operons was not studied. Another example is provided by transcriptome data for L. plantarum grown on short-chain fructooligosaccharides compared to glucose. As predicted, expression of the divergon associated with lp_0188 was induced under these conditions [65]. Nevertheless, the maltase/sucrase encoding gene lp_0174 that was predicted to be controlled by Lp_0188 (SacR) was not induced. This observation could very well relate to additional factors that are involved in the regulation of the particular gene. An example of the subtle differences between species is found for the regulation of the gal operon (galK, galT and galE) and lac operon (lacS and lacZ). In S. mutans [52], S. thermophilus CNRZ 302 [51] and S. salivarius [70] expression of the gal operon, as well as that of galM and the lac operon in S. thermophilus and S. salivarius was shown to be controlled by GalR and induced by galactose. Our predictions suggest that in L. plantarum the gal operon is similarly controlled by the GalR ortholog Lp_3479. The lac-operon in L. plantarum however, was predicted to be controlled by a paralogous LacI-family TF, Lp_3470 (LacR), that is absent from the Streptococci, but which is present in L. acidophilus where it was shown to regulate an integrated lac-gal operon [71]. At the same time, in some strains of another Lactobacillus species (L. delbrueckii [50]) the lac-operon was shown again to be controlled by an ortholog of Lp_3479 (GalR).
The mode of action: repression or activation
Although, the nomenclature of most LacI-family TFs hints that their main mode of operation is repression (hence: Repressor), for CcpA it has been shown that it can also act as activator [21, 23]. In Lactococcus lactis, activation by CcpA was observed when the central nucleotide of the CRE was located at position -31 or -21 with respect to the -35-sequence of the promoter and repression by CcpA, when it was located around positions -9, -4, +9, +19, +40, +50 and further downstream [25]. The characteristic intervals of 5 or 10.5 bases were ascribed to a helix-face dependence of the regulatory activity. A similar dependence had been observed before in the activation of ackA transcription in B. subtilis [72]. The footprints accumulated in Figure 5, indicated that most LacI-family TFs in L. plantarum are indeed expected to act as repressor. Moreover, in all cases the predicted operators are found at the expected characteristic positions with respect to the promoter.

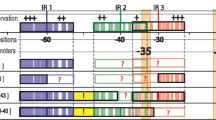
Operators present in the neighborhood of the genes encoding the LacI-family TFs of the 'CcpA-like' subfamily. For most TFs an alignment of the upstream region is shown for the sequences related to one GOOFE. In the case of CcpA, CcpB and Lp_0172 (MalR), no proper alignment could be made with regions from other organisms. Potential CREs are indicated by orange bordered boxes and the LacI-family TF specific operators are indicated by differently colored boxes. The -35/-10 regions of the putative promoters are underlined in purple and pink, respectively. The translation start is positioned at the right end and is indicated in green, as is the putative ribosome binding site.
Regulatory overlap between LacI-TFs
Another interesting aspect of the predicted regulatory connections that became apparent from inspection of the footprints in Figure 5 was that many operons appear to be preceded by multiple LacI-specific putative operators. For example, the two neighboring operons involved in sucrose/oligofructose transport and catabolism are preceded by two putative Lp_0188 (SacR) operators. It was shown that transcription of these operons is indeed induced simultaneously [65]. This observation fits the assumption that one of the operators controls the transcription in one direction and the other in the opposite direction. Conversely, two divergently transcribed genes can in principle also be controlled by a single operator. The latter was shown to be the case in the transcriptional control of the gene levR and the operon levABCDX in Lactobacillus casei [73] and in the transcriptional control of the genes pepQ and ccpA in L. delbrueckii [74] and L. lactis [25]. The genes pepQ and ccpA are similarly organized in L. plantarum. Furthermore, upstream of the ccpA gene three different putative promoter sites can be distinguished and every promoter seems to be connected to its own CcpA operator. This finding is in line with the experimental evidence provided by [49]. Nevertheless, based on the relative positions of the putative CREs, the effect of CcpA on its own expression is extremely difficult to predict of hand. CcpA seems to act as activator as well as repressor depending on the actual promoter.
The role of TF concentration
The molecular nature of the interaction between TF and operator dictates that the actual binding of the two will be dependent upon the activity (in the thermodynamic sense) of both. Consequently, the occupancy of any binding site by a certain TF can be raised by raising TF concentration. In fact, a relatively high TF concentration is anticipated for CcpA [21]. To get some idea of the relative concentrations of the LacI-family TFs in L. plantarum, transcript levels obtained under different growth conditions were inspected (see Figure 6). The observed transcript levels suggested that except for Lp_3625, all LacI-family TFs are under some conditions expressed to relatively intermediate levels and Lp_0172 (MalR), Lp_0188 (SacR), Lp_3531, and CcpB even to levels as high as, or even higher than CcpA.

The relative absolute levels of LacI-family TF related mRNAs in various microarray experiments with L. plantarum. The related array data were used before by [17]. Information on the determination of these levels can be found in the Materials and methods.
Discussion
A generic method to identify TF-specific operators
Every line of evidence sustains the validity of the approach we have formulated to identify LacI-family TF specific operator motifs. In all cases where a LacI-family TF operator has been characterized experimentally, our prediction is in full agreement (see Tables 1, 2 and 3). And likewise, several correlations between the TF sequence (protein) and operator sequence (DNA) that are anticipated on basis of structural information [56, 57] were retrieved perfectly. Moreover, the fact that a specific operator motif could be identified for every LacI-family TF and that relatively few proper hits for these operator motifs were found on the complete genome, is proof by itself. More non-specific methods inevitably would have yielded more degenerate motifs and more false-positive identifications.
In lactic acid bacteria many operons involved in carbohydrate catabolism are associated on the genome by the gene encoding the respective regulator [75]. In fact, this observation may be generalized for all TFs that are considered 'local' regulators. Our results indicate that especially for these TFs, a distribution into Groups of Orthologous Functional Equivalents will reduce the noise in the motif prediction significantly. In contrast, as current automated methods generate more degenerate motifs [17] these methods are better suited for the recovery of binding sites for 'global' regulators.
Characteristic motifs and the implications of degeneracy
As the interaction between TF and DNA allows for a certain structural freedom, a TF-specific operator is not necessarily a unique sequence but merely a collection of sequences which can be represented as a motif or consensus sequence. The molecular nature of the interactions dictates a distinct relationship between the affinity of the TF protein for the operator DNA and their respective sequence. As a consequence, a more degenerate operator motif relates to reduced affinity. For example, the LacI-family TF CytR in E. coli exhibits a more versatile binding of operator sequences than LacI (i.e. has a higher motif degeneracy). At the same time it was observed that its affinity for the operator is much reduced when compared to LacI [76, 77]. Likewise, the affinity of the TF for the operator was shown to be affected significantly by subtle changes in the protein sequence [78] as well as in the nucleotide sequence of the operator (for LacI: [79–81]; for CcpA: [29]). Lehming et al. therefore [78] assumed explicitly that the interaction between TF and operator should be concentration dependent. Ultimately, it is the relation between the concentration (or better: activity) of active TF and the rate of expression that determines key features of the dynamics of the cellular response to internal and external signals [82].
The predicted specific operator motifs of the LacI-family TFs in L. plantarum exhibit relatively little degeneracy (>8 nucleotides fully conserved for the 'CcpA-like' subfamily; see Figure 2) with one exception: the operator motif of CcpA itself. Considering the above, and based on the fact that in the 3D-structures of CcpA and LacI bound to their respective operators the same residues are involved in the interaction of TF with DNA [56, 57], the degeneracy of the CcpA operator motif indicates it should act at relatively higher concentrations with respect to LacI and other relatives. Concomitantly, variable regulation of ccpA expression would represent a way to control the differential binding of CcpA to CREs [21].
'Local' versus 'global' regulation
The identified CcpA operator motif (CRE) of L. plantarum is very similar to the consensus CRE that was initially defined for B. subtilis on basis of a site-directed mutagenesis study [29] and later refined on basis of the experimental identification of additional CREs [22, 30] (CRE consensus sequences are summarized in Table 1). Remarkably, the DNA-binding domain of CcpA on the protein level is considerably more conserved compared to that of the other LacI-family TFs (see Figure 2 right panel), whereas in contrast, the operator motif is the most degenerate. Both facts reflect and emphasize the 'global' role of CcpA. We observed that the CcpA regulon that was defined on basis of a genome wide search with the specific operator motif was relatively small. The same observation was made by [22] when the genome of B. subtilis was searched for potential CREs for the first time. The authors concluded that this related to the lack of degeneracy in the search motif and they proved experimentally that this was indeed the case.
It is generally assumed that transcription and translation are connected processes in bacteria [83] and as a consequence proteins should be produced in the physical vicinity of where they are encoded. A major implication of an intended local role of a TF would then be that the number of TF molecules necessary to effectively control expression can be minimized in case the affinity for the operator is relatively high (signified by a less degenerate motif). As mentioned in the previous section, all but one of the predicted operators indeed show a relatively high degree of conservation over different, sometimes even distantly related, species. A low TF concentration will keep in check non-local interactions as the TF will be virtually absent in the rest of the cell and, as a result, even operators that are very similar will not be affected. In fact, it was shown for carbohydrate utilization by Lactobacillus acidophilus that induction of catabolic operons is highly specific for distinct sugars [71]. Vice versa, a higher TF concentration, like anticipated for CcpA [21], would relax the sensitivity towards the composition of the operator and thus enable binding to sites for which the TF has less affinity. However, transcript levels that are observed in L. plantarum under different growth conditions are not completely conclusive (see Figure 6). Nevertheless, based on the observed transcript levels one should expect that Lp_0172 (MalR), Lp_0188 (SacR), Lp_3221, Lp_3531, Lp_3661 (RbsR), CcpB and CcpA in principle could regulate multiple and also distant operons.
Regulon boundaries and induced response
Searching the genome of L. plantarum with the identified specific operator motifs yielded a list of potential binding-sites for every LacI-family TF. To avoid many false predictions, we have used two conservative criteria to reduce the list of putative TF-specific binding sites. They related to the position of the site with respect to the translation start, as there is experimental data showing certain boundaries for that distance [21, 23], and to a maximum number of 2 deviating nucleotides. The genes/operons preceded by the putative binding sites thus should constitute putative minimal regulons. In principle, more degenerate motifs should lead to a longer list of compliant sites, as was indeed observed. This observation, which was earlier made by others [22], reveals a key point in regulon predictions based on operator motifs, namely motif degeneracy complicates a straightforward decision about the authenticity of the recovered sites. Moreover, as described in the above sections, binding will by necessity be influenced by TF concentration (activity). Therefore, experimental data on gene expression and TF concentration (activity) will be essential to refine the predictions. At the same time, in most cases, a proper interpretation of experimental transcription data will require motif and regulon predictions because of the fact that the activity of many TFs is intertwined and the number of conditions tested or testable too limited to untwine these. Although the extrapolation of the predictions to experimental data is non-trivial, several of the predicted associations could be confirmed on basis of data obtained in L. plantarum and related species (see Results). Moreover, a comparison of the predicted regulons depicted in Figure 3 with the environmental signals that are expected to govern the specific LacI-family TF activities (see Figure 4) shows that the recovered connection make perfect biological sense. This finding strongly supports the assertion that the predictions provide a valid coupling between the LacI-family TFs and functionalities encoded by the putative regulons.
Conclusion
We have formulated a sequence-based approach that enables the identification of TF-specific binding motifs. One of the major advantages of the approach is that it is generic and thus, in principle, can be applied to any TF family without prior knowledge of the actual composition of the binding motif. In fact, we are in the process of performing similar analyses for various TF-families, including two component systems, and the preliminary results confirm the assertion. The method appears perfectly suited to identify binding sites on the genome connected to local regulators in contrast to current automated procedures that yield mostly sites connected to global regulators.
The presented data substantiate the successful identification of specific operator motifs related to the LacI-family TFs in the model organism L. plantarum. The recovered motifs differ in at least one position but at the same time their similarity is considerable. As the composition of the operator motif is tightly related to the affinity of the TF for the DNA this finding implicates that some of the LacI-family TFs could potentially bind to the operators of another. In fact, the observed competition in B. subtilis TF knock outs, between CcpA and CcpB in the repression of the gnt and xyl operon [84], exemplifies this phenomenon. Simultaneously, higher TF (or binding site) concentration (activity) will result in regulation at degenerate sites (i.e. lower affinity) (see [1]), a conclusion that correlates well with the mechanism of control of TF-activity itself as this involves a change in affinity of the TF for the operator upon induction [34, 85]. An important corollary is that regulons, and especially those related to global regulators, will vary in size depending on the environmental conditions.
Finally, potential binding sites can be identified based on the operator motif predictions and from those the functionalities that are regulated in response to a given stimulus can be reconstructed. In principle, the coupling of putative regulons with potential TF inducers thus provides insight in the prioritization of the functionalities within a certain organism. Nevertheless, our data on LacI-family TFs in L. plantarum makes perfectly clear that in order to arrive at a complete reconstruction of the encoded transcriptional response to environmental stimuli, experimental data on transcription as well as TF and inducer concentration under different environmental conditions is adamant.
Methods
Resources and tools
All genomic information was obtained from the ERGO genome analysis and discovery system [86] and updated until the 1st of July 2007. Nevertheless, the presented results do not depend on the use of this particular resource and the methods described in this paper can as well be applied using publicly accessible resources (like those at NCBI [87]). The genome sequence of L. plantarum WCFS1 and the functional annotation of its genes was taken from our in-house annotation database [38]. Potentially homologous sequences were collected from the database using the BLAST algorithm [88], with a typical cut-off between 10-2 and 10-10. Multiple sequence alignments were made with MUSCLE [89] (default settings). Alignments were visually inspected and aberrant sequences were removed (characterized by many gaps and a distinctly different conservation pattern). BioEdit [90] and Jalview [91] were used to edit sequences, and ClustalW [92] was used to create (domain-) specific bootstrapped neighbor-joining trees (with 'correction for multiple substitutions' [93]). The resulting trees were analyzed using LOFT, a tool that automatically divides the sequences into orthologous groups based on the hierarchy of the tree and the duplication and speciation events implied by that hierarchy [62]. Overrepresented DNA sequences in a selected set of upstream regions (300 bases) were identified automatically using MEME [94] and MAST [95] was used to detect other potential TF-binding sites on the genome (default cut-off p-value < 10-5).
Identification of TF-specific operator motifs
A generic phylogenetic footprinting/shadowing approach was formulated to improve the identification of TF-specific operator motifs. Compared to other methods the specificity of the motif prediction is increased by the identification of orthologs proper and by taking into account the modular organization of the bacterial genome. The approach was applied to a model family of TFs (LacI) in the model organism L. plantarum WCFS1. The related flow scheme is depicted in Figure 7 and described in detail below:

The TF-specific operator motif identification workflow. 1) First a particular TF-family was selected and 2) a prominent representative of that family was chosen. 3) The related sequence was used to search the genome of a particular species for intra-species homologs. This search was iterated until no new sequences are recovered. A high e-value cut-off was employed to ensure the recovery of all homologs. The sequences were aligned and a NJ-tree was generated. Both the alignment and the NJ-tree were used to determine the family or sub-family boundaries. 4) The procedure was repeated to retrieve all inter-species homologs and the general features of the intra-species homologs were used to determine the sequences that were taken into consideration. Orthologous relations between sequences were established on basis of clustering in the NJ-tree and a sufficient bootstrap support (in green) for the clustering. In the case of Lp_0172 and Lp_0173 the orthologous clusters are color-coded in brown and orange, respectively, and the other TFs of L. plantarum are indicated in red. 5) The genomic context of the various orthologs was inspected (legend bottom left) and in case clear differences existed, the orthologous groups were sub-divided into different Groups of orthologous functional equivalents (GOOFEs), as illustrated. Then, upstream regions of the conserved gene(s) in context were selected and inspected for potential regulatory sequences (the selected regions are indicated by colored triangles). The potential regulatory sequences were compared and those that showed similar features were selected. In fact, only those sequences that showed the highest conservation were selected to determine a specific operator motif. In the case of Lp_0172 and Lp_0173, a 'CcpA-like' operator motif was found up to 3 times in the upstream regions. The sequences that were selected to determine the Lp_0172 and Lp_0173 specific operator motifs are displayed (Px indicates the relative position of the selected sequence with respect to other similar sequences and relative to the translation start). 6) The selected sequences were used to create a GOOFE specific operator motif. The thus identified specific motifs related to the orthologous groups containing Lp_0172 and Lp_0173 demonstrate that the division into GOOFEs was essential to arrive at highly specific operator motifs. Although the motifs within both orthologous groups are highly similar, they differ distinctly in one position depending on the GOOFE. In the case of the TFs orthologous to Lp_0172, the motifs are strikingly different at position +5, with a fully conserved guanine in the GOOFE containing Lp_0172 and a fully conserved thymidine in the other. And in the case of the TFs orthologous to Lp_0173 the motifs are strikingly different at position -5, with a fully conserved thymidine in the GOOFE containing Lp_0173 and a fully conserved adenine in the other. remark: The gene/protein identifiers in the figure are derived from the ERGO resource [86]. A conversion to other identifiers can be found in [Additional file 2]. The functional annotation of the depicted genes were taken from the in-house annotation database of L. plantarum WCFS1 ([38] and C. Francke unpublished results) and the ERGO resource. See [Additional file 9] for a detailed description of the functional annotation in L. plantarum.
- Selection of a TF family, the collection of homologs and the derivation of orthology (Figure 7 1–4)
Intra-species and inter-species homologs were collected from the database using BLAST and the search was iterated until no additional sequences were found. This search was not only performed on the level of the complete sequence but also with individual functional domains. The sequences were aligned, aberrant sequences were removed, a bootstrapped NJ-tree was generated, and the hierarchy of the branching together with the bootstrap support were considered to identify orthologs. In the case of LacI-family members, the complete sequence of CcpA from L. plantarum was used as a starting sequence, as well as the N-terminal (first 90 residues; DNA binding domain) and C-terminal (other residues; inducer binding domain) sequence. To restrict the size of the final collection, only Firmicutes genomes were analyzed. The examined species included well-studied organisms such as B. subtilis, L. lactis and S. thermophilus (see Additional file 1 for a complete list of analyzed genomes). To improve the potential for functional identification the genome sequences of several E. coli strains and Salmonella species were also included. A striking feature of the NJ tree of the Firmicutes LacI-family TF homologs was that the representation of the 'early' branching events came out very unreliable, as signified by the extremely low bootstrap support (several values were as low as 1). In contrast, most branches related to supposed more recent evolutionary events had high bootstrap values in the NJ-tree and, as a result, the LacI-family TF homologs could be separated reliably into groups of orthologous sequences (see Additional files 4 and 5). The set of homologs identified by us was compared to the entries in the PFAM database [96].
- Definition of functional equivalents (Figure 7 4,5)
Orthologous clusters can often be further subdivided to obtain putative Groups of Orthologous Functional Equivalent s or GOOFEs. The homogeneity of the sequence alignment (as indicated by conserved stretches of residues and the absence of large gaps or inserts), a high bootstrap-value at the branching point that separates the orthologous cluster from the other sequences (Figure 7 4), and most importantly, a clear difference in conserved gene-context within the group were used to evaluate the necessity of such sub-division (Figure 7 5). In the case of many of the LacI-family TFs of L. plantarum, the subdivision into GOOFEs resulted in clearly distinct operator motifs even within an orthologous group (as illustrated in Figure 7). The protein sequences, alignments and trees can be found in Additional files 1, 2, 3, 4, 5.
- Selection of upstream regions containing putative operators (Figure 7 5)
The observation that most genes encoding TFs seem to be associated on the genome with the genes whose transcription they control may guide the selection of upstream regions. The upstream regions of the conserved operons within a GOOFE were used to search putative operator sites (selected regions (see Additional file 6)). Only, in case the TF encoding gene lay solitary on the genome the upstream regions of the TFs from one GOOFE were used, based on the notion that autoregulation is a common feature of many TFs.
- Motif definition (Figure 7 6)
Potential TF binding regions on the DNA (i.e. operators) were searched automatically in the selected set of upstream regions (300 bases) using MEME. As motif prediction tools often produce multiple motifs including many false positives, an alignment of the regions was made and the observed conservations were compared to the automatically recovered motifs to remove most false positives. The final collection of motifs was then compared within the complete TF-family and the TF-specific motifs were defined based on conserved features, like characteristic residues, spacing and motif length. The LacI-family TFs are known to form functional dimers and as a consequence the reported binding sequence motifs for these proteins are palindromes of lengths varying between 10 and 16 basepairs [55, 57, 81]. Therefore MEME was tuned to find inverted repeats (-pal option) with a maximum width of 20 bases and the detection of 4 different motifs with zero or one occurrence per sequence (-ZOOPS option). The resulting motifs were compared and for each set of upstream regions (related to a certain TF) an operator region of 16 ('CcpA-like') or 17 ('EbgR-like') bases was defined.
- Identification of putative TF binding sites
A specific operator and a position-specific scoring matrix were created for each TF by application of MEME to the defined operator regions. To avoid base preferences in the scoring, a background file in which the probability of finding an A, T, C or G at a certain position at random was set at 0.25. The final position-specific scoring matrices were used as input for an automated genome-wide motif search using MAST. Two additional criteria were used to filter out potential false positives. Firstly, the vast majority of LacI operators that have been identified to date can be found in the range of -250 to +50 nucleotides from the translation start, with no instances further upstream [21, 23]. Therefore, identified sites located more than 250 nucleotides upstream and more than 50 nucleotides downstream of the translation start site were not considered. Secondly, all sites that deviated at more than two positions in the central 14 nucleotides with respect to the operators in the vicinity of the LacI-family TFs, were not considered. The tables that resulted from the MAST search have been deposited in Additional file 8.
Prediction of the inducer of TF activity
A bootstrapped NJ-tree was generated on basis of a multiple sequence alignment of all LacI-family TF homologs of L. plantarum, together with orthologous sequences for which experimental confirmation about the nature of the inducer could be retrieved. TFs were considered equivalent in case they were clearly orthologous (strong bootstrap support), were syntenous and provided the alignment was homogeneous (i.e. the absence of gaps and several clear conservations).
Reconstruction of the mode of regulation
In principle, TFs can act both as transcriptional activator and as repressor depending on the position of the operator relative to the promoter, upstream or inside/downstream, respectively [18, 25, 58, 97]. To resolve whether the TF acts as an activator or repressor, phylogenetic footprints were made for various upstream regions containing an operator and its position relative to that of the potential promoter was determined. In case the alignment was not clear, the predicted operators were used as an anchor to realign the flanking regions for promoter detection.
Determination of relative mRNA levels for the LacI-family TFs
Absolute expression data was obtained from 35 independent micro-array experiments with custom Agilent oligo-based arrays of L. plantarum WCFS1 (this yielded 70 semi-independent datasets). The experimental conditions tested varied from stress to over-expression of certain metabolic genes to growth on different oligosaccharides (D. Molenaar, unpublished data; see also [17]). The raw data were adapted as follows. The absolute signals of the spots related to individual proteins were averaged and then the signals were ranked independently for the two individual channels. Per experiment and per channel, the 50 lowest signals were discarded and the signals of the 200 proteins ranked lowest in the remaining list were averaged. The average was interpreted as basal signal and subtracted from the signals related to the LacI-family TFs. Finally, the resulting signals were made relative by dividing all signals by the highest signal displayed by a LacI-family TF representative.
Abbreviations
- CCR:
-
Carbon Catabolite Repression
- CRE:
-
CcpA-Responsive Element
- GOOFE:
-
Group of Orthologous Functional Equivalents
- TF:
-
Transcription Factor
References
Stormo GD: DNA binding sites: representation and discovery. Bioinformatics. 2000, 16 (1): 16-23. 10.1093/bioinformatics/16.1.16.
Bulyk ML: Computational prediction of transcription-factor binding site locations. Genome Biol. 2003, 5 (1): 201-10.1186/gb-2003-5-1-201.
Thompson W, Rouchka EC, Lawrence CE: Gibbs Recursive Sampler: finding transcription factor binding sites. Nucleic Acids Res. 2003, 31 (13): 3580-3585. 10.1093/nar/gkg608.
Kim JT, Gewehr JE, Martinetz T: Binding matrix: a novel approach for binding site recognition. J Bioinform Comput Biol. 2004, 2 (2): 289-307. 10.1142/S0219720004000569.
Osada R, Zaslavsky E, Singh M: Comparative analysis of methods for representing and searching for transcription factor binding sites. Bioinformatics. 2004, 20 (18): 3516-3525. 10.1093/bioinformatics/bth438.
Yellaboina S, Seshadri J, Kumar MS, Ranjan A: PredictRegulon: a web server for the prediction of the regulatory protein binding sites and operons in prokaryote genomes. Nucleic Acids Res. 2004, 32 (Web Server issue): W318-320. 10.1093/nar/gkh364.
Yan B, Lovley DR, Krushkal J: Genome-wide similarity search for transcription factors and their binding sites in a metal-reducing prokaryote Geobacter sulfurreducens. Biosystems. 2006
Rodionov DA: Comparative genomic reconstruction of transcriptional regulatory networks in bacteria. Chem Rev. 2007, 107 (8): 3467-3497. 10.1021/cr068309+.
Moses AM, Chiang DY, Pollard DA, Iyer VN, Eisen MB: MONKEY: identifying conserved transcription-factor binding sites in multiple alignments using a binding site-specific evolutionary model. Genome Biol. 2004, 5 (12): R98-10.1186/gb-2004-5-12-r98.
Carmack CS, McCue LA, Newberg LA, Lawrence CE: PhyloScan: identification of transcription factor binding sites using cross-species evidence. Algorithms Mol Biol. 2007, 2: 1-10.1186/1748-7188-2-1.
Okumura T, Makiguchi H, Makita Y, Yamashita R, Nakai K: Melina II: a web tool for comparisons among several predictive algorithms to find potential motifs from promoter regions. Nucleic Acids Res. 2007, 35 (Web Server issue): W227-231. 10.1093/nar/gkm362.
Kaplan T, Friedman N, Margalit H: Ab initio prediction of transcription factor targets using structural knowledge. PLoS Comput Biol. 2005, 1 (1): e1-10.1371/journal.pcbi.0010001.
Yan B, Núñez C, Ueki T, Esteve-Núñez A, Puljic M, Adkins RM, Methé BA, Lovley DR, Krushkal J: Computational prediction of RpoS and RpoD regulatory sites in Geobacter sulfurreducens using sequence and gene expression information. Gene. 2006, 384: 73-95. 10.1016/j.gene.2006.06.025.
Monsieurs P, Thijs G, Fadda AA, De Keersmaecker SC, Vanderleyden J, De Moor B, Marchal K: More robust detection of motifs in coexpressed genes by using phylogenetic information. BMC Bioinformatics. 2006, 7: 160-10.1186/1471-2105-7-160.
Alkema WB, Lenhard B, Wasserman WW: Regulog analysis: detection of conserved regulatory networks across bacteria: application to Staphylococcus aureus. Genome Res. 2004, 14 (7): 1362-1373. 10.1101/gr.2242604.
Van Hellemont R, Monsieurs P, Thijs G, de Moor B, Van de Peer Y, Marchal K: A novel approach to identifying regulatory motifs in distantly related genomes. Genome Biol. 2005, 6 (13): R113-10.1186/gb-2005-6-13-r113.
Wels M, Francke C, Kerkhoven R, Kleerebezem M, Siezen RJ: Predicting cis-acting elements of Lactobacillus plantarum by comparative genomics with different taxonomic subgroups. Nucleic Acids Res. 2006, 34 (7): 1947-1958. 10.1093/nar/gkl138.
Grundy FJ, Waters DA, Allen SH, Henkin TM: Regulation of the Bacillus subtilis acetate kinase gene by CcpA. J Bacteriol. 1993, 175 (22): 7348-7355.
Stülke J, Hillen W: Carbon catabolite repression in bacteria. Curr Opin Microbiol. 1999, 2 (2): 195-201. 10.1016/S1369-5274(99)80034-4.
Brückner R, Titgemeyer F: Carbon catabolite repression in bacteria: choice of the carbon source and autoregulatory limitation of sugar utilization. FEMS Microbiol Lett. 2002, 209 (2): 141-148.
Deutscher J, Francke C, Postma PW: How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol Mol Biol Rev. 2006, 70 (4): 939-1031. 10.1128/MMBR.00024-06.
Miwa Y, Nakata A, Ogiwara A, Yamamoto M, Fujita Y: Evaluation and characterization of catabolite-responsive elements (cre) of Bacillus subtilis. Nucleic Acids Res. 2000, 28 (5): 1206-1210. 10.1093/nar/28.5.1206.
Deutscher J, Galinier A, Martin-Verstraete I: Carbohydrate uptake and metabolism. Bacillus subtilis and its closest relatives; From genes to cells. Edited by: Sonenshein AL, Hoch JA, Losick R. 2002, Washington DC , ASM Press, 129-150.
Babu MM, Teichmann SA, Aravind L: Evolutionary dynamics of prokaryotic transcriptional regulatory networks. J Mol Biol. 2006, 358 (2): 614-633. 10.1016/j.jmb.2006.02.019.
Zomer AL, Buist G, Larsen R, Kok J, Kuipers OP: Time-resolved determination of the CcpA regulon of Lactococcus lactis subsp. cremoris MG1363. J Bacteriol. 2007, 189 (4): 1366-1381. 10.1128/JB.01013-06.
Kraus A, Hueck C, Gärtner D, Hillen W: Catabolite repression of the Bacillus subtilis xyl operon involves a cis element functional in the context of an unrelated sequence, and glucose exerts additional xylR-dependent repression. J Bacteriol. 1994, 176 (6): 1738-1745.
Leboeuf C, Leblanc L, Auffray Y, Hartke A: Characterization of the ccpA gene of Enterococcus faecalis: identification of starvation-inducible proteins regulated by ccpA. J Bacteriol. 2000, 182 (20): 5799-5806. 10.1128/JB.182.20.5799-5806.2000.
Yoshida K, Kobayashi K, Miwa Y, Kang CM, Matsunaga M, Yamaguchi H, Tojo S, Yamamoto M, Nishi R, Ogasawara N, Nakayama T, Fujita Y: Combined transcriptome and proteome analysis as a powerful approach to study genes under glucose repression in Bacillus subtilis. Nucleic Acids Res. 2001, 29 (3): 683-692. 10.1093/nar/29.3.683.
Weickert MJ, Chambliss GH: Site-directed mutagenesis of a catabolite repression operator sequence in Bacillus subtilis. Proc Natl Acad Sci USA. 1990, 87 (16): 6238-6242. 10.1073/pnas.87.16.6238.
Hueck CJ, Hillen W, Saier MH: Analysis of a cis-active sequence mediating catabolite repression in gram-positive bacteria. Res Microbiol. 1994, 145 (7): 503-518. 10.1016/0923-2508(94)90028-0.
Reidl J, Römisch K, Ehrmann M, Boos W: MalI, a novel protein involved in regulation of the maltose system of Escherichia coli, is highly homologous to the repressor proteins GalR, CytR, and LacI. J Bacteriol. 1989, 171 (9): 4888-4899.
Weickert MJ, Adhya S: A family of bacterial regulators homologous to Gal and Lac repressors. J Biol Chem. 1992, 267 (22): 15869-15874.
Spronk CA, Bonvin AM, Radha PK, Melacini G, Boelens R, Kaptein R: The solution structure of Lac repressor headpiece 62 complexed to a symmetrical lac operator. Structure. 1999, 7 (12): 1483-1492. 10.1016/S0969-2126(00)88339-2.
Lewis M: The lac repressor. C R Biol. 2005, 328 (6): 521-548. 10.1016/j.crvi.2005.04.004.
Makita Y, Nakao M, Ogasawara N, Nakai K: DBTBS: database of transcriptional regulation in Bacillus subtilis and its contribution to comparative genomics. Nucleic Acids Res. 2004, 32 (Database issue): D75-77. 10.1093/nar/gkh074.
Kummerfeld SK, Teichmann SA: DBD: a transcription factor prediction database. Nucleic Acids Res. 2006, 34 (Database issue): D74-81. 10.1093/nar/gkj131.
Salgado H, Santos-Zavaleta A, Gama-Castro S, Peralta-Gil M, Peñaloza-Spínola MI, Martínez-Antonio A, Karp PD, Collado-Vides J: The comprehensive updated regulatory network of Escherichia coli K-12. BMC Bioinformatics. 2006, 7 (1): 5-10.1186/1471-2105-7-5.
Kleerebezem M, Boekhorst J, van Kranenburg R, Molenaar D, Kuipers OP, Leer R, Tarchini R, Peters SA, Sandbrink HM, Fiers MW, Stiekema W, Klein Lankhorst RM, Bron PA, Hoffer SM, Nierop Groot M, Kerkhoven R, de Vries M, Ursing B, de Vos WM, Siezen RJ: Complete genome sequence of Lactobacillus plantarum WCFS1. Proc Natl Acad Sci USA. 2003, 100 (4): 1990-1995. 10.1073/pnas.0337704100.
Siezen R, Boekhorst J, Muscariello L, Molenaar D, Renckens B, Kleerebezem M: Lactobacillus plantarum gene clusters encoding putative cell-surface protein complexes for carbohydrate utilization are conserved in specific gram-positive bacteria. BMC Genomics. 2006, 7: 126-10.1186/1471-2164-7-126.
Fitch WM: Homology a personal view on some of the problems. Trends Genet. 2000, 16 (5): 227-231. 10.1016/S0168-9525(00)02005-9.
Koonin EV: Orthologs, paralogs, and evolutionary genomics. Annu Rev Genet. 2005, 39: 309-338. 10.1146/annurev.genet.39.073003.114725.
Huynen MA, Gabaldón T, Snel B: Variation and evolution of biomolecular systems: searching for functional relevance. FEBS Lett. 2005, 579 (8): 1839-1845. 10.1016/j.febslet.2005.02.004.
Tagle DA, Koop BF, Goodman M, Slightom JL, Hess DL, Jones RT: Embryonic epsilon and gamma globin genes of a prosimian primate (Galago crassicaudatus). Nucleotide and amino acid sequences, developmental regulation and phylogenetic footprints. J Mol Biol. 1988, 203 (2): 439-455. 10.1016/0022-2836(88)90011-3.
Boffelli D, McAuliffe J, Ovcharenko D, Lewis KD, Ovcharenko I, Pachter L, Rubin EM: Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science. 2003, 299 (5611): 1391-1394. 10.1126/science.1081331.
Hall BG: The EBG system of E. coli: origin and evolution of a novel beta-galactosidase for the metabolism of lactose. Genetica. 2003, 118 (2-3): 143-156. 10.1023/A:1024149508376.
Makarova K, Slesarev A, Wolf Y, Sorokin A, Mirkin B, Koonin E, Pavlov A, Pavlova N, Karamychev V, Polouchine N, Shakhova V, Grigoriev I, Lou Y, Rohksar D, Lucas S, Huang K, Goodstein DM, Hawkins T, Plengvidhya V, Welker D, Hughes J, Goh Y, Benson A, Baldwin K, Lee JH, Díaz-Muñiz I, Dosti B, Smeianov V, Wechter W, Barabote R, Lorca G, Altermann E, Barrangou R, Ganesan B, Xie Y, Rawsthorne H, Tamir D, Parker C, Breidt F, Broadbent J, Hutkins R, O'Sullivan D, Steele J, Unlu G, Saier M, Klaenhammer T, Richardson P, Kozyavkin S, Weimer B, Mills D: Comparative genomics of the lactic acid bacteria. Proc Natl Acad Sci USA. 2006, 103 (42): 15611-15616. 10.1073/pnas.0607117103.
Molenaar D, Bringel F, Schuren FH, de Vos WM, Siezen RJ, Kleerebezem M: Exploring Lactobacillus plantarum genome diversity by using microarrays. J Bacteriol. 2005, 187 (17): 6119-6127. 10.1128/JB.187.17.6119-6127.2005.
Mahr K, Hillen W, Titgemeyer F: Carbon catabolite repression in Lactobacillus pentosus: analysis of the ccpA region. Appl Environ Microbiol. 2000, 66 (1): 277-283.
Muscariello L, Marasco R, De Felice M, Sacco M: The functional ccpA gene is required for carbon catabolite repression in Lactobacillus plantarum. Appl Environ Microbiol. 2001, 67 (7): 2903-2907. 10.1128/AEM.67.7.2903-2907.2001.
Lapierre L, Mollet B, Germond JE: Regulation and adaptive evolution of lactose operon expression in Lactobacillus delbrueckii. J Bacteriol. 2002, 184 (4): 928-935. 10.1128/jb.184.4.928-935.2002.
Vaughan EE, van den Bogaard PT, Catzeddu P, Kuipers OP, de Vos WM: Activation of silent gal genes in the lac-gal regulon of Streptococcus thermophilus. J Bacteriol. 2001, 183 (4): 1184-1194. 10.1128/JB.183.4.1184-1194.2001.
Ajdić D, Ferretti JJ: Transcriptional regulation of the Streptococcus mutans gal operon by the GalR repressor. J Bacteriol. 1998, 180 (21): 5727-5732.
Nieto C, Puyet A, Espinosa M: MalR-mediated regulation of the Streptococcus pneumoniae malMP operon at promoter PM. Influence of a proximal divergent promoter region and competition between MalR and RNA polymerase proteins. J Biol Chem. 2001, 276 (18): 14946-14954. 10.1074/jbc.M010911200.
Mekjian KR, Bryan EM, Beall BW, Moran CP: Regulation of hexuronate utilization in Bacillus subtilis. J Bacteriol. 1999, 181 (2): 426-433.
Bell CE, Lewis M: The Lac repressor: a second generation of structural and functional studies. Curr Opin Struct Biol. 2001, 11 (1): 19-25. 10.1016/S0959-440X(00)00180-9.
Kalodimos CG, Biris N, Bonvin AM, Levandoski MM, Guennuegues M, Boelens R, Kaptein R: Structure and flexibility adaptation in nonspecific and specific protein-DNA complexes. Science. 2004, 305 (5682): 386-389. 10.1126/science.1097064.
Schumacher MA, Allen GS, Diel M, Seidel G, Hillen W, Brennan RG: Structural basis for allosteric control of the transcription regulator CcpA by the phosphoprotein HPr-Ser46-P. Cell. 2004, 118 (6): 731-741. 10.1016/j.cell.2004.08.027.
Kim JH, Yang YK, Chambliss GH: Evidence that Bacillus catabolite control protein CcpA interacts with RNA polymerase to inhibit transcription. Mol Microbiol. 2005, 56 (1): 155-162. 10.1111/j.1365-2958.2005.04496.x.
Becskei A, Serrano L: Engineering stability in gene networks by autoregulation. Nature. 2000, 405 (6786): 590-593. 10.1038/35014651.
Roy S, Sahu A, Adhya S: Evolution of DNA binding motifs and operators. Gene. 2002, 285 (1-2): 169-173. 10.1016/S0378-1119(02)00413-4.
Maheshri N, O'Shea EK: Living with noisy genes: how cells function reliably with inherent variability in gene expression. Annu Rev Biophys Biomol Struct. 2007, 36: 413-434. 10.1146/annurev.biophys.36.040306.132705.
van der Heijden RT, Snel B, van Noort V, Huynen MA: Orthology prediction at scalable resolution by phylogenetic tree analysis. BMC Bioinformatics. 2007, 8: 83-10.1186/1471-2105-8-83.
Quentin Y, Fichant G, Denizot F: Inventory, assembly and analysis of Bacillus subtilis ABC transport systems. J Mol Biol. 1999, 287 (3): 467-484. 10.1006/jmbi.1999.2624.
Le Breton Y, Pichereau V, Sauvageot N, Auffray Y, Rincé A: Maltose utilization in Enterococcus faecalis. J Appl Microbiol. 2005, 98 (4): 806-813. 10.1111/j.1365-2672.2004.02468.x.
Saulnier DM, Molenaar D, de Vos WM, Gibson GR, Kolida S: Identification of prebiotic fructooligosaccharide metabolism in Lactobacillus plantarum WCFS1 through microarrays. Appl Environ Microbiol. 2007, 73 (6): 1753-1765. 10.1128/AEM.01151-06.
Stentz R, Cornet M, Chaillou S, Zagorec M: Adaptation of Lactobacillus sakei to meat: a new regulatory mechanism of ribose utilization?. Lait. 2001, 81: 131-138. 10.1051/lait:2001117.
Hiratsuka K, Wang B, Sato Y, Kuramitsu H: Regulation of sucrose-6-phosphate hydrolase activity in Streptococcus mutans: characterization of the scrR gene. Infect Immun. 1998, 66 (8): 3736-3743.
Luesink EJ, Marugg JD, Kuipers OP, de Vos WM: Characterization of the divergent sacBK and sacAR operons, involved in sucrose utilization by Lactococcus lactis. J Bacteriol. 1999, 181 (6): 1924-1926.
Barrangou R, Altermann E, Hutkins R, Cano R, Klaenhammer TR: Functional and comparative genomic analyses of an operon involved in fructooligosaccharide utilization by Lactobacillus acidophilus. Proc Natl Acad Sci USA. 2003, 100 (15): 8957-8962. 10.1073/pnas.1332765100.
Vaillancourt K, Moineau S, Frenette M, Lessard C, Vadeboncoeur C: Galactose and lactose genes from the galactose-positive bacterium Streptococcus salivarius and the phylogenetically related galactose-negative bacterium Streptococcus thermophilus: organization, sequence, transcription, and activity of the gal gene products. J Bacteriol. 2002, 184 (3): 785-793.
Barrangou R, Azcarate-Peril MA, Duong T, Conners SB, Kelly RM, Klaenhammer TR: Global analysis of carbohydrate utilization by Lactobacillus acidophilus using cDNA microarrays. Proc Natl Acad Sci USA. 2006, 103 (10): 3816-3821. 10.1073/pnas.0511287103.
Turinsky AJ, Grundy FJ, Kim JH, Chambliss GH, Henkin TM: Transcriptional activation of the Bacillus subtilis ackA gene requires sequences upstream of the promoter. J Bacteriol. 1998, 180 (22): 5961-5967.
Maze A, Boel G, Poncet S, Mijakovic I, Le Breton Y, Benachour A, Monedero V, Deutscher J, Hartke A: The Lactobacillus casei ptsHI47T mutation causes overexpression of a LevR-regulated but RpoN-independent operon encoding a mannose class phosphotransferase system. J Bacteriol. 2004, 186 (14): 4543-4555. 10.1128/JB.186.14.4543-4555.2004.
Schick J, Weber B, Klein JR, Henrich B: PepR1, a CcpA-like transcription regulator of Lactobacillus delbrueckii subsp. lactis. Microbiology. 1999, 145: 3147-3154.
Andersson U, Molenaar D, Rådström P, de Vos WM: Unity in organisation and regulation of catabolic operons in Lactobacillus plantarum, Lactococcus lactis and Listeria monocytogenes. Syst Appl Microbiol. 2005, 28 (3): 187-195. 10.1016/j.syapm.2004.11.004.
Pedersen H, Valentin-Hansen P: Protein-induced fit: the CRP activator protein changes sequence-specific DNA recognition by the CytR repressor, a highly flexible LacI member. EMBO J. 1997, 16 (8): 2108-2118. 10.1093/emboj/16.8.2108.
Falcon CM, Matthews KS: Operator DNA sequence variation enhances high affinity binding by hinge helix mutants of lactose repressor protein. Biochemistry. 2000, 39 (36): 11074-11083. 10.1021/bi000924z.
Lehming N, Sartorius J, Kisters-Woike B, von Wilcken-Bergmann B, Müller-Hill B: Mutant lac repressors with new specificities hint at rules for protein-DNA recognition. EMBO J. 1990, 9 (3): 615-621.
Sadler JR, Sasmor H, Betz JL: A perfectly symmetric lac operator binds the lac repressor very tightly. Proc Natl Acad Sci USA. 1983, 80 (22): 6785-6789. 10.1073/pnas.80.22.6785.
Betz JL, Sasmor HM, Buck F, Insley MY, Caruthers MH: Base substitution mutants of the lac operator: in vivo and in vitro affinities for lac repressor. Gene. 1986, 50 (1-3): 123-132. 10.1016/0378-1119(86)90317-3.
Spronk CA, Folkers GE, Noordman AM, Wechselberger R, van den Brink N, Boelens R, Kaptein R: Hinge-helix formation and DNA bending in various lac repressor-operator complexes. EMBO J. 1999, 18 (22): 6472-6480. 10.1093/emboj/18.22.6472.
Rosenfeld N, Young JW, Alon U, Swain PS, Elowitz MB: Gene regulation at the single-cell level. Science. 2005, 307 (5717): 1962-1965. 10.1126/science.1106914.
Robinow C, Kellenberger E: The bacterial nucleoid revisited. Microbiol Rev. 1994, 58 (2): 211-232.
Chauvaux S, Paulsen IT, Saier MH: CcpB, a novel transcription factor implicated in catabolite repression in Bacillus subtilis. J Bacteriol. 1998, 180 (3): 491-497.
Barkley MD, Bourgeois S: Repressor recognition of operator and effectors. The operon. Edited by: Miller JH, Reznikoff WS. 1978, Cold Spring Harbor, NY , Cold Spring Harbor Laboratory, 177-220.
Overbeek R, Larsen N, Walunas T, D'Souza M, Pusch G, Selkov E, Liolios K, Joukov V, Kaznadzey D, Anderson I, Bhattacharyya A, Burd H, Gardner W, Hanke P, Kapatral V, Mikhailova N, Vasieva O, Osterman A, Vonstein V, Fonstein M, Ivanova N, Kyrpides N: The ERGO genome analysis and discovery system. Nucleic Acids Res. 2003, 31 (1): 164-171. 10.1093/nar/gkg148.
Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Edgar R, Federhen S, Geer LY, Kapustin Y, Khovayko O, Landsman D, Lipman DJ, Madden TL, Maglott DR, Ostell J, Miller V, Pruitt KD, Schuler GD, Sequeira E, Sherry ST, Sirotkin K, Souvorov A, Starchenko G, Tatusov RL, Tatusova TA, Wagner L, Yaschenko E: Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2007, 35 (Database issue): D5-12. 10.1093/nar/gkl1031.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25 (17): 3389-3402. 10.1093/nar/25.17.3389.
Edgar RC: MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004, 5: 113-10.1186/1471-2105-5-113.
Tippmann HF: Analysis for free: comparing programs for sequence analysis. Brief Bioinform. 2004, 5 (1): 82-87. 10.1093/bib/5.1.82.
Clamp M, Cuff J, Searle SM, Barton GJ: The Jalview Java alignment editor. Bioinformatics. 2004, 20 (3): 426-427. 10.1093/bioinformatics/btg430.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG: The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25 (24): 4876-4882. 10.1093/nar/25.24.4876.
Kimura M: Estimation of evolutionary distances between homologous nucleotide sequences. Proc Natl Acad Sci USA. 1981, 78 (1): 454-458. 10.1073/pnas.78.1.454.
Bailey TL, Elkan C: Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994, 2: 28-36.
Bailey TL, Gribskov M: Combining evidence using p-values: application to sequence homology searches. Bioinformatics. 1998, 14 (1): 48-54. 10.1093/bioinformatics/14.1.48.
Finn RD, Mistry J, Schuster-Böckler B, Griffiths-Jones S, Hollich V, Lassmann T, Moxon S, Marshall M, Khanna A, Durbin R, Eddy SR, Sonnhammer EL, Bateman A: Pfam: clans, web tools and services. Nucleic Acids Res. 2006, 34 (Database issue): D247-251. 10.1093/nar/gkj149.
Tojo S, Satomura T, Morisaki K, Deutscher J, Hirooka K, Fujita Y: Elaborate transcription regulation of the Bacillus subtilis ilv-leu operon involved in the biosynthesis of branched-chain amino acids through global regulators of CcpA, CodY and TnrA. Mol Microbiol. 2005, 56 (6): 1560-1573.
Wolf M, Müller T, Dandekar T, Pollack JD: Phylogeny of Firmicutes with special reference to Mycoplasma (Mollicutes) as inferred from phosphoglycerate kinase amino acid sequence data. Int J Syst Evol Microbiol. 2004, 54 (Pt 3): 871-875. 10.1099/ijs.0.02868-0.
Crooks GE, Hon G, Chandonia JM, Brenner SE: WebLogo: a sequence logo generator. Genome Res. 2004, 14 (6): 1188-1190. 10.1101/gr.849004.
Andersson U, Rådström P: Physiological function of the maltose operon regulator, MalR, in Lactococcus lactis. BMC Microbiol. 2002, 2: 28-10.1186/1471-2180-2-28.
Daniel RA, Haiech J, Denizot F, Errington J: Isolation and characterization of the lacA gene encoding beta-galactosidase in Bacillus subtilis and a regulator gene, lacR. J Bacteriol. 1997, 179 (17): 5636-5638.
Inaoka T, Takahashi K, Yada H, Yoshida M, Ochi K: RNA polymerase mutation activates the production of a dormant antibiotic 3,3'-neotrehalosadiamine via an autoinduction mechanism in Bacillus subtilis. J Biol Chem. 2004, 279 (5): 3885-3892. 10.1074/jbc.M309925200.
Jobe A, Bourgeois S: lac Repressor-operator interaction. VI. The natural inducer of the lac operon. J Mol Biol. 1972, 69 (3): 397-408. 10.1016/0022-2836(72)90253-7.
Müller W, Horstmann N, Hillen W, Sticht H: The transcription regulator RbsR represents a novel interaction partner of the phosphoprotein HPr-Ser46-P in Bacillus subtilis. FEBS J. 2006, 273 (6): 1251-1261. 10.1111/j.1742-4658.2006.05148.x.
Nieto C, Espinosa M, Puyet A: The maltose/maltodextrin regulon of Streptococcus pneumoniae. Differential promoter regulation by the transcriptional repressor MalR. J Biol Chem. 1997, 272 (49): 30860-30865. 10.1074/jbc.272.49.30860.
Schönert S, Seitz S, Krafft H, Feuerbaum EA, Andernach I, Witz G, Dahl MK: Maltose and maltodextrin utilization by Bacillus subtilis. J Bacteriol. 2006, 188 (11): 3911-3922. 10.1128/JB.00213-06.
Silvestroni A, Connes C, Sesma F, Savoy de Giori G, Piard JC: Characterization of the melA locus for alpha-galactosidase in Lactobacillus plantarum. Appl Environ Microbiol. 2002, 68 (11): 5464-5471.
Stentz R, Zagorec M: Ribose utilization in Lactobacillus sakei: analysis of the regulation of the rbs operon and putative involvement of a new transporter. J Mol Microbiol Biotechnol. 1999, 1 (1): 165-173.
Woodson K, Devine KM: Analysis of a ribose transport operon from Bacillus subtilis. Microbiology. 1994, 140 ( Pt 8): 1829-1838.
Marasco R, Muscariello L, Rigano M, Sacco M: Mutational analysis of the bglH catabolite-responsive element (cre) in Lactobacillus plantarum. FEMS Microbiol Lett. 2002, 208 (1): 143-146. 10.1111/j.1574-6968.2002.tb11074.x.
NC-IUB: Nomenclature for incompletely specified bases in nucleic acid sequences. Recommendations 1984. Eur J Biochem. 1985, 150 (1): 1-5. 10.1111/j.1432-1033.1985.tb08977.x.
Rodionov DA, Mironov AA, Gelfand MS: Transcriptional regulation of pentose utilisation systems in the Bacillus/Clostridium group of bacteria. FEMS Microbiol Lett. 2001, 205 (2): 305-314. 10.1111/j.1574-6968.2001.tb10965.x.
Acknowledgements
RK acknowledges the support from the Netherlands Ministry of Economic Affairs via the IOP Program, grant IGE01018, and CF the support of NBIC/the Netherlands Genomics Initiative via the Kluyver Centre for Genomics of Industrial Fermentations and the BioRange program.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
CF conceived, designed and coordinated the study, carried out the motif and functional analyses and drafted and revised the manuscript; RK conceived and designed the study, carried out the motif analysis, wrote the scripts to convert MEME and MAST output into tabular format and helped revising the manuscript; MW carried out and helped interpret the genome-wide motif searches and helped revising the manuscript; RJS conceived and coordinated the study and helped drafting and revising the manuscript. All authors have read and approved the final manuscript.
Christof Francke, Robert Kerkhoven contributed equally to this work.
Electronic supplementary material
12864_2007_1339_MOESM1_ESM.xls
Additional file 1: The number of LacI-family members in sequenced Firmicutes. The file lists the species whose genome was studied, their abbreviation and the number of LacI-family TFs recovered for each genome. (XLS 59 KB)
12864_2007_1339_MOESM2_ESM.xls
Additional file 2: IDs and sequences of LacI-family TFs in sequenced Firmicutes. The file contains the sequences that were considered in this study with their different IDs. (XLS 448 KB)
12864_2007_1339_MOESM3_ESM.aln
Additional file 3: Multiple sequence alignment of LacI-family TFs of sequenced Firmicutes. The file gives the sequence alignment of the TFs considered in this study. (ALN 684 KB)
12864_2007_1339_MOESM4_ESM.phb
Additional file 4: Neighbor Joining tree for the LacI-family TFs of sequenced Firmicutes. The file gives the bootstrapped (n = 250) NJ-tree for the LacI-family TF homologs of the Firmicutes, Salmonella and E. coli. (PHB 28 KB)
12864_2007_1339_MOESM5_ESM.ppt
Additional file 5: Multiple sequence alignments and Neighbor joining trees for the two functional domains of the LacI-family TF homologs in L. plantarum. The file contains images of the sequence alignments and the bootstrapped (n = 1000) NJ-trees for the two TF functional domains. (PPT 94 KB)
12864_2007_1339_MOESM6_ESM.ppt
Additional file 6: Gene context conservation of the LacI-family TF homologs in L. plantarum WCFS1. The file provides a visualization of context information that was used to define the GOOFEs and contains the motifs used to perform the MAST searches. (PPT 3 MB)
12864_2007_1339_MOESM7_ESM.xls
Additional file 7: LacI-family homologs deviant at position 24 and their putative operators. The file lists the putative binding motifs for several LacITFs with a conserved substitution of the conserved arginine at position 24 (in Figure 2). (XLS 88 KB)
12864_2007_1339_MOESM8_ESM.xls
Additional file 8: MAST output. The file contains the parsed output of the MAST searches. A new sheet is provided for each criterion that was applied to constrain the list. The results are color-coded. (XLS 1 MB)
12864_2007_1339_MOESM9_ESM.rtf
Additional file 9: The functional annotation of the genes and operons regulated by LacI-TFs in L. plantarum. The file contains a functional description of the genes and operons that putatively constitute the minimal regulons depicted in Figure 3 (with relevant references). (RTF 101 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Francke, C., Kerkhoven, R., Wels, M. et al. A generic approach to identify Transcription Factor-specific operator motifs; Inferences for LacI-family mediated regulation in Lactobacillus plantarum WCFS1. BMC Genomics 9, 145 (2008). https://doi.org/10.1186/1471-2164-9-145
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-9-145